1. Introduction
Helicobacter pylori is a gram-negative bacterium that infects about half of the world’s human population. Unless treated, colonization in the human gastrointestinal tract persists lifelong.
H. pylori infection represents a key factor in the aetiology of various gastroduodenal diseases, including chronic active gastritis, peptic or duodenal ulcers, gastric adenocarcinoma, and mucosa associated tissue lymphoma [
1,
2,
3]. The current first-choice empiric regimen for
H. pylori eradication in countries with clarithromycin resistance <15% consists of a clarithromycin-based triple therapy with a proton pump inhibitor (PPI) in combination with metronidazole or amoxicillin [
4,
5]. However, in the last years, the effectiveness of this empiric first-line therapy has been steadily reduced due to increasing clarithromycin and/or metronidazole resistance in
H. pylori [
6,
7,
8]. Alternatively, non-bismuth or bismuth containing quadruple therapy combined with tetracycline, levofloxacin, or rifabutin-based antibiotic regimens can be administered to patients [
5,
9]. However, the increase in quinolone resistant
H. pylori strains [
10,
11], and the emergence of quadruple-resistant
H.
pylori clinical isolates in Western Europe [
8,
12] emphasize the need for more rapid and cost effective molecular methods that enable reliable prediction of antibiotic resistance phenotypes prior to the administration of antimicrobial therapy.
Diagnosis of
H. pylori infection and determination of the pathogen’s antibiotic susceptibility mainly relies on bacterial culture and phenotypic drug susceptibility testing (DST), typically delivering results within two weeks. However, visual inspection of minimum inhibitory concentrations (MICs) on the E-Test
® strips leaves great scope for personal interpretation and potentially leads to high inter-observer variability due to the fastidious growth of
H. pylori [
13].
Recently, whole-genome sequencing (WGS) from cultured bacterial isolates has emerged as an important tool for surveillance and antibiotic resistance control. This primarily owes to improvements in sequencing technologies, affordable instrument pricing, user friendly and simple workflows requiring little hands on time, availability of standardized protocols and reagents, and reasonable per sample costs (150 EUR per 5 MB genome in the case of the Illumina MiSeq), and are hence an attractive choice as a diagnostic tool to detect drug resistance in diagnostic microbiology laboratories. Within a clinically relevant timeframe (24 to 48 h), WGS provides a comprehensive view of the genotype of a bacterial isolate [
14,
15]. In principle, the genome sequence contains all the information required to determine the antibiotic resistance phenotype. The biggest challenge, however, lies in the identification of high-confidence single nucleotide polymorphisms (SNPs) associated with resistance against a certain drug. Genotype-based prediction of phenotypic resistance is less complex for
H. pylori than for other bacteria as antibiotic resistance is primarily based on point mutations in the genome and resistance determinants in
H. pylori seem not to be encoded on plasmids, transposons, or integrons [
16].
In detail, clarithromycin resistance in
H. pylori has been associated with point mutations in domain V of the 23S rRNA gene, namely at the nucleotide position, A2146 and A2147 (positions according to
H. pylori reference strain 26695; correspond to nucleotides A2058 and A2059 in
Escherichia coli) [
17]. Some studies have reported point mutations outside these positions, but their association with clarithromycin resistance is still a matter of debate [
18,
19,
20]. Amino acid (aa) exchanges in the quinolone resistance-determining region (QRDR) of the
H. pylori gyrA gene at codons 87 and 91 (positions according to
H. pylori reference strain 26695; corresponds to codons 83 and 87 in
Escherichia coli) alone or in combination with mutations in
gyrB have been reported to lead to resistance to fluoroquinolones [
10,
11,
21]. Rifampicin resistance in
H. pylori has been associated with aa exchanges in the rifampicin resistance-determining region (RRDR) of
rpoB, mainly at codons 525 to 545, 547, and 586 (positions according to
H. pylori reference strain 26695; corresponds to codons 512 to 573 in
Escherichia coli) [
22]. The mechanism of resistance to metronidazole is less clear. Frameshift mutations and truncations in genes that encode electron transfer proteins, such as the nicotinamide adenine dinucleotide phosphate hydrogen (NADPH)-flavin nitroreductase (
frxA) and oxygen-insensitive NADPH nitroreductase (
rdxA), have been implicated in elevated MIC values to metronidazole [
23,
24]. Thus far, it is unclear, whether elevated metronidazole MIC values lead to therapeutic failure [
25]. For tetracycline, one resistance mechanism appears to be single, double, or triple base pair substitutions in the primary binding site of tetracycline (at nucleotide positions 926 to 928) in the 16S rRNA gene [
26].
Here, we report on the first sequencing study that systematically applied WGS to H. pylori clinical isolates to detect specific point mutations in the 23S rRNA, gyrA, rpoB, frxA, rdxA, and 16S rRNA genes. We aimed at correlating the occurrence of SNPs in these target genes to phenotypic DST results for each drug investigated, thereby allowing the calculation of predictive sensitivities and specificities of each set of SNPs.
4. Discussion
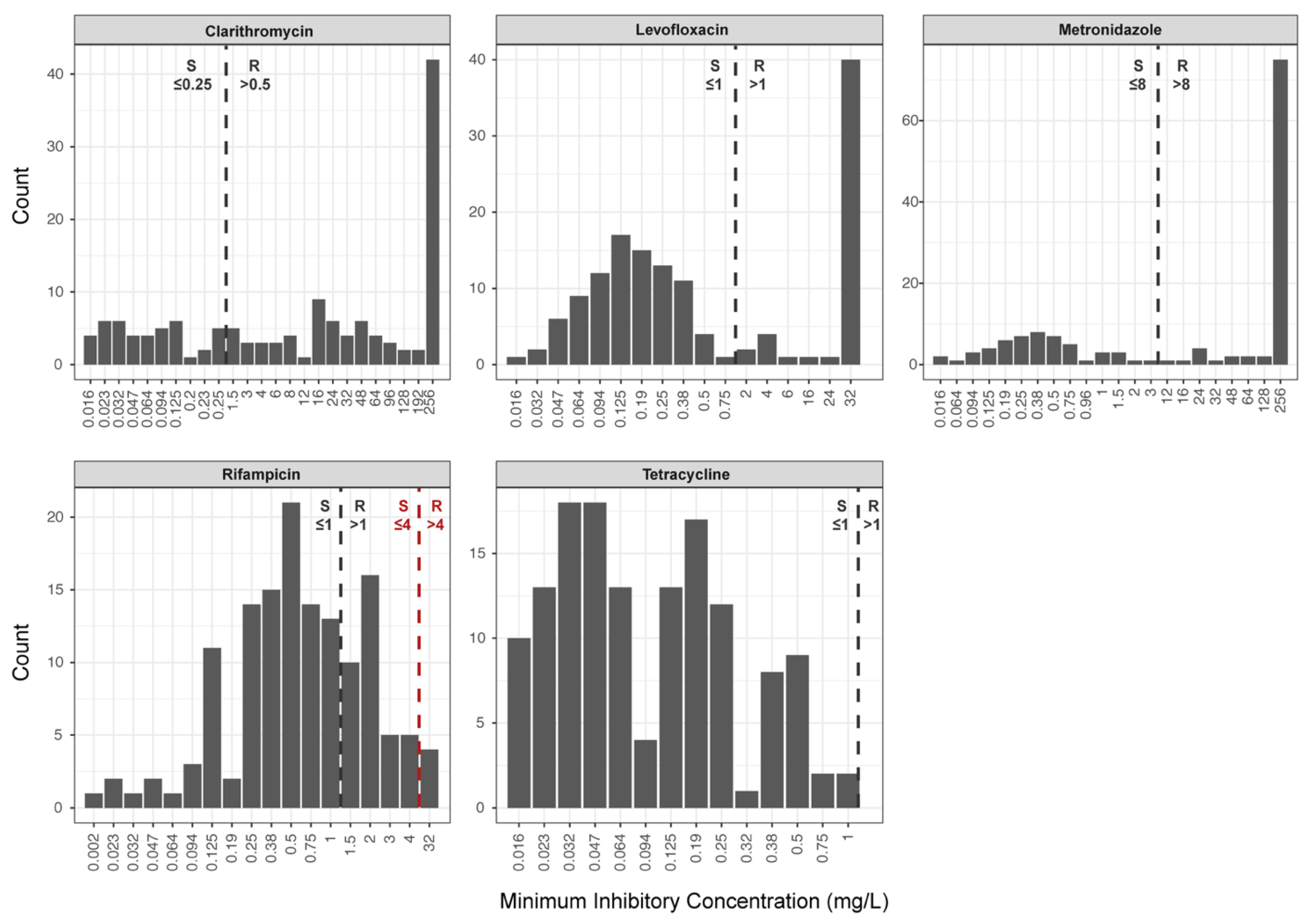
In the present study, we have applied massive parallel Illumina-based WGS on clinical H. pylori isolates to define the relationship between the occurrence of SNPs in selected target genes and phenotypic antimicrobial susceptibility. We could show a clear correlation between the occurrence of point mutations in the 23S rRNA, gyrA, and rpoB genes of H. pylori and macrolide, fluoroquinolone, and rifamycin resistance, respectively. To our knowledge, this is the first study showing that genetic determinants of antimicrobial resistance identified by WGS can be used for the prediction of drug resistance phenotypes in H. pylori. In contrast, there was no clear association between identified SNPs in frxA and rdxA and phenotypic metronidazole resistance.
During this study, we realized that assessment of antimicrobial susceptibility in
H. pylori by culture based phenotypic DST can be challenging due to the pathogen’s fastidious growth requirements. Moreover, it was not always possible to determine an explicit MIC of a drug by E-Test
® because of the small and transparent colonies that
H. pylori forms. Variations in the redox potential of the test medium caused a broad range of MIC distributions, especially for metronidazole, when performing the E-Test
®, thereby making standardized and reproducible interpretation difficult. Phenotypic DST may also be challenged when a mixed population of resistant and susceptible
H. pylori strains is present in the same patient [
38]. In two samples included in this study, phenotypic DST initially did not detect the resistant sub-population. This is problematic, as antimicrobial therapy is based on in vitro phenotypic susceptibly information. In contrast, the presence of susceptible and resistant
H. pylori subpopulations can be predicted from the WGS sequencing data through the occurrence of hetero-resistance at specific nucleotide positions [
38].
Molecular based methods, like polymerase chain reaction (PCR) and line probe assays, that enable specific detection of point mutations in the 23S rRNA (nucleotide positions, A2146 and A2147) or
gyrA gene (codons 87 and 91) can be directly applied on clinical specimens [
39,
40]. Our data indicate that molecular assays targeting these point mutations would be sufficient to monitor clarithromycin and levofloxacin resistance in
H. pylori. However, one advantage of WGS is that it delivers a more complete picture of resistance determinants present in a clinical isolate than targeted molecular approaches that can only examine a limited number of nucleotide positions. Thus, false negative results may occur, when new polymorphisms, potentially conferring drug resistance, arise that are not covered by the assay. In contrast, the relevance of these polymorphisms can easily be assessed by retrospective analysis of WGS data, whereas targeted molecular assays would need to be redesigned and samples retested.
In this study, we found clarithromycin resistance in
H. pylori to be highly correlated with the presence of mutations A2146C, A2146G or A2147G in the domain V of the 23S rRNA gene. Recent studies reported additional point mutations outside the domain V and an active drug efflux mechanism to be involved in clarithromycin resistance [
35,
41]. Also, mutations in other target genes, like
rpl22 (encodes a ribosomal protein that interacts with the 23S rRNA domains) and
infB (encodes translation initiation factor, IF-2), were identified to induce low level clarithromycin resistance (MICs of 0.5 to 4 mg/L). Interestingly, these mutations led to a high level of clarithromycin resistance (MICs >256 mg/L) in combination with A2146 and A2147 mutations [
42]. In a recent study, it was reported that mutations in multidrug efflux transporter genes may be involved in clarithromycin resistance [
43]. In this study, however, we found that the occurrence of point mutations at nucleotide positions, A2146 and A2147, were sufficient for the reliable prediction of phenotypic clarithromycin resistance in
H. pylori.
For metronidazole, there was no clear correlation between the isolates’ observed phenotype and their genotype. Firstly, frameshift mutations in
rdxA and
frxA that were reported to confer metronidazole resistance were also found in
H. pylori strains assessed as metronidazole susceptible by phenotypic DST. Secondly, in contrast to recent reports suggesting that metronidazole resistance requires mutations in both genes,
frxA and
rdxA, one isolate was found to be resistant (MIC of 26 mg/L) though carrying only a mutation in
frxA [
44,
45]. Some
rdxA and
frxA mutations were identified in metronidazole resistant
H. pylori isolates; however, several metronidazole resistant strains harboured no mutations at all in
rdxA and/or
frxA. Recently, it has been reported that additional genes, such as
mdaB, omp11, and
rpsU, may be involved in metronidazole resistance in
H. pylori [
46,
47]. However, consensual data are still lacking and a relationship between SNPs detected in one of these five genes (
rdxA,
frxA,
mdaB,
omp11, and
rpsU) and phenotypic metronidazole resistance was not found in the
H. pylori isolates investigated in this study. In sum, it remains difficult to predict metronidazole susceptibility based merely on genotypic data, as the correlation between detected SNPs and the phenotype to date is very poor.
On the basis of the presented data, we therefore suggest to reconsider the usefulness of phenotypic metronidazole susceptibility testing in
H. pylori as it is stated in the CASFM/EUCAST recommendations for
H. pylori [
29]. Moreover, in vitro and in vivo resistance data are not congruent [
48] as
H. pylori strains tested resistant in vitro can still be eradicated with a combination therapy that contains an increased dosage of metronidazole [
25].
Resistance to fluoroquinolones could be attributed to mutations in the QRDR of
gyrA at codons 87 and 91 in 98% of the fluoroquinolone resistant
H. pylori strains. One levofloxacin resistant
H. pylori strain with an MIC of 1.5 mg/L showed no codon exchange in the QRDR of
gyrA. The relevance of mutations in other target genes, like
gyrB, in quinolone resistant
H. pylori isolates without
gyrA mutations is not consensual [
49,
50]. The aforementioned isolate harboured various SNPs in
gyrB; however, none matched any mutations recently reported to be associated with quinolone resistance in
H. pylori [
49,
51]. Notably,
H. pylori lacks genes encoding topoisomerase IV (encoded by
parC and
parE genes), which is targeted by quinolones in other bacteria [
52]. Moreover, active efflux pumps seem not to be involved in mediating fluoroquinolone resistance [
53].
In our study,
H. pylori strains with a rifampicin susceptible phenotype (i.e., MICs ≤ 4 mg/L) showed no mutation in the RRDR of
rpoB. Amino acid substitutions, L525P and H540N, were associated with resistance and MICs of >32 mg/L. Noteworthy, application of the current EUCAST clinical breakpoint for rifampicin of 1 mg/L would split the wild-type population (
Figure 1). This in turn introduces serious interpretation problems, since some
H. pylori strains that do not carry point mutations in the RRDR of
rpoB would be classified as rifampicin resistant, representing a major error. Given the rifampicin MIC distribution of
H. pylori isolates analysed in this study, we recommend using the clinical breakpoint of 4 mg/L proposed by Hays et al. [
22] and the CASFM/EUCAST guidelines [
29].
Findings gained in this study can be useful for future research that may aim at performing culture-independent WGS directly from clinical specimens for the prediction of phenotypic drug susceptibility in
H. pylori. Some studies have already successfully applied WGS directly on gastric biopsies for the detection of
H. pylori [
54,
55]. Moreover, in-house developed and commercial protocols (e.g., [
56,
57]) are becoming available for the depletion of human DNA or the enrichment of bacterial DNA prior to performing WGS, thereby increasing the efficiency and cost-effectiveness of NGS due to less human DNA background in samples.
Our study has several limitations: It was designed as a single centre laboratory-based study using clinical
H. pylori isolates. For some antibiotics, there were only a few (i.e., rifampicin) or no resistant
H. pylori isolates (i.e., tetracycline) available. Thus, the analytical sensitivity of WGS in determining phenotypic rifampicin resistance based on mutations in the RRDR of
rpoB might have been overestimated as rifampicin resistance has also been reported in
H. pylori isolates lacking these mutations [
8]. For tetracycline, calculation of the analytical sensitivity of WGS was not possible due to a lack of resistant
H. pylori isolates. Moreover, genotype-based prediction of tetracycline resistance in
H. pylori is additionally hampered since isolates without mutations at nucleotide positions, 926 to 928, in the 16S rRNA gene display a resistant phenotype [
58]. Therefore, tetracycline resistance seems to be multifactorial, involving alterations in ribosomal binding, enzymatic degradation of antibiotics, a reduction of membrane permeability, and an active efflux [
59,
60,
61].
In this study, we have not examined the possible role of active efflux mechanisms. Their involvement in intrinsic antibiotic resistance in H. pylori is still a matter of debate and would require transcriptional analyses of the genes encoding drug efflux systems (e.g., by using a meta-transcriptomics approach), which was beyond the scope of this study.

 ,
, 

{kind=link}