Identification and Characterization of New Variants in FOXRED1 Gene Expands the Clinical Spectrum Associated with Mitochondrial Complex I Deficiency

 , , ,
, , ,  , and
, and 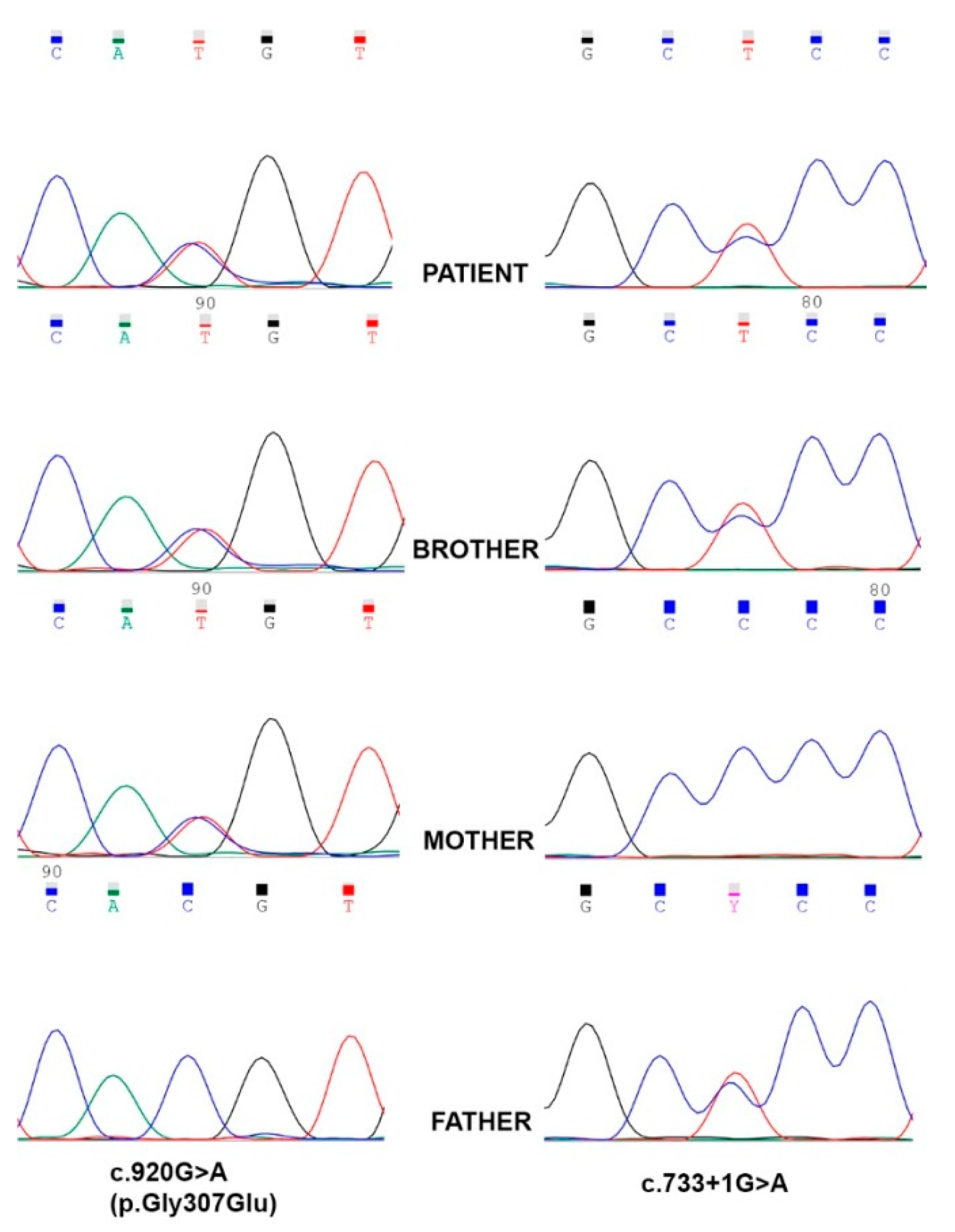{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Clinical Profile
2.2. Targeted Next-Generation Sequencing
2.3. Protein Modeling
2.4. Cell Culture
2.5. Mitochondrial Isolation
2.6. Respirometry and OXPHOS Activity
2.7. SDS-PAGE and BN-PAGE Immunoblot Analysis
3. Results
3.1. Molecular Genetics and in Silico Analysis of FOXRED1 Variants
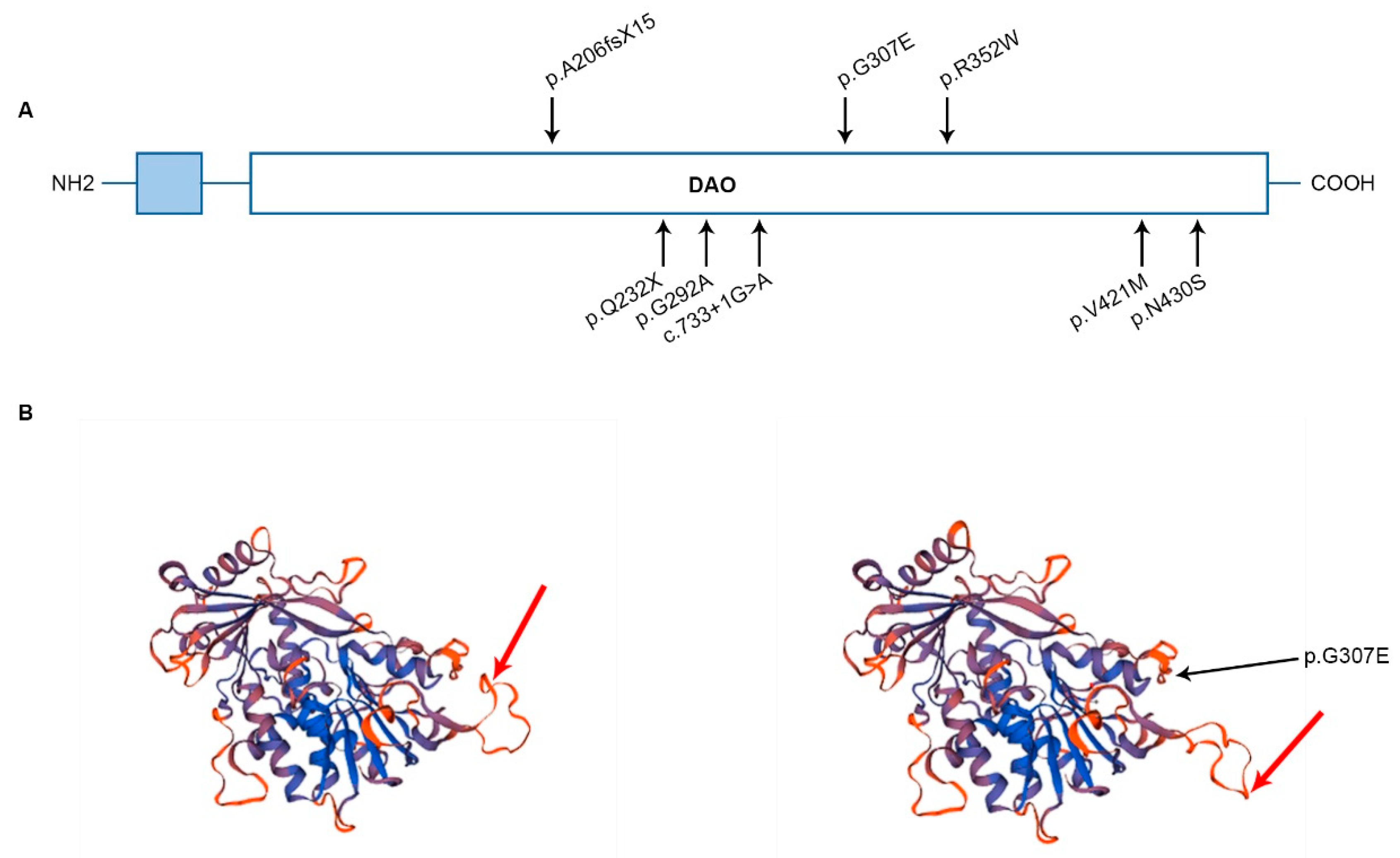
3.2. FOXRED1 Protein Modeling
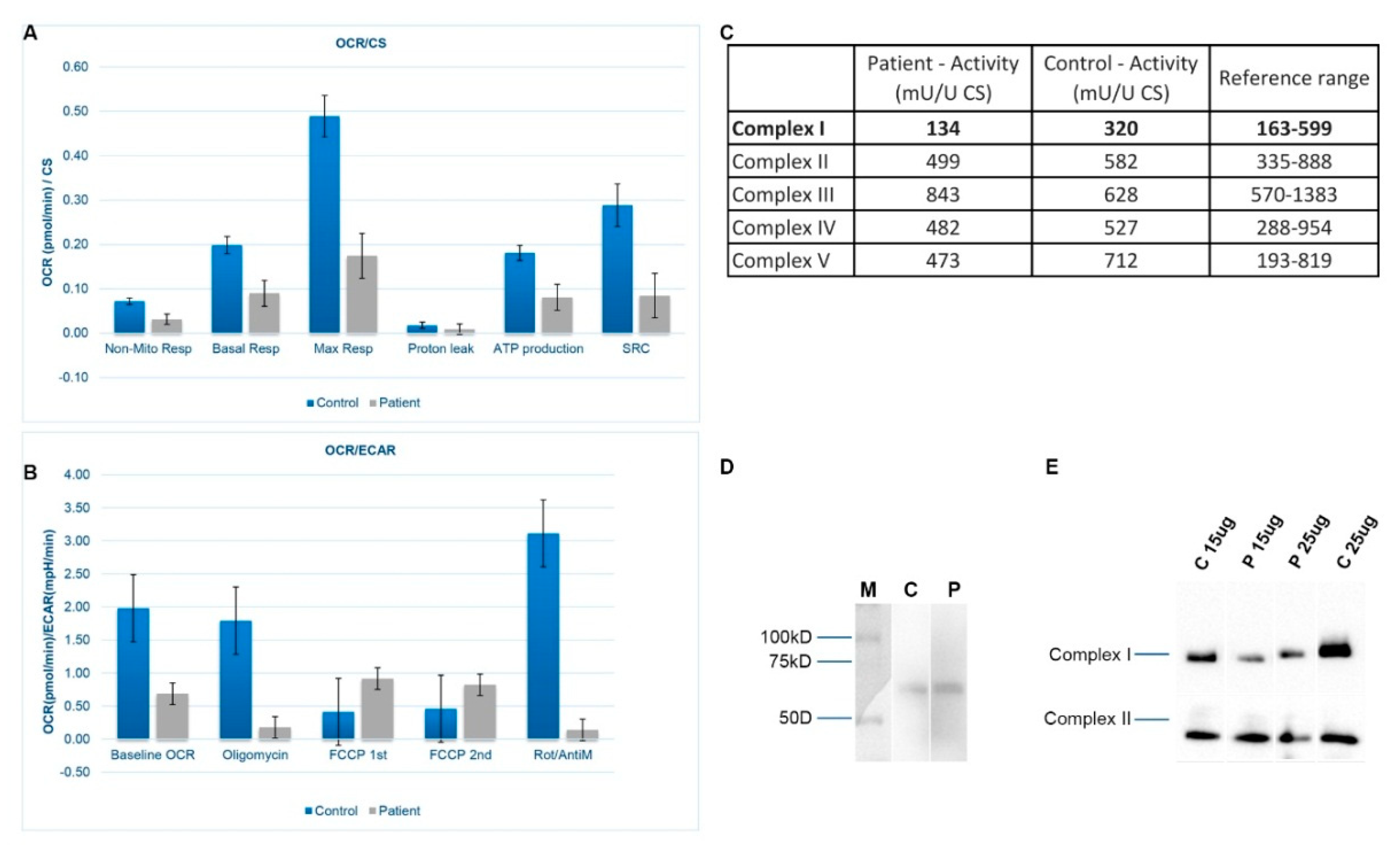
3.3. Mitochondrial Respiration and OXPHOS Activity
3.4. SDS-PAGE and BN-PAGE Immunoblot Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Valsecchi, F.; Koopman, W.J.H.; Manjeri, G.R.; Rodenburg, R.J.; Smeitink, J.A.M.; Willems, P.H.G.M. Complex I disorders: Causes, mechanisms, and development of treatment strategies at the cellular level. Dev. Disabil. Res. Rev. 2010, 16, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Tucker, E.J.; Compton, A.G.; Calvo, S.E.; Thorburn, D.R. The molecular basis of human complex I deficiency. IUBMB Life 2011, 63, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M. The Mechanism of Oxidative Phosphorylation. In The Cell: A Molecular Approach, 2nd ed.; National Center for Biotechnology Information: Bethesda, MD, USA, 2000. [Google Scholar]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial respiratory complex I: Structure, function and implication in human diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Brandt, U. Energy converting NADH: Quinone oxidoreductase (complex I). Annu. Rev. Biochem. 2006, 75, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Rodenburg, R.J. Mitochondrial complex I-linked disease. Biochim. Biophys. Acta 2016, 1857, 938–945. [Google Scholar] [CrossRef]
- Ugalde, C.; Janssen, R.J.R.J.; van den Heuvel, L.P.; Smeitink, J.A.M.; Nijtmans, L.G.J. Differences in assembly or stability of complex I and other mitochondrial OXPHOS complexes in inherited complex I deficiency. Hum. Mol. Genet. 2004, 13, 659–667. [Google Scholar] [CrossRef]
- Mimaki, M.; Wang, X.; McKenzie, M.; Thorburn, D.R.; Ryan, M.T. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta 2012, 1817, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Schultz, B.E.; Chan, S.I. Structures and proton-pumping strategies of mitochondrial respiratory enzymes. Annu. Rev. Biophys. Biomol. Struct 2001, 30, 23–65. [Google Scholar] [CrossRef]
- Nouws, J.; Nijtmans, L.G.J.; Smeitink, J.A.; Vogel, R.O. Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: Cause, pathology and treatment options. Brain 2012, 135, 12–22. [Google Scholar] [CrossRef]
- Simon, M.T.; Eftekharian, S.S.; Stover, A.E.; Osborne, A.F.; Braffman, B.H.; Chang, R.C.; Wang, R.Y.; Steenari, M.R.; Tang, S.; Wu, P.W.L.; et al. Novel mutations in the mitochondrial complex I assembly gene NDUFAF5 reveal heterogeneous phenotypes. Mol. Genet. Metab. 2019, 126, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Formosa, L.E.; Mimaki, M.; Frazier, A.E.; McKenzie, M.; Stait, T.L.; Thorburn, D.R.; Stroud, D.A.; Ryan, M.T. Characterization of mitochondrial FOXRED1 in the assembly of respiratory chain complex I. Hum. Mol. Genet. 2015, 24, 2952–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distelmaier, F.; Koopman, W.J.H.; van den Heuvel, L.P.; Rodenburg, R.J.; Mayatepek, E.; Willems, P.H.G.M.; Smeitink, J.A.M. Mitochondrial complex I deficiency: From organelle dysfunction to clinical disease. Brain 2009, 132, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Kirby, D.M.; Crawford, M.; Cleary, M.A.; Dahl, H.H.; Dennett, X.; Thorburn, D.R. Respiratory chain complex I deficiency: An underdiagnosed energy generation disorder. Neurology 1999, 52, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Tucker, E.J.; Compton, A.G.; Kirby, D.M.; Crawford, G.; Burtt, N.P.; Rivas, M.; Guiducci, C.; Bruno, D.L.; Goldberger, O.A.; et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet. 2010, 42, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Fassone, E.; Duncan, A.J.; Taanman, J.W.; Pagnamenta, A.T.; Sadowski, M.I.; Holand, T.; Qasim, W.; Rutland, P.; Calvo, S.E.; Mootha, V.K.; et al. FOXRED1, encoding an FAD-dependent oxidoreductase complex-I-specific molecular chaperone, is mutated in infantile-onset mitochondrial encephalopathy. Hum. Mol. Genet. 2010, 19, 4837–4847. [Google Scholar] [CrossRef] [Green Version]
- Zurita Rendón, O.; Antonicka, H.; Horvath, R.; Shoubridge, E.A. A Mutation in the Flavin Adenine Dinucleotide-Dependent Oxidoreductase FOXRED1 Results in Cell-Type-Specific Assembly Defects in Oxidative Phosphorylation Complexes I and II. Mol. Cell Biol. 2016, 36, 2132–2140. [Google Scholar] [CrossRef] [Green Version]
- Apatean, D.; Rakic, B.; Brunel-Guitton, C.; Hendson, G.; Bai, R.; Sargent, M.A.; Lavoie, P.M.; Patel, M.; Stockler-Ipsiroglu, S. Congenital lactic acidosis, cerebral cysts and pulmonary hypertension in an infant with FOXRED1 related complex I deficiency. Mol. Genet. Metab. Rep. 2019, 18, 32–38. [Google Scholar] [CrossRef]
- Haack, T.B.; Madignier, F.; Herzer, M.; Lamantea, E.; Danhauser, K.; Invernizzi, F.; Koch, J.; Freitag, M.; Drost, R.; Hillier, I.; et al. Mutation screening of 75 candidate genes in 152 complex I deficiency cases identifies pathogenic variants in 16 genes including NDUFB9. J. Med. Genet. 2012, 49, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Agilent SureDesign. Available online: https://earray.chem.agilent.com/suredesign/ (accessed on 15 March 2019).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NGSrich. Available online: https://sourceforge.net/projects/ngsrich/ (accessed on 11 October 2018).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Picard Tools by Broad Institute. Available online: http://broadinstitute.github.io/picard/ (accessed on 15 March 2019).
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-Throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; et al. On behalf of the ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar]
- Lemire, B.D. Evolution of FOXRED1, an FAD-dependent oxidoreductase necessary for NADH: Ubiquinone oxidoreductase (Complex I) assembly. Biochim. Biophys. Acta 2015, 1847, 451–457. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef]
- Srere, P.A. Citrate synthase: [EC 4.1.3.7. Citrate oxaloacetate-lyase (CoA-acetylating)]. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1969; Volume 13, pp. 3–11. [Google Scholar]
- Rodenburg, R.J.T. Biochemical diagnosis of mitochondrial disorders. J. Inherit. Metab. Dis. 2011, 34, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Lemire, B.D. A structural model for FOXRED1, an FAD-dependent oxidoreductase necessary for NADH: Ubiquinone oxidoreductase (complex I) assembly. Mitochondrion 2015, 22, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Lemire, B.D. Glutathione metabolism links FOXRED1 to NADH: Ubiquinone oxidoreductase (complex I) deficiency: A hypothesis. Mitochondrion 2015, 24, 105–112. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbosa-Gouveia, S.; González-Vioque, E.; Borges, F.; Gutiérrez-Solana, L.; Wintjes, L.; Kappen, A.; van den Heuvel, L.; Leis, R.; Rodenburg, R.; Couce, M.L. Identification and Characterization of New Variants in FOXRED1 Gene Expands the Clinical Spectrum Associated with Mitochondrial Complex I Deficiency. J. Clin. Med. 2019, 8, 1262. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8081262
Barbosa-Gouveia S, González-Vioque E, Borges F, Gutiérrez-Solana L, Wintjes L, Kappen A, van den Heuvel L, Leis R, Rodenburg R, Couce ML. Identification and Characterization of New Variants in FOXRED1 Gene Expands the Clinical Spectrum Associated with Mitochondrial Complex I Deficiency. Journal of Clinical Medicine. 2019; 8(8):1262. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8081262
Chicago/Turabian StyleBarbosa-Gouveia, Sofia, Emiliano González-Vioque, Filipa Borges, Luis Gutiérrez-Solana, Liesbeth Wintjes, Antonia Kappen, Lambert van den Heuvel, Rosaura Leis, Richard Rodenburg, and María Luz Couce. 2019. "Identification and Characterization of New Variants in FOXRED1 Gene Expands the Clinical Spectrum Associated with Mitochondrial Complex I Deficiency" Journal of Clinical Medicine 8, no. 8: 1262. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8081262