The Evolving Landscape of Biomarkers for Anti-PD-1 or Anti-PD-L1 Therapy

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Frequency and Phenotype of Tumor-Infiltrating Immune Cells
2.2. PD-L1 Expression by Tumor Cells and Tumor-Infiltrating Immune Cells
2.3. Tumor Mutational and Neoantigen Burden
2.4. Frequency and Phenotype of Blood-Circulating Immune Cells and Soluble Molecules
2.5. Microbiome
3. Conclusions
Summary
Author Contributions
Conflicts of Interest
References
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Swallow, M.M.; Wallin, J.J.; Sha, W.C. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by TNFalpha. Immunity 1999, 11, 423–432. [Google Scholar] [CrossRef]
- Park, J.-J.; Omiya, R.; Matsumura, Y.; Sakoda, Y.; Kuramasu, A.; Augustine, M.M.; Yao, S.; Tsushima, F.; Narazaki, H.; Anand, S.; et al. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood 2010, 116, 1291–1298. [Google Scholar] [CrossRef]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Pauken, K.E.; Eagar, T.N.; Obu, T.; Wu, J.; Tang, Q.; Azuma, M.; Krummel, M.F.; Bluestone, J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009, 10, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef] [PubMed]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yao, L.-C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.-X.; Shi, W.; Ma, A.-H.; De Vere White, R.W.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. KEYNOTE-001 Investigators Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. PACIFIC Investigators Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Redman, B.G.; Kuzel, T.M.; Harrison, M.R.; Vaishampayan, U.N.; Drabkin, H.A.; George, S.; Logan, T.F.; et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J. Clin. Oncol. 2015, 33, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. CheckMate 025 Investigators Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.-O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- Massard, C.; Gordon, M.S.; Sharma, S.; Rafii, S.; Wainberg, Z.A.; Luke, J.; Curiel, T.J.; Colon-Otero, G.; Hamid, O.; Sanborn, R.E.; et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J. Clin. Oncol. 2016, 34, 3119–3125. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Sosman, J.A.; Atkins, M.B.; Leming, P.D.; et al. Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non–Small Cell Lung Cancer Treated With Nivolumab. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. CheckMate 214 Investigators Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. KEYNOTE-407 Investigators Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. IMmotion151 Study Group Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet (Lond. Engl.) 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Kohrt, H.E.; Tumeh, P.C.; Benson, D.; Bhardwaj, N.; Brody, J.; Formenti, S.; Fox, B.A.; Galon, J.; June, C.H.; Kalos, M.; et al. Cancer Immunotherapy Trials Network (CITN) Immunodynamics: A cancer immunotherapy trials network review of immune monitoring in immuno-oncology clinical trials. J. Immunother. Cancer 2016, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring immune-checkpoint blockade: Response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet (Lond. Engl.) 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Thommen, D.S.; Koelzer, V.H.; Herzig, P.; Roller, A.; Trefny, M.; Dimeloe, S.; Kiialainen, A.; Hanhart, J.; Schill, C.; Hess, C.; et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018, 24, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e20. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.-A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Shin, D.S.; Zaretsky, J.; Frederiksen, J.; Cornish, A.; Avramis, E.; Seja, E.; Kivork, C.; Siebert, J.; Kaplan-Lefko, P.; et al. PD-1 Blockade Expands Intratumoral Memory T Cells. Cancer Immunol. Res. 2016, 4, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Zappasodi, R.; Budhu, S.; Hellmann, M.D.; Postow, M.A.; Senbabaoglu, Y.; Manne, S.; Gasmi, B.; Liu, C.; Zhong, H.; Li, Y.; et al. Non-conventional Inhibitory CD4+Foxp3−PD-1hi T Cells as a Biomarker of Immune Checkpoint Blockade Activity. Cancer Cell 2018, 33, 1017–1032.e7. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Shrimali, R.K.; Ahmad, S.; Dai, W.; Wang, H.; Lu, S.; Nandre, R.; Gaur, P.; Lopez, J.; Sade-Feldman, M.; et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1+CD38hi cells and anti-PD-1 resistance. Nat. Immunol. 2019, 20, 1231–1243. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). Available online: https://www.fda.gov/medical-devices/vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-vitro-and-imaging-tools/ (accessed on 18 August 2019).
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M. Unleashing the immune system: PD-1 and PD-Ls in the pre-treatment tumor microenvironment and correlation with response to PD-1/PD-L1 blockade. Oncoimmunology 2014, 3, e963413. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. IMpassion130 Trial Investigators Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Alerts Health Care Professionals and Oncology Clinical Investigators about an Efficacy Issue Identified in Clinical Trials for Some Patients Taking Keytruda (pembrolizumab) or Tecentriq (atezolizumab) as Monotherapy to Treat Urothelial Cancer with Low Expression of PD-L1. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-alerts-health-care-professionals-and-oncology-clinical-investigators-about-efficacy-issue (accessed on 18 August 2019).
- European Medicines Agency EMA. Available online: https://www.ema.europa.eu/en/news/ema-restricts-use-keytruda-tecentriq-bladder-cancer (accessed on 18 August 2019).
- Gourd, E. EMA restricts use of anti-PD-1 drugs for bladder cancer. Lancet. Oncol. 2018, 19, e341. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Horn, L.; Gandhi, L.; Spigel, D.R.; Antonia, S.J.; Rizvi, N.A.; Powderly, J.D.; Heist, R.S.; Carvajal, R.D.; Jackman, D.M.; et al. Overall Survival and Long-Term Safety of Nivolumab (Anti–Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2015, 33, 2004–2012. [Google Scholar] [CrossRef]
- Weichert, W.; Schwamborn, K.; Boxberg, M.; Scheel, A.; Büttner, R.; Schildhaus, H.; Schirmacher, P.; Lasitschka, F.; Kirchner, T.; Reu, S.; et al. QUIP-Ringversuch zur PD-L1 Expressionsbestimmung in nicht kleinzelligen Lungenkarzinomen (NSCLC)-German. Der Pathol. 2017, 6, 563–564. [Google Scholar]
- VENTANA. PD-L1 (SP142) Assay Interpretation Manual, TNBC. Available online: https://diagnostics.roche.com/content/dam/diagnostics/us/en/resource-center/VENTANA-PD-L1-(SP142)-Assay-Interpretation-Guide.pdf (accessed on 18 August 2019).
- Agilent. PD-L1 IHC 22C3 pharmDx, Interpretation Manual, Urothelial Carcinoma. Available online: https://www.agilent.com/en-us/library/usermanuals?N=135&Ntt=22C3&redirect=0 (accessed on 18 August 2019).
- Rimm, D.L.; Han, G.; Taube, J.M.; Yi, E.S.; Bridge, J.A.; Flieder, D.B.; Homer, R.; West, W.W.; Wu, H.; Roden, A.C.; et al. A Prospective, Multi-institutional, Pathologist-Based Assessment of 4 Immunohistochemistry Assays for PD-L1 Expression in Non-Small Cell Lung Cancer. JAMA Oncol. 2017, 3, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Schwamborn, K.; Ammann, J.U.; Knüchel, R.; Hartmann, A.; Baretton, G.; Lasitschka, F.; Schirmacher, P.; Braunschweig, T.; Tauber, R.; Erlmeier, F.; et al. Multicentric analytical comparability study of programmed death-ligand 1 expression on tumor-infiltrating immune cells and tumor cells in urothelial bladder cancer using four clinically developed immunohistochemistry assays. Virchows Arch. 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sommer, U.; Eckstein, M.; Ammann, J.; Braunschweig, T.; Macher-Goeppinger, S.; Schwamborn, K.; Hieke-Schulz, S.; Wullich, B.; Wirth, M.; Roth, W.; et al. Analytical and inter-observer comparability of 4 clinically developed programmed death-ligand 1 (PD-L1) immunohistochemistry assays in clear cell renal cell carcinoma (CCRCC). In Proceedings of the AACR Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Eckstein, M.; Erben, P.; Kriegmair, M.C.; Worst, T.S.; Weiß, C.-A.; Wirtz, R.M.; Wach, S.; Stoehr, R.; Sikic, D.; Geppert, C.I.; et al. Performance of the Food and Drug Administration/EMA-approved programmed cell death ligand-1 assays in urothelial carcinoma with emphasis on therapy stratification for first-line use of atezolizumab and pembrolizumab. Eur. J. Cancer 2019, 106, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Scheel, A.H.; Dietel, M.; Heukamp, L.C.; Jöhrens, K.; Kirchner, T.; Reu, S.; Rüschoff, J.; Schildhaus, H.-U.; Schirmacher, P.; Tiemann, M.; et al. Harmonized PD-L1 immunohistochemistry for pulmonary squamous-cell and adenocarcinomas. Mod. Pathol. 2016, 29, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Scheel, A.H.; Baenfer, G.; Baretton, G.; Dietel, M.; Diezko, R.; Henkel, T.; Heukamp, L.C.; Jasani, B.; Jöhrens, K.; Kirchner, T.; et al. Interlaboratory concordance of PD-L1 immunohistochemistry for non-small-cell lung cancer. Histopathology 2018, 72, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Eroglu, Z.; Zaretsky, J.M.; Hu-Lieskovan, S.; Kim, D.W.; Algazi, A.; Johnson, D.B.; Liniker, E.; Kong, B.; Munhoz, R.; Rapisuwon, S.; et al. High response rate to PD-1 blockade in desmoplastic melanomas. Nature 2018, 553, 347–350. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bossé, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. V STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łuksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017, 551, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef] [Green Version]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Tanemura, A.; Tada, Y.; Katayama, I.; Kumanogoh, A.; Nishikawa, H. Clinical response to PD-1 blockade correlates with a sub-fraction of peripheral central memory CD4+ T cells in patients with malignant melanoma. Int. Immunol. 2018, 30, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, M.; CelliMarchett, G.; van Zyl, B.; Tang, D.; Vilain, R.E.; van der Westhuizen, A.; Bowden, N.A. Monitoring Patient Response to Pembrolizumab With Peripheral Blood Exhaustion Marker Profiles. Front. Med. 2019, 6, 113. [Google Scholar] [CrossRef] [PubMed]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Weide, B.; Martens, A.; Hassel, J.C.; Berking, C.; Postow, M.A.; Bisschop, K.; Simeone, E.; Mangana, J.; Schilling, B.; Di Giacomo, A.M.; et al. Baseline Biomarkers for Outcome of Melanoma Patients Treated with Pembrolizumab. Clin. Cancer Res. 2016, 22, 5487–5496. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zou, J.; Hu, Y.; Li, S.; Zhou, T.; Gong, J.; Li, J.; Zhang, X.; Zhou, J.; Lu, M.; et al. Serological Markers Associated With Response to Immune Checkpoint Blockade in Metastatic Gastrointestinal Tract Cancer. JAMA Netw. Open 2019, 2, e197621. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.T.; Ozga, A.J.; Servis, R.L.; Frederick, D.T.; Lo, J.A.; Fisher, D.E.; Freeman, G.J.; Boland, G.M.; Luster, A.D. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 2019, 50, 1498–1512.e5. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Elkrief, A.; El Raichani, L.; Richard, C.; Messaoudene, M.; Belkaid, W.; Malo, J.; Belanger, K.; Miller, W.; Jamal, R.; Letarte, N.; et al. Antibiotics are associated with decreased progression-free survival of advanced melanoma patients treated with immune checkpoint inhibitors. Oncoimmunology 2019, 8, e1568812. [Google Scholar] [CrossRef] [PubMed]


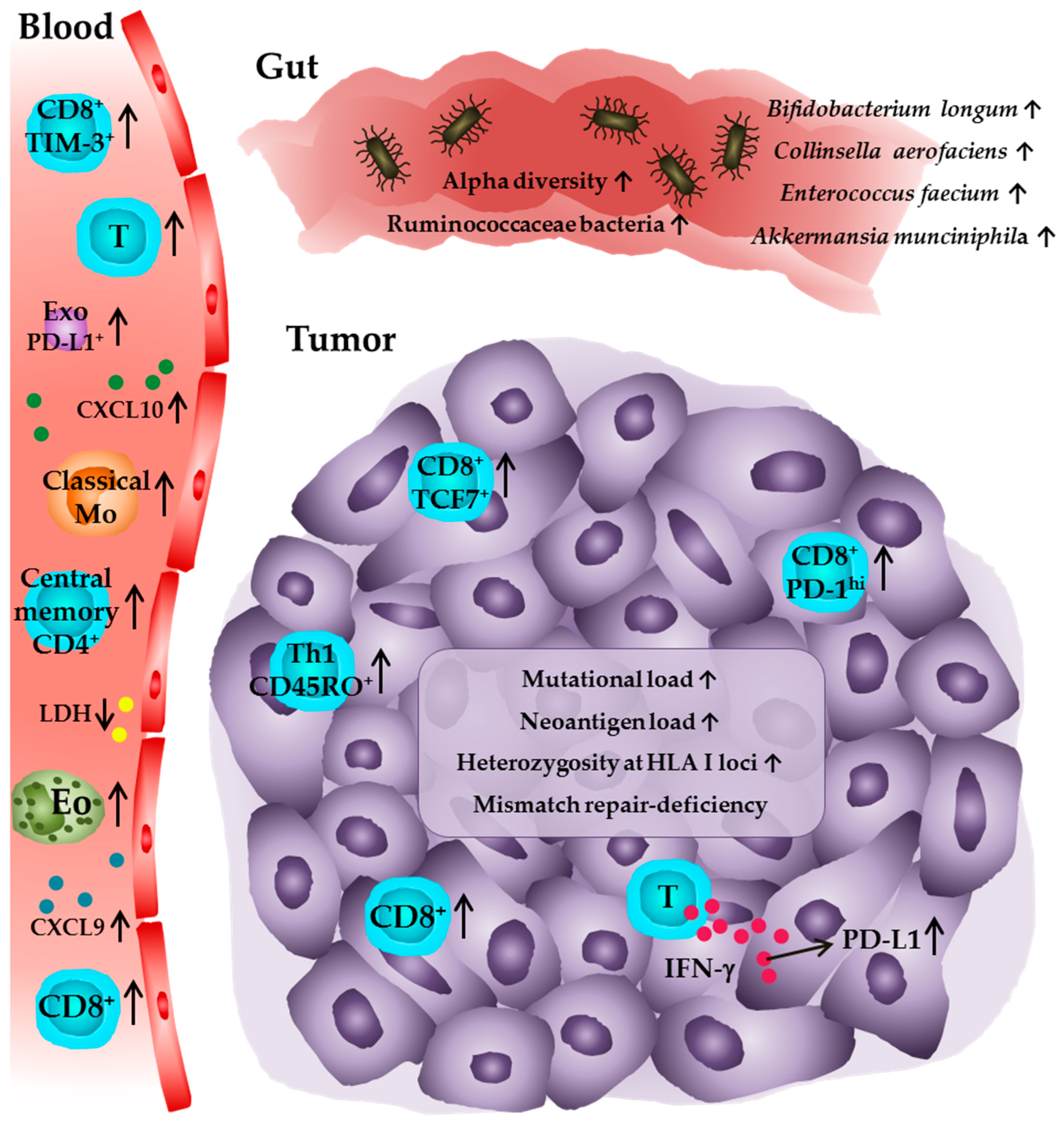{kind=link}
| Biomarker | Association with Clinical Outcome | Malignancy | Treatment | Tissue Type for Biomarker Assessment | References |
|---|---|---|---|---|---|
| Pre-existing and highly frequent tumor-infiltrating CD8+ T cells | Positive | Melanoma | Anti-PD-1 | Tumor tissue | [47,52] |
| High T effector gene signature expression | Positive | RCC | Anti-PD-L1 and anti-VEGF | Tumor tissue | [48] |
| High myeloid inflammation gene signature expression | Negative | RCC | Anti-PD-L1/Anti-PD-L1 and anti-VEGF | Tumor tissue | [48] |
| PD-1hiCD8+ T cells | Positive | NSCLC | Anti-PD-1 | Tumor tissue | [49] |
| Ratio memory-like to exhausted CD8+ T cells | Positive | Melanoma | Anti-PD-1 and/or anti-CTLA-4 | Tumor tissue | [50] |
| TCF7+CD8+ T cells | Positive | Melanoma | Anti-PD-1 | Tumor tissue | [50] |
| CD4+FoxP3-PD-1hi T cells | Negative | NSCLC | Anti-PD-1 | Tumor tissue | [53] |
| PD-1+CD38hiCD8+ T cells | Negative | Melanoma | Anti-PD-1 | Tumor tissue, blood | [54] |
| PD-L1 expression | Positive | Multiple cancer types | Anti-PD-1/Anti-PD-L1 | Tumor tissue | [56,57,58] |
| Tumor mutational and neoantigen load | Positive | NSCLC | Anti-PD-1/Anti-PD-L1 | Tumor tissue, blood | [73,79,95] |
| Mismatch repair-deficiency | Positive | Multiple cancer types | Anti-PD-1 | Tumor tissue | [74,75,76] |
| Loss-of-function mutations in PBRM1 | Positive | RCC | Anti-PD-1/Anti-PD-L/Anti-PD-1 or anti-PD-L1 and anti-CTLA-4 | Tumor tissue | [77] |
| STK11/LKB1 mutations | Negative | KRAS-mutant lung adeno-carcinoma | Anti-PD-1 | Tumor tissue | [78] |
| Reduction of mutational and neoantigen load under therapy | Positive | Melanoma | Anti-PD-1 | Tumor tissue | [81] |
| HLA class I heterozygosity | Positive | Melanoma, NSCLC | Anti-PD-1 and/or anti-CTLA-4 | Blood | [82] |
| JAK1/JAK2 mutations | Negative | Melanoma | Anti-PD-1 | Tumor tissue | [83] |
| B2M mutations | Negative | Melanoma, NSCLC | Anti-PD-1 | Tumor tissue | [83,84,85] |
| PD-1+CD8+ T cells | Positive | NSCLC | Anti-PD-1 | Blood | [86] |
| CD27+Fas-CD45RA-CCR7+CD4+ T cells | Positive | Melanoma | Anti-PD-1 | Blood | [87] |
| PD-1+CD4+ T cells | Positive | Melanoma | Anti-PD-1 | Blood | [89] |
| TIM-3+CD8+ T cells | Positive | Melanoma | Anti-PD-1 | Blood | [89] |
| Classical monocytes | Positive | Melanoma | Anti-PD-1 | Blood | [90] |
| High relative eosinophil counts | Positive | Melanoma | Anti-PD-1 | Blood | [91] |
| High relative lymphocyte counts | Positive | Melanoma | Anti-PD-1 | Blood | [91] |
| Low LDH | Positive | Melanoma | Anti-PD-1 | Blood | [91] |
| Low MCP1, LIF, CTLA-4 | Negative | Gastro-intestinal cancer | Anti-PD-1/Anti-PD-L1/Anti-CTLA-4 | Blood | [92] |
| High CXCL9 and CXCL10 | Positive | Melanoma | Anti-PD-1/Anti-PD-1 and anti-CTLA-4 | Blood | [93] |
| PD-L1+ Exosomes | Positive | Melanoma | Anti-PD-1 | Blood | [94] |
| High alpha diversity | Positive | Melanoma | Anti-PD-1 | Gut | [97] |
| Bacteria of the Ruminococcaceae family | Positive | Melanoma | Anti-PD-1 | Gut | [97] |
| Bacteria of the Bacteroidales order | Negative | Melanoma | Anti-PD-1 | Gut | [97] |
| Bifidobacterium longum, Collinsella aerofaciens, Enterococcus faecium | Positive | Melanoma | Anti-PD-1 | Gut | [99] |
| Akkermansia munciniphila | Positive | NSCLC, RCC, urothelial carcinoma | Anti-PD-1 | Gut | [98] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tunger, A.; Sommer, U.; Wehner, R.; Kubasch, A.S.; Grimm, M.-O.; Bachmann, M.P.; Platzbecker, U.; Bornhäuser, M.; Baretton, G.; Schmitz, M. The Evolving Landscape of Biomarkers for Anti-PD-1 or Anti-PD-L1 Therapy. J. Clin. Med. 2019, 8, 1534. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8101534
Tunger A, Sommer U, Wehner R, Kubasch AS, Grimm M-O, Bachmann MP, Platzbecker U, Bornhäuser M, Baretton G, Schmitz M. The Evolving Landscape of Biomarkers for Anti-PD-1 or Anti-PD-L1 Therapy. Journal of Clinical Medicine. 2019; 8(10):1534. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8101534
Chicago/Turabian StyleTunger, Antje, Ulrich Sommer, Rebekka Wehner, Anne Sophie Kubasch, Marc-Oliver Grimm, Michael Philipp Bachmann, Uwe Platzbecker, Martin Bornhäuser, Gustavo Baretton, and Marc Schmitz. 2019. "The Evolving Landscape of Biomarkers for Anti-PD-1 or Anti-PD-L1 Therapy" Journal of Clinical Medicine 8, no. 10: 1534. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8101534