The Role of Neutrophils and Neutrophil Extracellular Traps in Vascular Damage in Systemic Lupus Erythematosus

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. SLE and Atherosclerosis: Epidemiology
Lupus-Specific Mechanisms of Enhanced Atherosclerosis
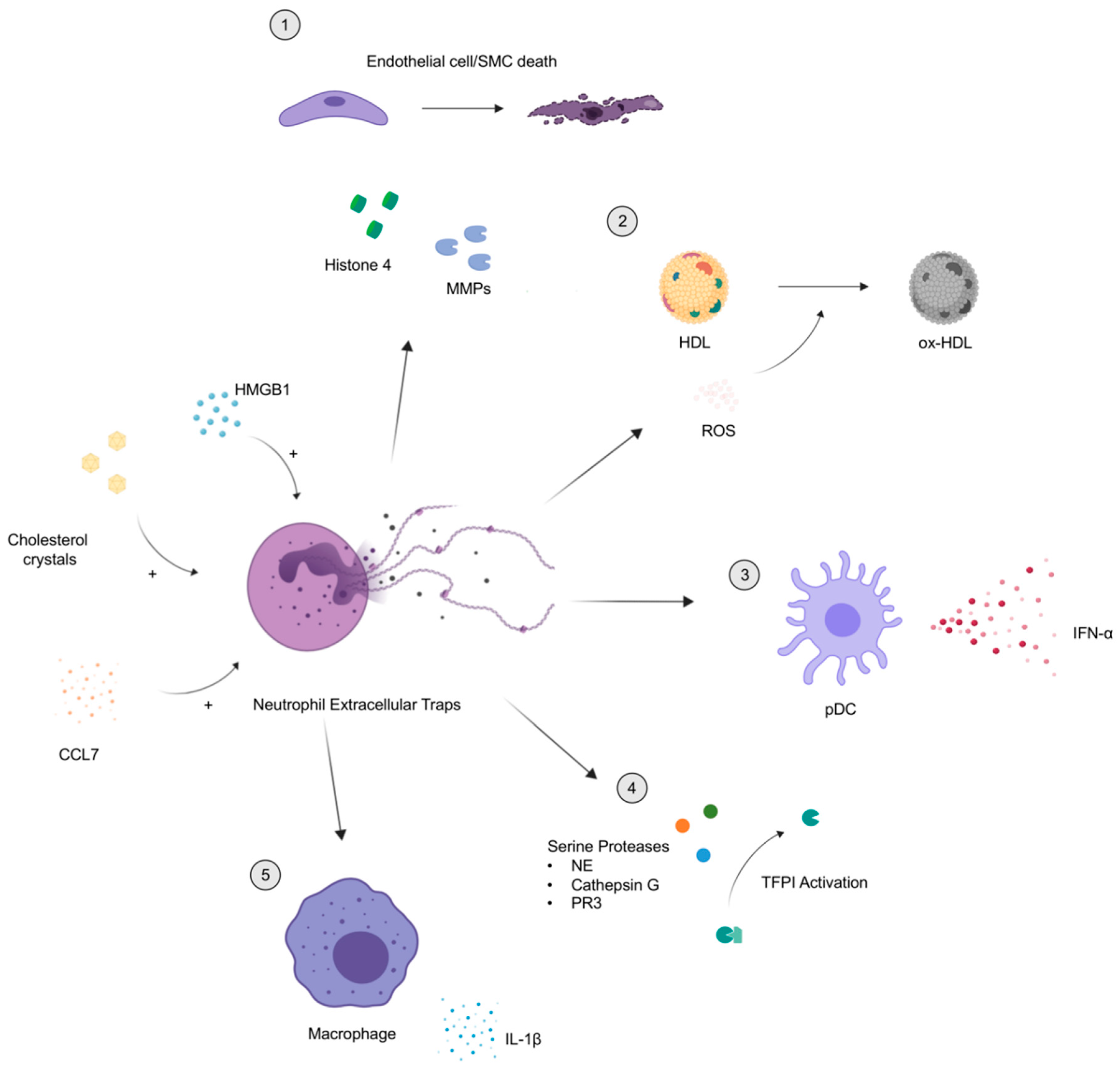
3. Inflammation, Neutrophil Extracellular Traps (NETs), and Atherosclerosis
4. Neutrophils Facilitate Atherosclerosis Development and Progression
4.1. The Role of Serine Proteases
4.2. Cellular Toxicity by Histone H4 and Matrix Metalloproteinases
4.3. PAD-4, a Potential Target for Therapy
4.4. Mitochondrial DNA and Reactive Oxygen Species
4.5. NETs as a Biomarker for Subclinical Atherosclerosis
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Gistera, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Sima, P.; Vannucci, L.; Vetvicka, V. Atherosclerosis as autoimmune disease. Ann. Transl. Med. 2018, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Frieri, M.; Stampfl, H. Systemic lupus erythematosus and atherosclerosis: Review of the literature. Autoimmun. Rev. 2016, 15, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Turano, L. Premature atherosclerotic cardiovascular disease in systemic lupus erythematosus: Understanding management strategies. J. Cardiovasc. Nurs. 2013, 28, 48–53. [Google Scholar] [CrossRef]
- Kaplan, M.J. Neutrophils in the pathogenesis and manifestations of SLE. Nat. Rev. Rheumatol. 2011, 7, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, J.T.; Herlitz Lindberg, M.; Gunnarsson, I.; Pettersson, S.; Elvin, K.; Ohrvik, J.; Larsson, A.; Jensen-Urstad, K.; Svenungsson, E. Excess atherosclerosis in systemic lupus erythematosus,-A matter of renal involvement: Case control study of 281 SLE patients and 281 individually matched population controls. PLoS ONE 2017, 12, e0174572. [Google Scholar] [CrossRef]
- Kiani, A.N.; Magder, L.S.; Post, W.S.; Szklo, M.; Bathon, J.M.; Schreiner, P.J.; O’Leary, D.; Petri, M. Coronary calcification in SLE: Comparison with the Multi-Ethnic Study of Atherosclerosis. Rheumatol. (Oxf.) 2015, 54, 1976–1981. [Google Scholar] [CrossRef]
- Manzi, S.; Meilahn, E.N.; Rairie, J.E.; Conte, C.G.; Medsger, T.A., Jr.; Jansen-McWilliams, L.; D’Agostino, R.B.; Kuller, L.H. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: Comparison with the Framingham Study. Am. J. Epidemiol. 1997, 145, 408–415. [Google Scholar] [CrossRef]
- Rubin, L.A.; Urowitz, M.B.; Gladman, D.D. Mortality in systemic lupus erythematosus: The bimodal pattern revisited. Q. J. Med. 1985, 55, 87–98. [Google Scholar]
- Bengtsson, C.; Ohman, M.L.; Nived, O.; Rantapaa Dahlqvist, S. Cardiovascular event in systemic lupus erythematosus in northern Sweden: Incidence and predictors in a 7-year follow-up study. Lupus 2012, 21, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Manzi, S.; Selzer, F.; Sutton-Tyrrell, K.; Fitzgerald, S.G.; Rairie, J.E.; Tracy, R.P.; Kuller, L.H. Prevalence and risk factors of carotid plaque in women with systemic lupus erythematosus. Arthritis Rheum 1999, 42, 51–60. [Google Scholar] [CrossRef]
- Haque, S.; Gordon, C.; Isenberg, D.; Rahman, A.; Lanyon, P.; Bell, A.; Emery, P.; McHugh, N.; Teh, L.S.; Scott, D.G.; et al. Risk factors for clinical coronary heart disease in systemic lupus erythematosus: The lupus and atherosclerosis evaluation of risk (LASER) study. J. Rheumatol. 2010, 37, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Sule, S.; Fivush, B.; Neu, A.; Furth, S. Increased risk of death in pediatric and adult patients with ESRD secondary to lupus. Pediatr. Nephrol. 2011, 26, 93–98. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Grossman, J.; FitzGerald, J.; Dahlin-Lee, E.; Wallace, D.J.; Thong, B.Y.; Badsha, H.; Kalunian, K.; Charles, C.; Navab, M.; et al. Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.G.; Giles, I.; Lambrianides, A.; Manson, J.; D’Cruz, D.; Schrieber, L.; March, L.M.; Latchman, D.S.; Isenberg, D.A.; Rahman, A. Antibodies to apolipoprotein A-I, high-density lipoprotein, and C-reactive protein are associated with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Tupin, E.; Nicoletti, A.; Elhage, R.; Rudling, M.; Ljunggren, H.G.; Hansson, G.K.; Berne, G.P. CD1d-dependent activation of NKT cells aggravates atherosclerosis. J. Exp. Med. 2004, 199, 417–422. [Google Scholar] [CrossRef]
- Smith, E.; Croca, S.; Waddington, K.E.; Sofat, R.; Griffin, M.; Nicolaides, A.; Isenberg, D.A.; Torra, I.P.; Rahman, A.; Jury, E.C. Cross-talk between iNKT cells and monocytes triggers an atheroprotective immune response in SLE patients with asymptomatic plaque. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef]
- Bonciani, D.; Antiga, E.; Bonciolini, V.; Verdelli, A.; Del Bianco, E.; Volpi, W.; Caproni, M. Homocysteine serum levels are increased and correlate with disease severity in patients with lupus erythematosus. Clin. Exp. Rheumatol. 2016, 34, 76–81. [Google Scholar]
- Clarke, R.; Daly, L.; Robinson, K.; Naughten, E.; Cahalane, S.; Fowler, B.; Graham, I. Hyperhomocysteinemia: An independent risk factor for vascular disease. N. Engl. J. Med. 1991, 324, 1149–1155. [Google Scholar] [CrossRef]
- Crow, M.K. Type I interferon in the pathogenesis of lupus. J. Immunol. 2014, 192, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Amisten, S.; Gullstrand, B.; Jonsen, A.; Allhorn, M.; Truedsson, L.; Sturfelt, G.; Erlinge, D.; Bengtsson, A.A. Platelet transcriptional profile and protein expression in patients with systemic lupus erythematosus: Up-regulation of the type I interferon system is strongly associated with vascular disease. Blood 2010, 116, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Lucke, C.; Rossig, L.; Fichtlscherer, S.; Vasa, M.; Britten, M.; Kamper, U.; Dimmeler, S.; Zeiher, A.M. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: Proof of concept for the clinical importance of endogenous vascular repair. Circulation 2005, 111, 2981–2987. [Google Scholar] [CrossRef] [PubMed]
- Anania, C.; Gustafsson, T.; Hua, X.; Su, J.; Vikstrom, M.; de Faire, U.; Heimburger, M.; Jogestrand, T.; Frostegard, J. Increased prevalence of vulnerable atherosclerotic plaques and low levels of natural IgM antibodies against phosphorylcholine in patients with systemic lupus erythematosus. Arthritis Res. 2010, 12, R214. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.D.; Manson, J.E.; Rossouw, J.E.; Siscovick, D.S.; Mouton, C.P.; Rifai, N.; Wallace, R.B.; Jackson, R.D.; Pettinger, M.B.; Ridker, P.M. Inflammatory biomarkers, hormone replacement therapy, and incident coronary heart disease: Prospective analysis from the Women’s Health Initiative observational study. JAMA 2002, 288, 980–987. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Taleb, S. Inflammation in atherosclerosis. Arch. Cardiovasc. Dis. 2016, 109, 708–715. [Google Scholar] [CrossRef]
- Moreno, J.A.; Ortega-Gomez, A.; Delbosc, S.; Beaufort, N.; Sorbets, E.; Louedec, L.; Esposito-Farese, M.; Tubach, F.; Nicoletti, A.; Steg, P.G.; et al. In vitro and in vivo evidence for the role of elastase shedding of CD163 in human atherothrombosis. Eur. Heart J. 2012, 33, 252–263. [Google Scholar] [CrossRef]
- Doring, Y.; Drechsler, M.; Soehnlein, O.; Weber, C. Neutrophils in atherosclerosis: From mice to man. Arter. Thromb. Vasc. Biol. 2015, 35, 288–295. [Google Scholar] [CrossRef]
- Pillay, J.; den Braber, I.; Vrisekoop, N.; Kwast, L.M.; de Boer, R.J.; Borghans, J.A.; Tesselaar, K.; Koenderman, L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 2010, 116, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, L.J.; Kaplan, M.J. Neutrophils in Rheumatoid Arthritis: Breaking Immune Tolerance and Fueling Disease. Trends Mol. Med. 2019, 25, 215–227. [Google Scholar] [CrossRef]
- Borregaard, N.; Cowland, J.B. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 1997, 89, 3503–3521. [Google Scholar] [PubMed]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, Y.L.; Kaplan, M.J. Disentangling the role of neutrophil extracellular traps in rheumatic diseases. Curr. Opin. Rheumatol. 2017, 29, 65–70. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; de Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindau, D.; Mussard, J.; Rabsteyn, A.; Ribon, M.; Kotter, I.; Igney, A.; Adema, G.J.; Boissier, M.C.; Rammensee, H.G.; Decker, P. TLR9 independent interferon alpha production by neutrophils on NETosis in response to circulating chromatin, a key lupus autoantigen. Ann. Rheum. Dis. 2014, 73, 2199–2207. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.C.; Semerano, L.; Decker, P. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Weil, B.R.; Neelamegham, S. Selectins and Immune Cells in Acute Myocardial Infarction and Post-infarction Ventricular Remodeling: Pathophysiology and Novel Treatments. Front. Immunol. 2019, 10, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drechsler, M.; Megens, R.T.; van Zandvoort, M.; Weber, C.; Soehnlein, O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 2010, 122, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, J.; Sela, S.; Cohen, H.I.; Chezar, J.; Kristal, B. Priming of polymorphonuclear leukocytes: A culprit in the initiation of endothelial cell injury. Am. J. Physiol. Heart. Circ. Physiol. 2006, 290, H2051–H2058. [Google Scholar] [CrossRef] [PubMed]
- Taekema-Roelvink, M.E.; Kooten, C.; Kooij, S.V.; Heemskerk, E.; Daha, M.R. Proteinase 3 enhances endothelial monocyte chemoattractant protein-1 production and induces increased adhesion of neutrophils to endothelial cells by upregulating intercellular cell adhesion molecule-1. J. Am. Soc. Nephrol. 2001, 12, 932–940. [Google Scholar]
- Wedmore, C.V.; Williams, T.J. Control of vascular permeability by polymorphonuclear leukocytes in inflammation. Nature 1981, 289, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Kai-Larsen, Y.; Frithiof, R.; Sorensen, O.E.; Kenne, E.; Scharffetter-Kochanek, K.; Eriksson, E.E.; Herwald, H.; Agerberth, B.; Lindbom, L. Neutrophil primary granule proteins HBP and HNP1-3 boost bacterial phagocytosis by human and murine macrophages. J. Clin. Investig. 2008, 118, 3491–3502. [Google Scholar] [CrossRef]
- Gombart, A.F.; Krug, U.; O’Kelly, J.; An, E.; Vegesna, V.; Koeffler, H.P. Aberrant expression of neutrophil and macrophage-related genes in a murine model for human neutrophil-specific granule deficiency. J. Leukoc. Biol. 2005, 78, 1153–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higazi, A.A.; Lavi, E.; Bdeir, K.; Ulrich, A.M.; Jamieson, D.G.; Rader, D.J.; Usher, D.C.; Kane, W.; Ganz, T.; Cines, D.B. Defensin stimulates the binding of lipoprotein (a) to human vascular endothelial and smooth muscle cells. Blood 1997, 89, 4290–4298. [Google Scholar] [PubMed]
- Leclercq, A.; Houard, X.; Philippe, M.; Ollivier, V.; Sebbag, U.; Meilhac, O.; Michel, J.B. Involvement of intraplaque hemorrhage in atherothrombosis evolution via neutrophil protease enrichment. J. Leukoc. Biol. 2007, 82, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Laxton, R.C.; Hu, Y.; Duchene, J.; Zhang, F.; Zhang, Z.; Leung, K.Y.; Xiao, Q.; Scotland, R.S.; Hodgkinson, C.P.; Smith, K.; et al. A role of matrix metalloproteinase-8 in atherosclerosis. Circ. Res. 2009, 105, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.; Jakowitsch, J.; Panzenbock, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Riegger, J.; Byrne, R.A.; Joner, M.; Chandraratne, S.; Gershlick, A.H.; Ten Berg, J.M.; Adriaenssens, T.; Guagliumi, G.; Godschalk, T.C.; Neumann, F.J.; et al. Histopathological evaluation of thrombus in patients presenting with stent thrombosis. A multicenter European study: A report of the prevention of late stent thrombosis by an interdisciplinary global European effort consortium. Eur. Heart J. 2016, 37, 1538–1549. [Google Scholar] [CrossRef] [PubMed]
- Pertiwi, K.R.; de Boer, O.J.; Mackaaij, C.; Pabittei, D.R.; de Winter, R.J.; Li, X.; van der Wal, A.C. Extracellular traps derived from macrophages, mast cells, eosinophils and neutrophils are generated in a time-dependent manner during atherothrombosis. J. Pathol. 2019, 247, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Megens, R.T.; Vijayan, S.; Lievens, D.; Doring, Y.; van Zandvoort, M.A.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; de Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef]
- De Boer, O.J.; Li, X.; Teeling, P.; Mackaay, C.; Ploegmakers, H.J.; van der Loos, C.M.; Daemen, M.J.; de Winter, R.J.; van der Wal, A.C. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 2013, 109, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.K.; Vivekanandan-Giri, A.; Tang, C.; Knight, J.S.; Mathew, A.; Padilla, R.L.; Gillespie, B.W.; Carmona-Rivera, C.; Liu, X.; Subramanian, V.; et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: An additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. 2014, 66, 2532–2544. [Google Scholar] [CrossRef] [PubMed]
- Doring, Y.; Manthey, H.D.; Drechsler, M.; Lievens, D.; Megens, R.T.; Soehnlein, O.; Busch, M.; Manca, M.; Koenen, R.R.; Pelisek, J.; et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 2012, 125, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Soehnlein, O.; Ortega-Gomez, A.; Doring, Y.; Weber, C. Neutrophil-macrophage interplay in atherosclerosis: Protease-mediated cytokine processing versus NET release. Thromb. Haemost. 2015, 114, 866–867. [Google Scholar] [CrossRef] [PubMed]
- Petretto, A.; Bruschi, M.; Pratesi, F.; Croia, C.; Candiano, G.; Ghiggeri, G.; Migliorini, P. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PLoS ONE 2019, 14, e0218946. [Google Scholar] [CrossRef]
- Perez-Cremades, D.; Bueno-Beti, C.; Garcia-Gimenez, J.L.; Ibanez-Cabellos, J.S.; Hermenegildo, C.; Pallardo, F.V.; Novella, S. Extracellular histones disarrange vasoactive mediators release through a COX-NOS interaction in human endothelial cells. J. Cell. Mol. Med. 2017, 21, 1584–1592. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Gordon, R.A.; Herter, J.M.; Rosetti, F.; Campbell, A.M.; Nishi, H.; Kashgarian, M.; Bastacky, S.I.; Marinov, A.; Nickerson, K.M.; Mayadas, T.N.; et al. Lupus and proliferative nephritis are PAD4 independent in murine models. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Q.; Venugopal, J.; Wang, J.; Kleiman, K.; Guo, C.; Eitzman, D.T. Obesity-induced Endothelial Dysfunction is Prevented by Neutrophil Extracellular Trap Inhibition. Sci. Rep. 2018, 8, 4881. [Google Scholar] [CrossRef] [PubMed]
- Sorvillo, N.; Mizurini, D.; Coxon, C.; Martinod, K.; Tilvawala, R.; Cherpokova, D.; Salinger, A.J.; Seward, R.J.; Staudinger, C.; Weerapana, E.; et al. Plasma Peptidylarginine Deiminase IV Promotes VWF-Platelet String Formation and Accelerates Thrombosis after Vessel Injury. Circ. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Carmona-Rivera, C.; Moore, E.; Seto, N.L.; Knight, J.S.; Pryor, M.; Yang, Z.H.; Hemmers, S.; Remaley, A.T.; Mowen, K.A.; et al. Myeloid-Specific Deletion of Peptidylarginine Deiminase 4 Mitigates Atherosclerosis. Front. Immunol. 2018, 9, 1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madamanchi, N.R.; Zhou, R.H.; Vendrov, A.E.; Niu, X.L.; Runge, M.S. Does oxidative DNA damage cause atherosclerosis and metabolic syndrome?: New insights into which came first: The chicken or the egg. Circ. Res. 2010, 107, 940–942. [Google Scholar] [CrossRef]
- Dorighello, G.G.; Paim, B.A.; Leite, A.C.R.; Vercesi, A.E.; Oliveira, H.C.F. Spontaneous experimental atherosclerosis in hypercholesterolemic mice advances with ageing and correlates with mitochondrial reactive oxygen species. Exp. Gerontol. 2018, 109, 47–50. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Wang, N.; Tall, A.R.; Tabas, I. Mitochondrial Oxidative Stress Promotes Atherosclerosis and Neutrophil Extracellular Traps in Aged Mice. Arter. Thromb. Vasc. Biol. 2017, 37, e99–e107. [Google Scholar] [CrossRef] [Green Version]
- Ronda, N.; Favari, E.; Borghi, M.O.; Ingegnoli, F.; Gerosa, M.; Chighizola, C.; Zimetti, F.; Adorni, M.P.; Bernini, F.; Meroni, P.L. Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 2014, 73, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, J.; White, P.C.; Milward, M.R.; Cooper, P.R.; Chapple, I.L.C. Modulation of Neutrophil Extracellular Trap and Reactive Oxygen Species Release by Periodontal Bacteria. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [PubMed]
- Carlucci, P.M.; Purmalek, M.M.; Dey, A.K.; Temesgen-Oyelakin, Y.; Sakhardande, S.; Joshi, A.A.; Lerman, J.B.; Fike, A.; Davis, M.; Chung, J.H.; et al. Neutrophil subsets and their gene signature associate with vascular inflammation and coronary atherosclerosis in lupus. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teague, H.L.; Varghese, N.J.; Tsoi, L.C.; Dey, A.K.; Garshick, M.S.; Silverman, J.I.; Baumer, Y.; Harrington, C.L.; Stempinski, E.; Elnabawi, Y.A.; et al. Neutrophil Subsets, Platelets, and Vascular Disease in Psoriasis. JACC Basic. Transl. Sci. 2019, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; Ten Cate, H.; Hofstra, L.; et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arter. Thromb. Vasc. Biol. 2013, 33, 2032–2040. [Google Scholar] [CrossRef]
- Thacker, S.G.; Berthier, C.C.; Mattinzoli, D.; Rastaldi, M.P.; Kretzler, M.; Kaplan, M.J. The detrimental effects of IFN-alpha on vasculogenesis in lupus are mediated by repression of IL-1 pathways: Potential role in atherogenesis and renal vascular rarefaction. J. Immunol. 2010, 185, 4457–4469. [Google Scholar] [CrossRef] [PubMed]
- Enocsson, H.; Sjowall, C.; Skogh, T.; Eloranta, M.L.; Ronnblom, L.; Wettero, J. Interferon-alpha mediates suppression of C-reactive protein: Explanation for muted C-reactive protein response in lupus flares? Arthritis Rheum. 2009, 60, 3755–3760. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Neil, L.J.; Kaplan, M.J.; Carmona-Rivera, C. The Role of Neutrophils and Neutrophil Extracellular Traps in Vascular Damage in Systemic Lupus Erythematosus. J. Clin. Med. 2019, 8, 1325. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8091325
O’Neil LJ, Kaplan MJ, Carmona-Rivera C. The Role of Neutrophils and Neutrophil Extracellular Traps in Vascular Damage in Systemic Lupus Erythematosus. Journal of Clinical Medicine. 2019; 8(9):1325. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8091325
Chicago/Turabian StyleO’Neil, Liam J., Mariana J. Kaplan, and Carmelo Carmona-Rivera. 2019. "The Role of Neutrophils and Neutrophil Extracellular Traps in Vascular Damage in Systemic Lupus Erythematosus" Journal of Clinical Medicine 8, no. 9: 1325. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8091325