AST-120 Reduces Neuroinflammation Induced by Indoxyl Sulfate in Glial Cells

 , ,
, ,
Abstract
:1. Introduction
2. Experimental Section
2.1. Reagents
2.2. In Vitro Studies on Glioma Cell Line (C6)
Glioma Cell Line (C6)
2.3. Studies on the Effect of Sera of CKD Patients on C6 Astrocytes
2.3.1. C6 Cells Treatment with Sera of CKD Patients
2.3.2. ROS Evaluation in C6 Cells
2.3.3. NO Determination in C6 Cells
2.3.4. TNF-α Determination in C6 Cells
2.4. Ex Vivo Studies on Primary Astocytes, Glial Cells
2.4.1. Primary Astrocytes and Glial Cells
2.4.2. Primary Astrocytes, Glial Cells Treatment
2.4.3. NO, TNF-α and IL-6, ROS Determination in Primary Astrocytes, Glial Cells
2.4.4. iNOS, COX-2, Nitrotyrosine and HO-1 Detection in Primary Astrocytes, Glial Cells
2.4.5. Immunofluorescence Analysis with Confocal Microscopy for Primary Astrocytes and Mixed Glial Cells
2.5. Ex Vivo Studies on Primary Cortical and Hippocampal Neuronal Cultures
2.5.1. Primary Neurons
2.5.2. Primary Cortical and Hippocampal Neuronal Cells Treatment
2.5.3. Cytotoxicity Assay on Primary Cortical and Hippocampal Neuronal Cultures
2.6. Data Analysis
3. Results
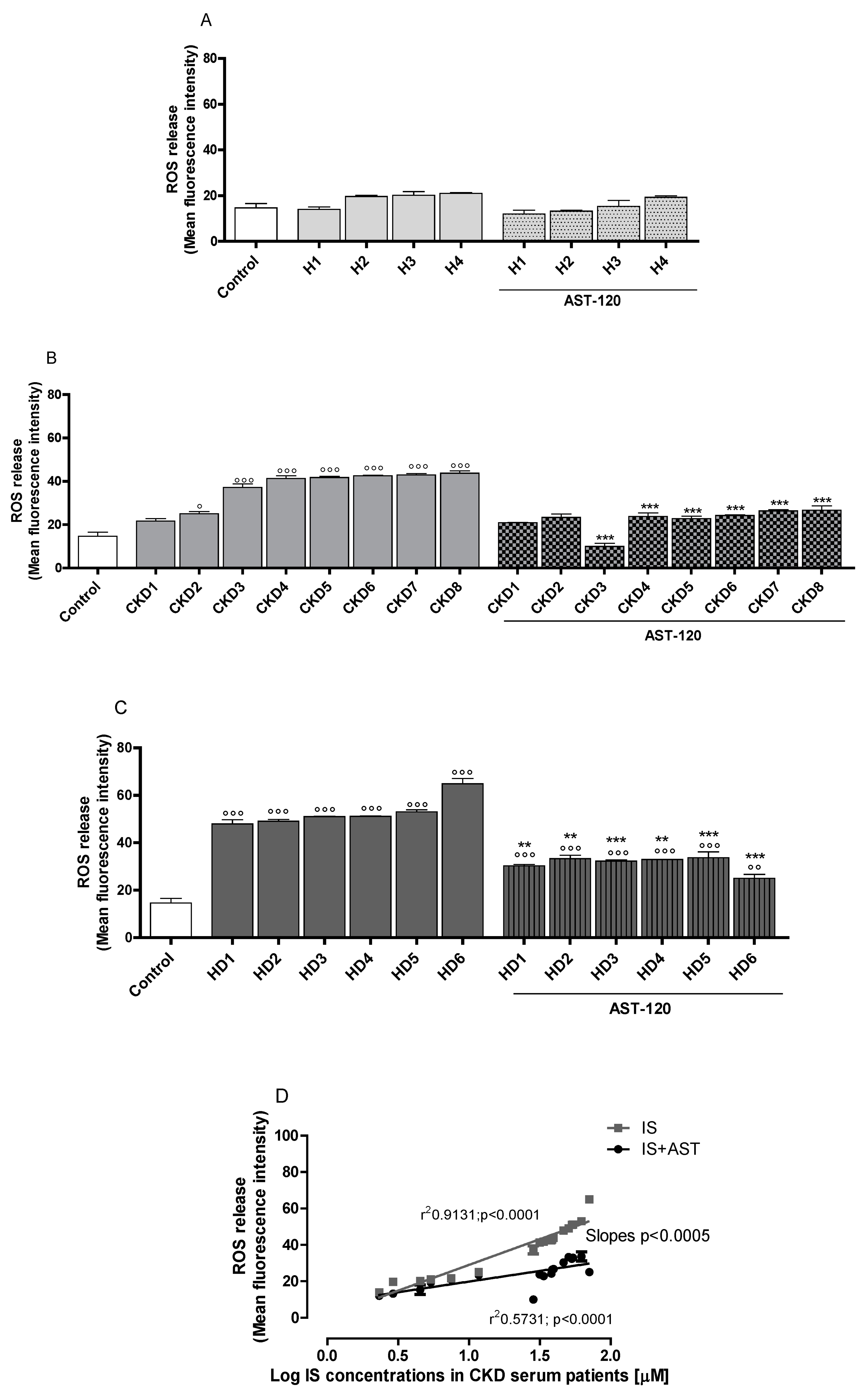
3.1. Sera of CDK Patients Increased ROS, Nitrite and TNF-α Production in a IS-Concentration Related Manner
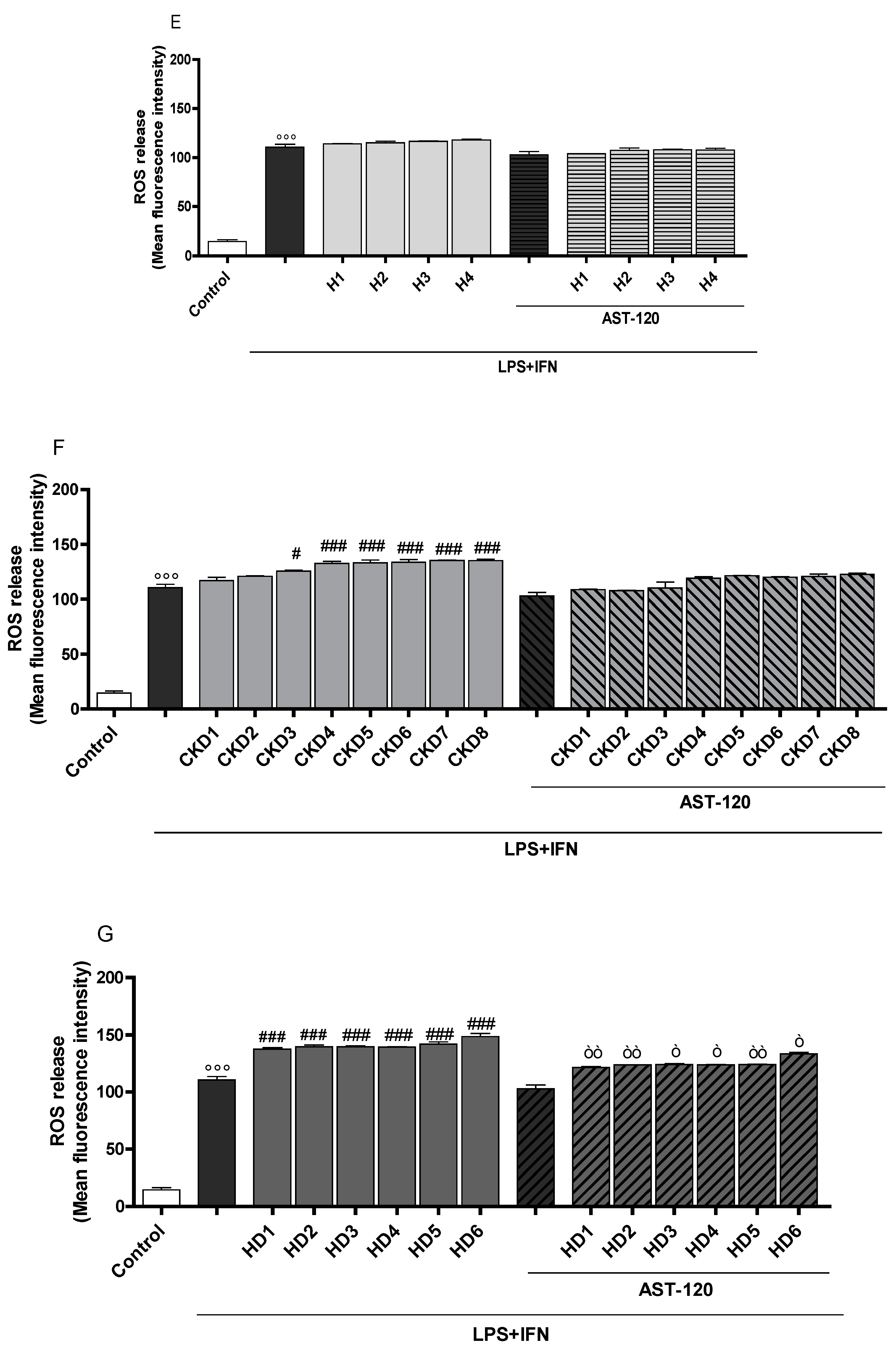
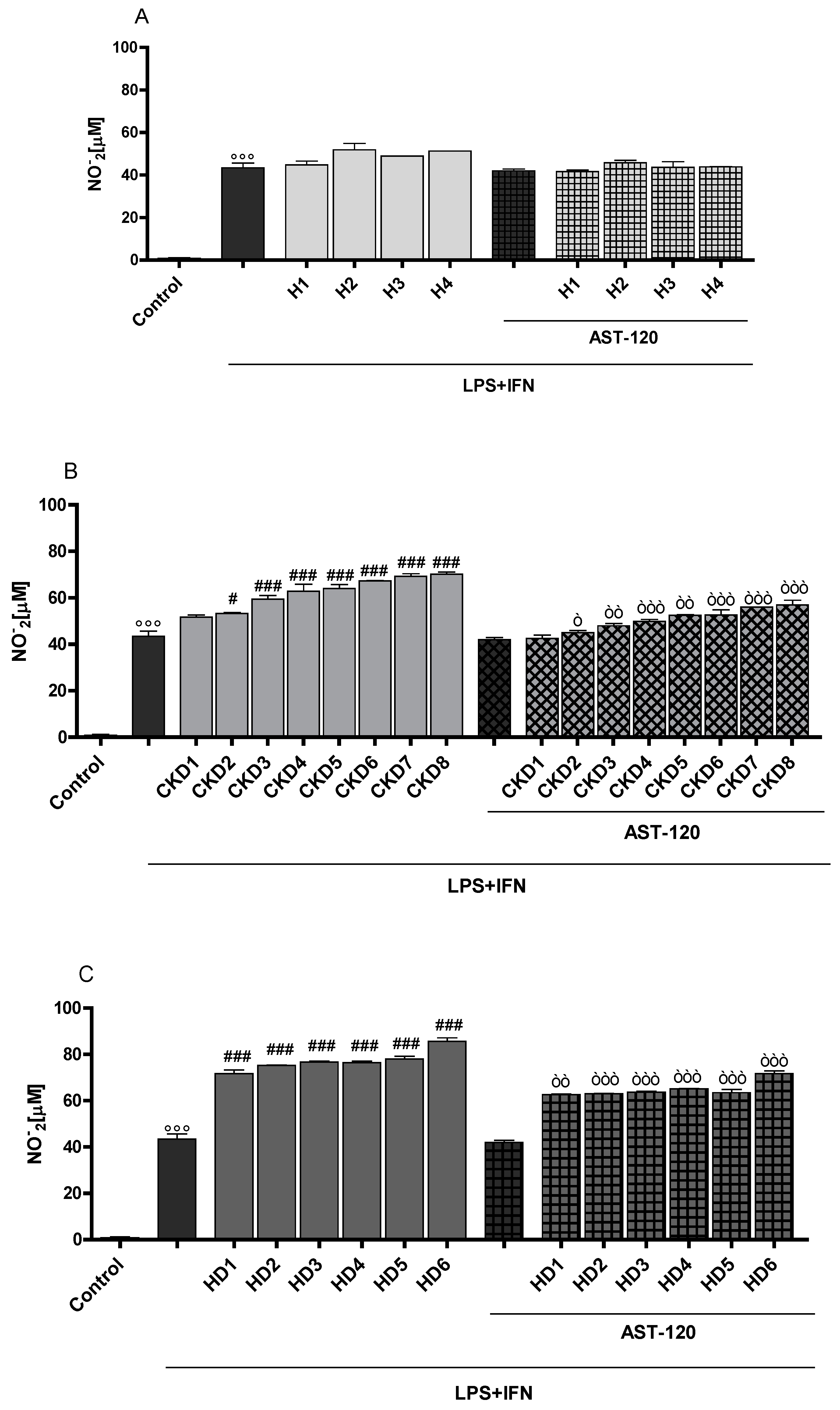
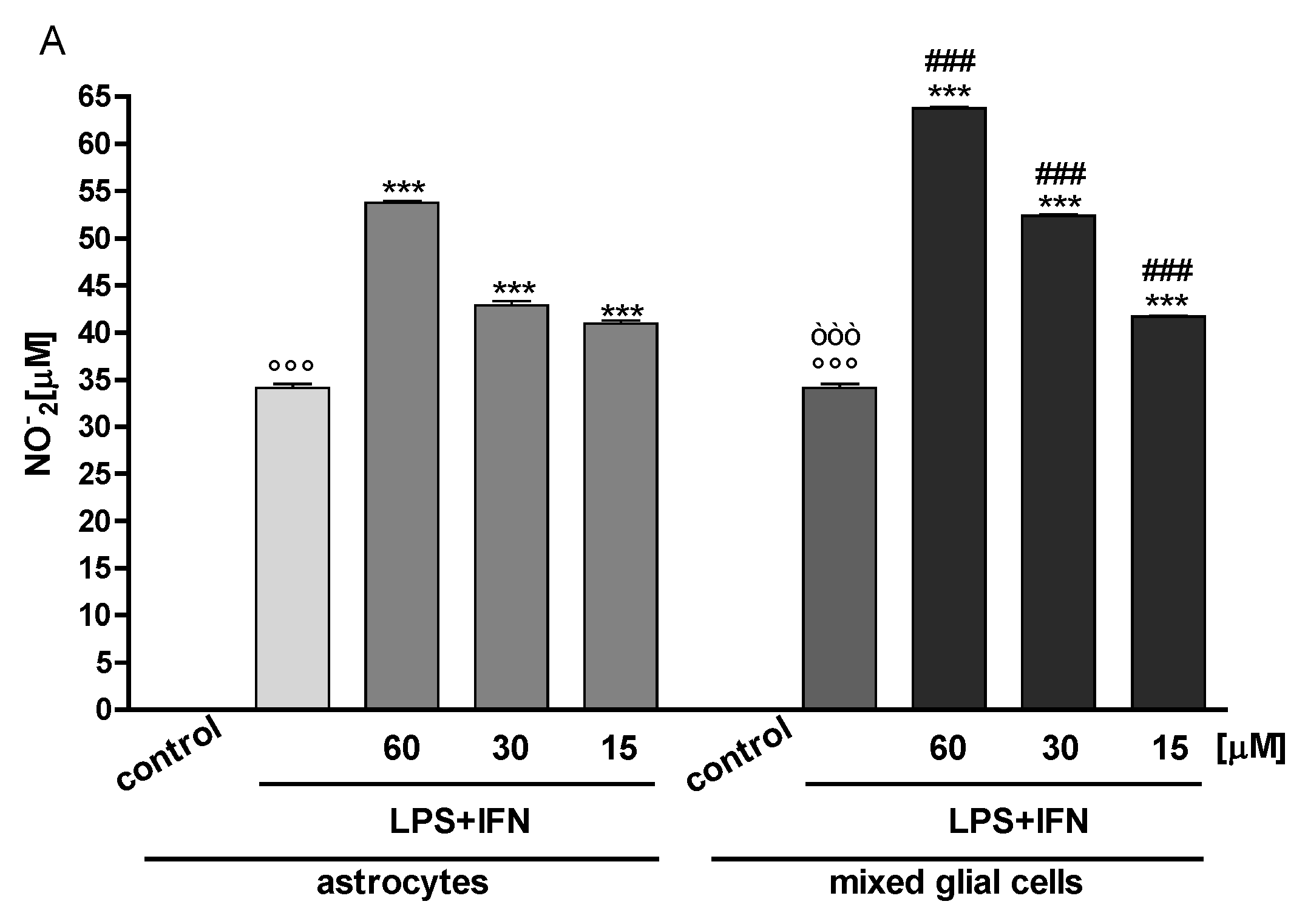
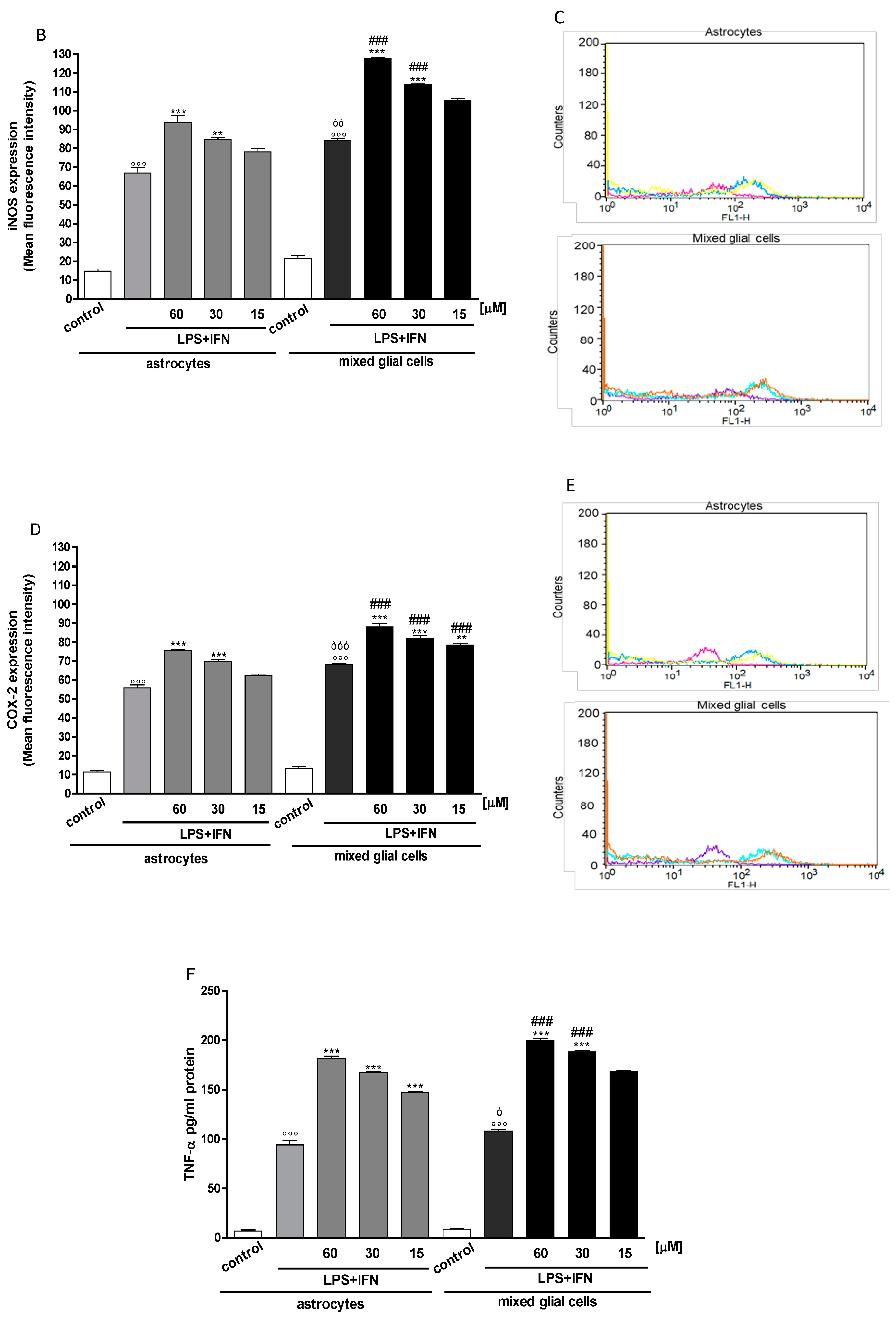
3.2. Indoxyl Sulphate Enhanced NO Release, iNOS and COX-2 Expression in Inflammatory Conditions in CNS Cells
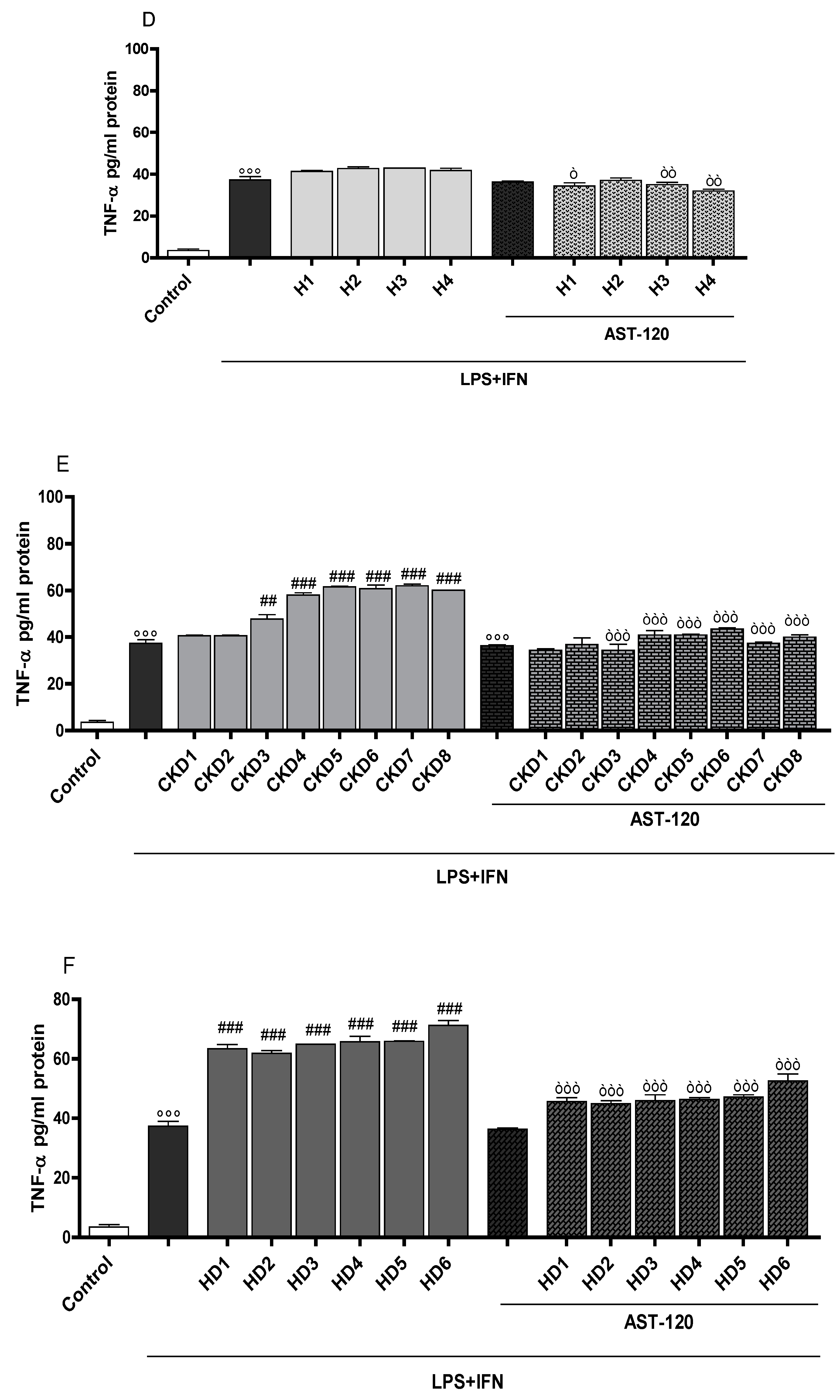
3.3. Indoxyl Sulphate Enhanced TNF-α, IL-6 Production in Inflammatory Conditions in CNS Cells
3.4. Indoxyl Sulphate Enhanced Nitrotyrosine Formation in Inflammatory Conditions in CNS Cells
3.5. Indoxyl Sulphate Enhanced ROS Release and HO-1 Expression in Inflammatory Conditions in CNS Cells
3.6. Indoxyl Sulphate Supernatant from Glial Cells and Increased Neuronal Death
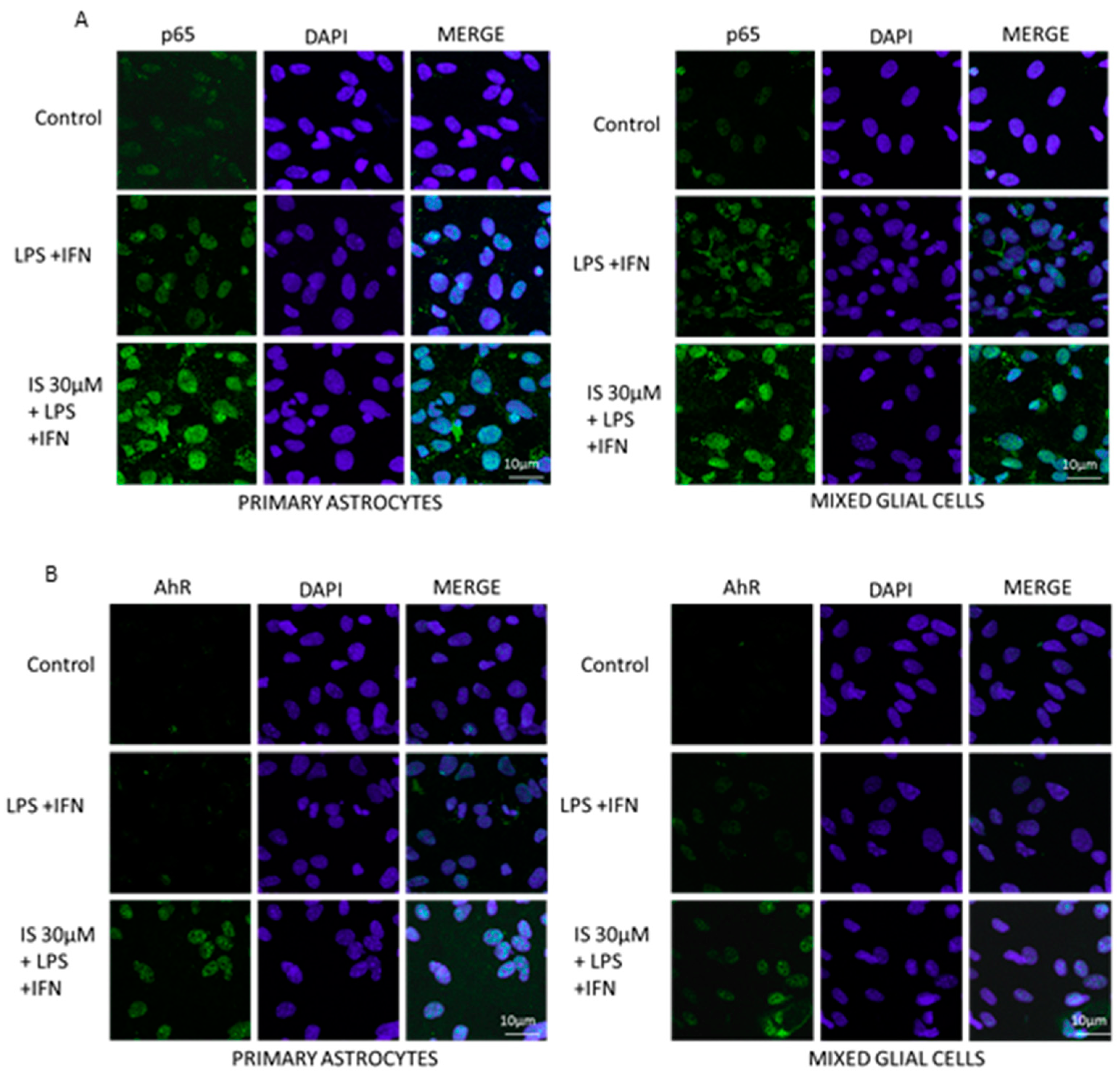
3.7. Indoxyl Sulphate Enhanced NF-κB p65 Nuclear Translocation and AhR Activation, Induced by LPS + IFN in CNS Cultures
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.L.; Yang, C.M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. BioMed Res. Int. 2013, 2013, 484613. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Kiernan, M.C. Neurological complications of chronic kidney disease. Nat. Rev. Neurol. 2009, 5, 542–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchman, A.S.; Tanne, D.; Boyle, P.A.; Shah, R.C.; Leurgans, S.E.; Bennett, D.A. Kidney function is associated with the rate of cognitive decline in the elderly. Neurology 2009, 73, 920–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl Sulfate Affects Glial Function Increasing Oxidative Stress and Neuroinflammation in Chronic Kidney Disease: Interaction between Astrocytes and Microglia. Front Pharmacol. 2017, 8, 370. [Google Scholar] [CrossRef] [PubMed]
- Kurella, M.; Chertow, G.M.; Luan, J.; Yaffe, K. Cognitive impairment in chronic kidney disease. J. Am. Geriatr. Soc. 2004, 52, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Thornton, W.L.; Shapiro, R.J.; Deria, S.; Gelb, S.; Hill, A. Differential impact of age on verbal memory and executive functioning in chronic kidney disease. J. Int. Neuropsychol. Soc. 2007, 13, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Nulsen, R.S.; Yaqoob, M.M.; Mahon, A.; Stoby-Fields, M.; Kelly, M.; Varagunam, M. Prevalence of cognitive impairment in patients attending pre-dialysis clinic. J. Ren. Care 2008, 34, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.M.; Tupper, D.E.; Knopman, D.S.; Gilbertson, D.T.; Pederson, S.L.; Li, S.; Smith, G.E.; Hochhalter, A.K.; Collins, A.J.; Kane, R.L. Cognitive impairment in hemodialysis patients is common. Neurology 2006, 67, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.M.; Kielstein, J.T.; Braverman, J.; Novak, M. Cognitive Function in Patients with Chronic Kidney Disease: Challenges in Neuropsychological Assessments. Semin. Nephrol. 2015, 35, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Ren. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aveles, P.R.; Criminacio, C.R.; Goncalves, S.; Bignelli, A.T.; Claro, L.M.; Siqueira, S.S.; Nakao, L.S.; Pecoits-Filho, R. Association between biomarkers of carbonyl stress with increased systemic inflammatory response in different stages of chronic kidney disease and after renal transplantation. Nephron Clin. Pract. 2010, 116, c294–c299. [Google Scholar] [CrossRef] [PubMed]
- Prats, M.; Font, R.; Garcia, C.; Munoz-Cortes, M.; Cabre, C.; Jariod, M.; Romeu, M.; Giralt, M.; Martinez- Vea, A. Oxidative stress markers in predicting response to treatment with ferric carboxymaltose in nondialysis chronic kidney disease patients. Clin. Nephrol. 2014, 81, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Akchurin, O.M.; Kaskel, F. Update on inflammation in chronic kidney disease. Blood Purif. 2015, 39, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Smith, G.E.; Waring, S.C.; Ivnik, R.J.; Kokmen, E.; Tangelos, E.G. Aging, memory, and mild cognitive impairment. Int. Psychogeriatr. 1997, 9, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Trollor, J.; Agars, E. Systemic inflammation andcognition in the Elderly. In Neuropsychiatric Disorders; Springer: Tokyo, Japan, 2010; pp. 177–198. [Google Scholar]
- Montinaro, V.; Iaffaldano, G.P.; Granata, S.; Porcelli, P.; Todarello, O.; Schena, F.P.; Pertosa, G. Emotional symptoms, quality of life and cytokine profile in hemodialysis patients. Clin. Nephrol. 2010, 73, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Cunnigham, C.; Hennesy, E. Co-morbidity and systemic inflammation as drivers of cognitive decline: New experimental models adopting a broader paradigm in dementia research. Alzheimer’s Res. Ther. 2015, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. European Uremic Toxin Work Group (EUTox): Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Bugnicourt, J.M.; Godefroy, O.; Chillon, J.C.; Choukroun, G.; Massy, Z.A. Cognitive disorders and dementia in CKD: The neglected kidney-brain axis. J. Am. Soc. Nephrol. 2013, 24, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T. Indoxyl sulfate is a nephro-vascular toxin. J. Ren. Nutr. 2010, 20, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adesso, S.; Popolo, A.; Bianco, G.; Sorrentino, R.; Pinto, A.; Autore, G.; Marzocco, S. The uremic toxin indoxyl sulphate enhances macrophage response to LPS. PLoS ONE 2013, 8, e76778. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Niwa, T. Roles of organic anion transporters in the progression of chronic renal failure. Ther. Apher. Dial. 2007, 11, S27–S31. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Asaba, H.; Takanaga, H.; Deguchi, T.; Hosoya, K.; Otagiri, M.; Terasaki, T. Role of blood-brain barrier organic anion transporter 3 (OAT3) in the efflux of indoxyl sulfate, a uremic toxin: Its involvement in neurotransmitter metabolite clearance from the brain. J. Neurochem. 2002, 83, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Watanabe, T.; Nakayama, M. Cerebro-renal interactions: Impact of uremic toxins on cognitive function. Neurotoxicology 2014, 44, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Benda, P.; Lightbody, J.; Sato, G.; Levine, L.; Sweet, W. Differentiated rat glial cell strain in tissue culture. Science 1968, 161, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Quincozes-Santos, A.; Nardin, P.; de Souza, D.F.; Gelain, D.P.; Moreira, J.C.; Latini, A.; Gonçalves, C.A.; Gottfried, C. The janus face of resveratrol in astroglial cells. Neurotox. Res. 2009, 16, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.K.; Norenberg, M.D.; Vernadakis, A. “Transdifferentiation’’ of C6 glial cells in culture. Science 1980, 208, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Dal Piaz, F.; Di Micco, L.; Torraca, S.; Sirico, M.L.; Tartaglia, D.; Autore, G.; Di Iorio, B. Very low protein diet reduces indoxyl sulfate levels in chronic kidney disease. Blood Purif. 2013, 35, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.; Di Micco, L.; Torraca, S.; Sirico, M.L.; Russo, L.; Pota, A.; Mirenghi, F.; Russo, D. Acute effects of very-low-protein diet on FGF23 levels: A randomized study. Clin. J. Am. Soc. Nephrol. 2012, 7, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Fazeli, G.; Di Micco, L.; Autore, G.; Adesso, S.; Dal Piaz, F.; Heidland, A.; Di Iorio, B. Supplementation of Short-Chain Fatty Acid, Sodium Propionate, in Patients on Maintenance Hemodialysis: Beneficial Effects on Inflammatory Parameters and Gut-Derived Uremic Toxins, A Pilot Study (PLAN Study). J. Clin. Med. 2018, 7, 315. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.; Marzocco, S.; Bellasi, A.; De Simone, E.; Dal Piaz, F.; Rocchetti, M.T.; Cosola, C.; Di Micco, L.; Gesualdo, L. Nutritional therapy reduces protein carbamylation through urea lowering in chronic kidney disease. Nephrol. Dial. Transplant. 2018, 33, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Luckman, S.P.; Skeie, G.O.; Helgeland, G.; Gilhus, N.E. Effects of myasthenia gravis patient sera on human myoblast cultures. Acta Neurol. Scand. Suppl. 2006, 183, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Dal Toso, R.; Pressi, G.; Bramanti, P.; Meli, R.; Cuzzocrea, S. Protective effect of verbascoside in activated C6 glioma cells: Possible molecular mechanisms. Naunyn Schmiedebergs Arch. Pharmacol. 2010, 381, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Adesso, S.; Alilou, M.; Stuppner, H.; Schwaiger, S. Anti-Inflammatory and Anti-Oxidant Potential of the Root Extract and Constituents of Doronicum austriacum. Molecules 2017, 22, 1003. [Google Scholar] [CrossRef] [PubMed]
- Shimoishi, K.; Anraku, M.; Kitamura, K.; Tasaki, Y.; Taguchi, K.; Hashimoto, M.; Fukunaga, E.; Maruyama, T.; Otagiri, M. An oral adsorbent, AST-120 protects against the progression of oxidative stress by reducing the accumulation of indoxyl sulfate in the systemic circulation in renal failure. Pharm. Res. 2007, 24, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Popolo, A.; Bianco, G.; Pinto, A.; Autore, G. Pro-apoptotic effect of methylguanidine on hydrogen peroxide-treated rat glioma cell line. Neurochem. Int. 2010, 57, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Weymar, A.; Bernreuther, C.; Velden, J.; Arunachalam, P.; Steinbach, K.; Orthey, E.; Arumugam, T.V.; Leypoldt, F.; Simova, O.; et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 2012, 120, 3793–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinelli, C.; Di Liddo, R.; Facci, L.; Bertalot, T.; Conconi, M.T.; Zusso, M.; Skaper, S.D.; Giusti, P. Ligand engagement of Toll-like receptors regulates their expression in cortical microglia and astrocytes. J. Neuroinflamm. 2015, 12, 244. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G.J. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Artieda, J.; Muruzabal, J.; Larumbe, R.; Garcia de Casasola, C.; Obeso, J.A. Cortical mechanisms mediating asterixis. Mov. Disord. 1992, 7, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, K.; Tachikawa, M. Roles of organic anion/cation transporters at the blood-brain and blood-cerebrospinal fluid barriers involving uremic toxins. Clin. Exp. Nephrol. 2011, 15, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Autore, G.; Quaroni, A.; Popolo, A.; Severino, L.; Marzocco, S. The Food Contaminants Nivalenol and Deoxynivalenol Induce Inflammation in Intestinal Epithelial Cells by Regulating Reactive Oxygen Species Release. Nutrients 2017, 9, 1343. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.; Lee, S.Y.; Manzanero, S.; Tang, S.C.; Gelderblom, M.; Chunduri, P.; Bernreuther, C.; Glatzel, M.; Cheng, Y.L.; Thundyil, J.; et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013, 4, e790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struzynska, L.; Dabrowska-Bouta, B.; Koza, K.; Sulkowski, G. Inflammation-like glial response in lead-exposed immature rat brain. Toxicol. Sci. 2007, 95, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Liu, X.Q.; Wang, W.; Shen, X.F.; Che, H.L.; Guo, Y.Y.; Zhao, M.G.; Chen, J.Y.; Luo, W.J. Involvement of microglia activation in the lead induced long-term potentiation impairment. PLoS ONE 2012, 7, e43924. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 7, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.C.; Dinatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Morena, M.; Cristol, J.P.; Senécal, L.; Leray-Moragues, H.; Krieter, D.; Canaud, B. Oxidative stress in hemodialysis patients: Is NADPH oxidase complex the culprit? Kidney Int. 2002, 61, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B. Cross-talk signals in the CNS: Role of neurotrophic and hormonal factors, adhesion molecules and intercellular signaling agents in luteinizing hormone-releasing hormone (LHRH)-astroglial interactive network. Front Biosci. 1997, 2, d88–d125. [Google Scholar] [CrossRef] [PubMed]
- Vernadakis, A. Glia-neuron intercommunications and synaptic plasticity. Prog. Neurobiol. 1996, 49, 185–214. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Olabarria, M.; Chvatal, A.; Verkhratsky, A. Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 2009, 16, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Erol, A. Are paradoxical cell cycle activities in neurons and glia related to the metabolic theory of Alzheimer’s disease? J. Alzheimers Dis. 2010, 19, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, M. A role for guanidino compounds in the brain. Mol. Cell Biochem. 2003, 244, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Marescau, B.; De Deyn, P.; Wiechert, P.; Van Gorp, L.; Lowenthal, A. Comparative study of guanidino compounds in serum and brain of mouse, rat, rabbit, and man. J. Neurochem. 1986, 46, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Marescau, B.; Deshmukh, D.R.; Kockx, M.; Possemiers, I.; Qureshi, I.A.; Wiechert, P.; De Deyn, P.P. Guanidino compounds in serum, urine, liver, kidney, and brain of man and some ureotelic animals. Metabolism 1992, 41, 526–532. [Google Scholar] [CrossRef]
- Mori, A. Biochemistry and neurotoxicology of guanidino compounds. Hist. Recent Adv. Pay. J. Biol. Sci. 1987, 22, 85–94. [Google Scholar]
- Álvarez-Lario, B.; Macarrón-Vicente, J. Is there anything good in uric acid? QJM 2011, 104, 1015–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindermayr, C.; Durner, J. Interplay of reactive oxygen species and nitric oxide: Nitric oxide coordinates reactive oxygen species homeostasis. Plant Physiol. 2015, 167, 1209–1210. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Vanholder, R.; Niwa, T. AST-120 for the management of progression of chronic kidney disease. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T. The role of the nitric oxide pathway in brain injury and its treatment—From bench to bedside. Exp. Neurol. 2014, 263, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.; Segura, T.; Brea, D.; Galindo, M.F.; Castillo, J. Inflammation as therapeutic objective in stroke. Curr. Pharm. Des. 2008, 14, 3549–3564. [Google Scholar] [CrossRef] [PubMed]
- Samdani, A.F.; Dawson, T.M.; Dawson, V.L. Nitric oxide synthase in models of focal ischemia. Stroke 1997, 28, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Galea, E.; Feinstein, D.L.; Reis, D.J. Induction of calcium-independent nitric oxide synthase activity in primary rat glial cultures. Proc. Natl. Acad. Sci. USA 1992, 89, 10945–10949. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Pahan, K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid. Redox. Signal 2006, 8, 929–947. [Google Scholar] [CrossRef] [PubMed]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Olmos, G.; Lladó, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Mozar, A.; Louvet, L.; Morlière, P.; Godin, C.; Boudot, C.; Kamel, S.; Drüeke, T.B.; Massy, Z.A. Uremic toxin indoxyl sulfate inhibits human vascular smooth muscle cell proliferation. Ther. Apher. Dial. 2011, 15, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Yisireyili, M.; Higashiyama, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate upregulates renal expression of ICAM-1 via production of ROS and activation of NF-κB and p53 in proximal tubular cells. Life Sci. 2013, 92, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Varley, J.; Brooks, D.J.; Edison, P. Imaging neuroinflammation in Alzheimer’s disease and other dementias: Recent advances and future directions. Alzheimer’s Dement. 2015, 11, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- D’Amelio, M.; Cavallucci, V.; Cecconi, F. Neuronal caspase-3 signaling: Not only cell death. Cell Death Differ. 2010, 17, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulphate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef] [PubMed]
- Masai, N.; Tatebe, J.; Yoshino, G.; Morita, T. Indoxyl sulfate stimulates monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells by inducing oxidative stress through activation of the NADPH oxidase-nuclear factor-κB pathway. Circ. J. 2010, 74, 2216–2224. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; Descamps-Latscha, B.; Henle, T.; et al. Uremic toxicity: Present state of the art. Int. J. Artif. Organs 2001, 24, 695–725. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Juan, S.H.; Wang, C.Y.; Sun, Y.Y.; Chou, C.M.; Chang, S.F.; Hu, S.Y.; Lee, W.S.; Lee, Y.H. Neuronal activity enhances aryl hydrocarbon receptor-mediated gene expression and dioxin neurotoxicity in cortical neurons. J. Neurochem. 2008, 104, 1415–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Yang, J.H. Neurotoxic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in cerebellar granule cells. Exp. Mol. Med. 2005, 37, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, M.A.; Gasiewicz, T.A.; Opanashuk, L.A. Aryl hydrocarbon receptorexpression and activity in cerebellar granule neuroblasts: Implications for development and dioxin neurotoxicity. Toxicol. Sci. 2005, 83, 340–348. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy Subjects (H) | CKD Patients (CKD) | CKD Dialysed Patients (HD) | |
|---|---|---|---|
| Number | 4 | 8 | 6 |
| Males | 2 | 5 | 3 |
| Age, years | 51.0 ± 5.8 | 58.9 ± 12.4 | 66.4 ± 5.5 |
| Body Weight, kg | 77.6 ± 7.9 | 75.2 ± 12.2 | 73.5 ± 16.3 |
| Creatinine, mg/dL | 0.9 ± 0.2 | 2.1 ± 0.5 | 9.8 ± 2.1 |
| Creatinine Clearance | 115.0 ± 11.0 | 37.0 ± 9.0 | Not Applicable |
| Sodium, mmol/L | 141.0 ± 4.0 | 139.0 ± 3.0 | 142.0 ± 6.0 |
| Kalium, mmol/L | 4.5 ± 0.7 | 4.8 ± 0.6 | 4.9 ± 0.9 |
| Haemoglobin, g/dL | 13.6 ± 0.4 | 11.2 ± 0.5 | 10.9 ± 0.6 |
| Albumin, g/dL | 4.2 ± 0.1 | 3.6 ± 0.23 | 3.5 ± 0.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adesso, S.; Paterniti, I.; Cuzzocrea, S.; Fujioka, M.; Autore, G.; Magnus, T.; Pinto, A.; Marzocco, S. AST-120 Reduces Neuroinflammation Induced by Indoxyl Sulfate in Glial Cells. J. Clin. Med. 2018, 7, 365. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm7100365
Adesso S, Paterniti I, Cuzzocrea S, Fujioka M, Autore G, Magnus T, Pinto A, Marzocco S. AST-120 Reduces Neuroinflammation Induced by Indoxyl Sulfate in Glial Cells. Journal of Clinical Medicine. 2018; 7(10):365. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm7100365
Chicago/Turabian StyleAdesso, Simona, Irene Paterniti, Salvatore Cuzzocrea, Masaki Fujioka, Giuseppina Autore, Tim Magnus, Aldo Pinto, and Stefania Marzocco. 2018. "AST-120 Reduces Neuroinflammation Induced by Indoxyl Sulfate in Glial Cells" Journal of Clinical Medicine 7, no. 10: 365. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm7100365