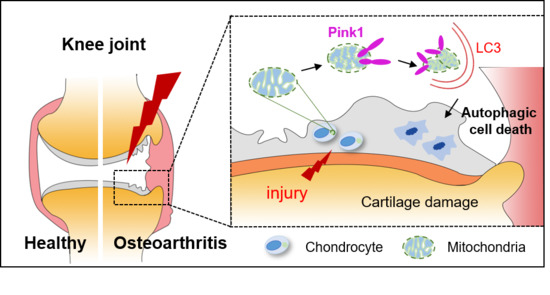
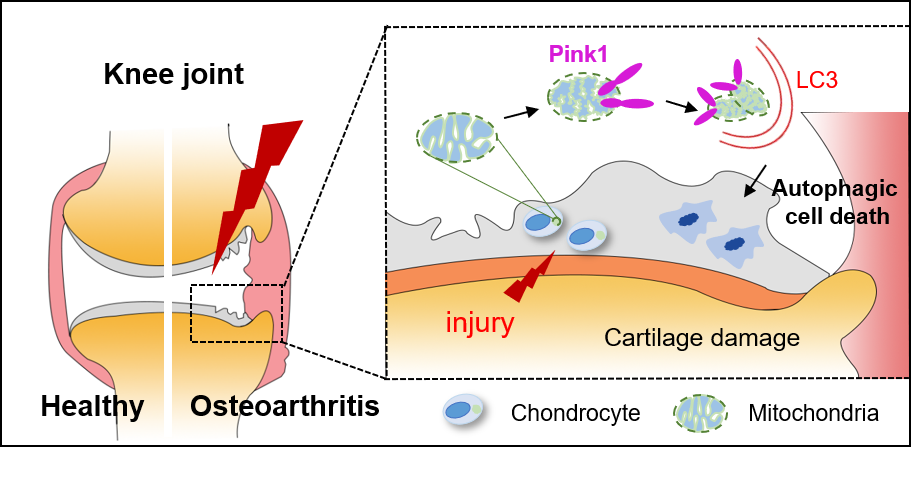
Pink1-Mediated Chondrocytic Mitophagy Contributes to Cartilage Degeneration in Osteoarthritis

 , , , , and
, , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
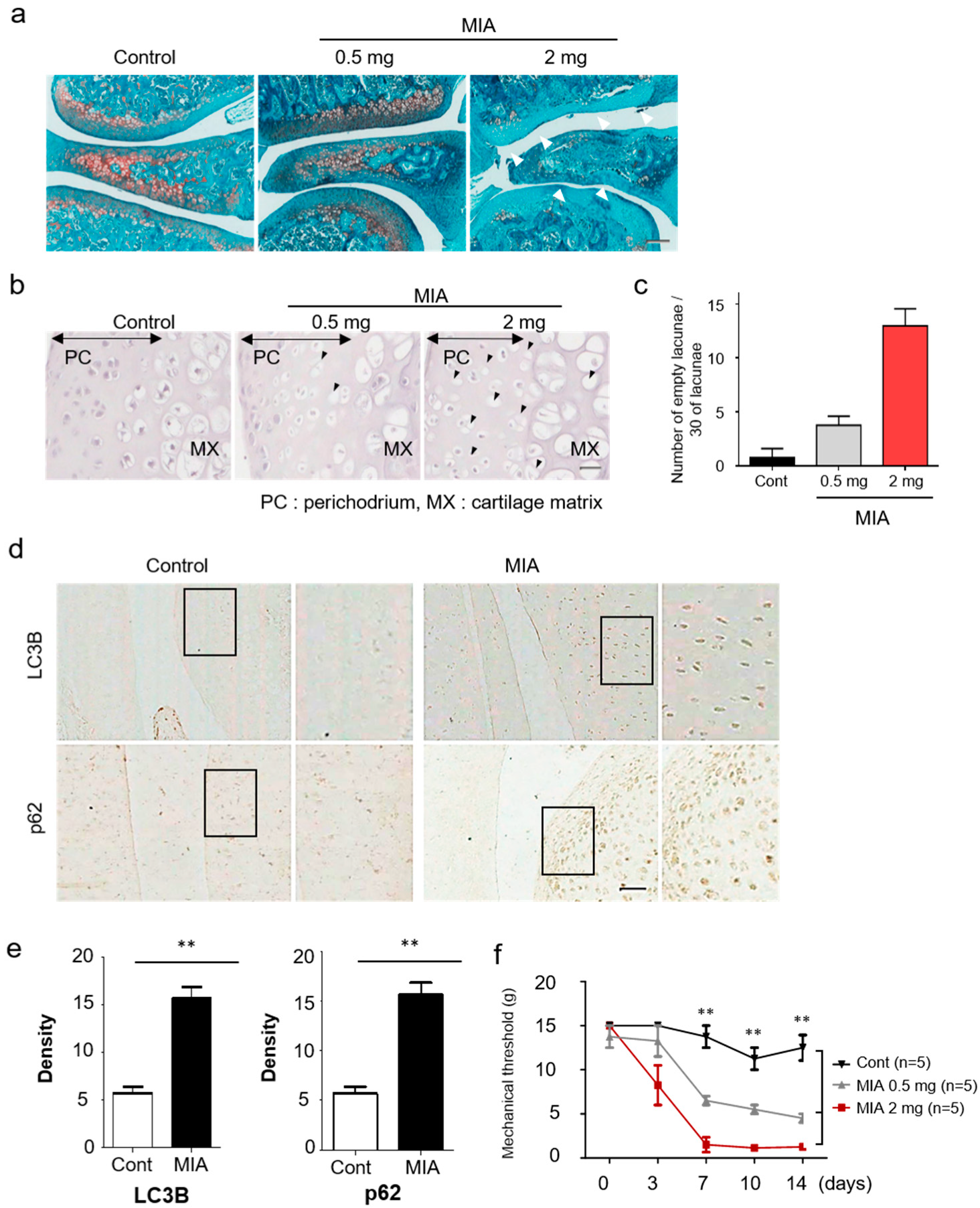
2.1. Animals and Arthritis Models
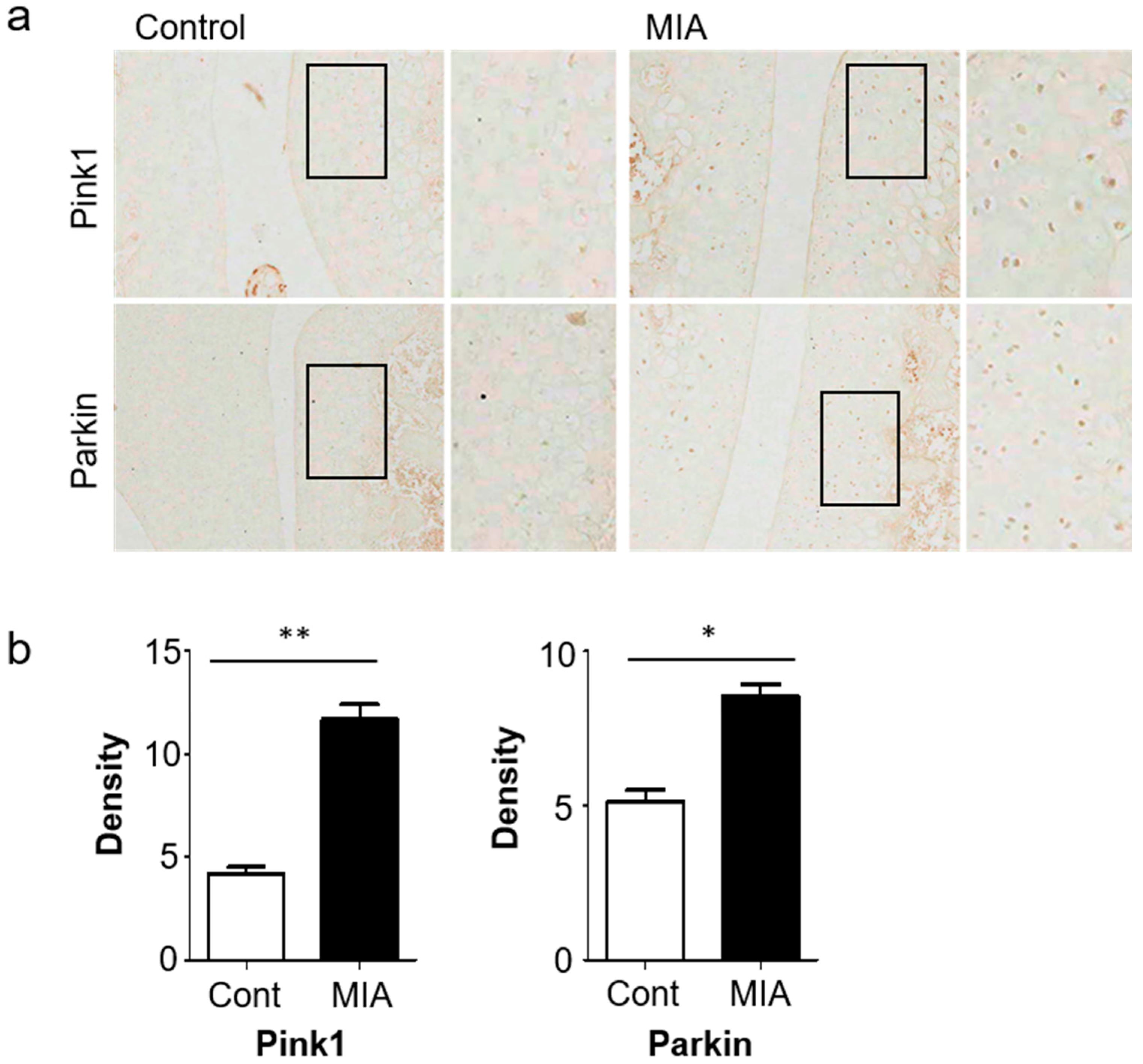
2.2. Histology and Immunohistochemistry
2.3. Chondrocyte Isolation and Culture Condition
2.4. Sucrose Gradient for Mitochondria Fraction
2.5. Behavioral Testing
2.6. Antibodies and Reagents
2.7. Quantitative Real-Time PCR
2.8. Statistical Analysis
3. Results
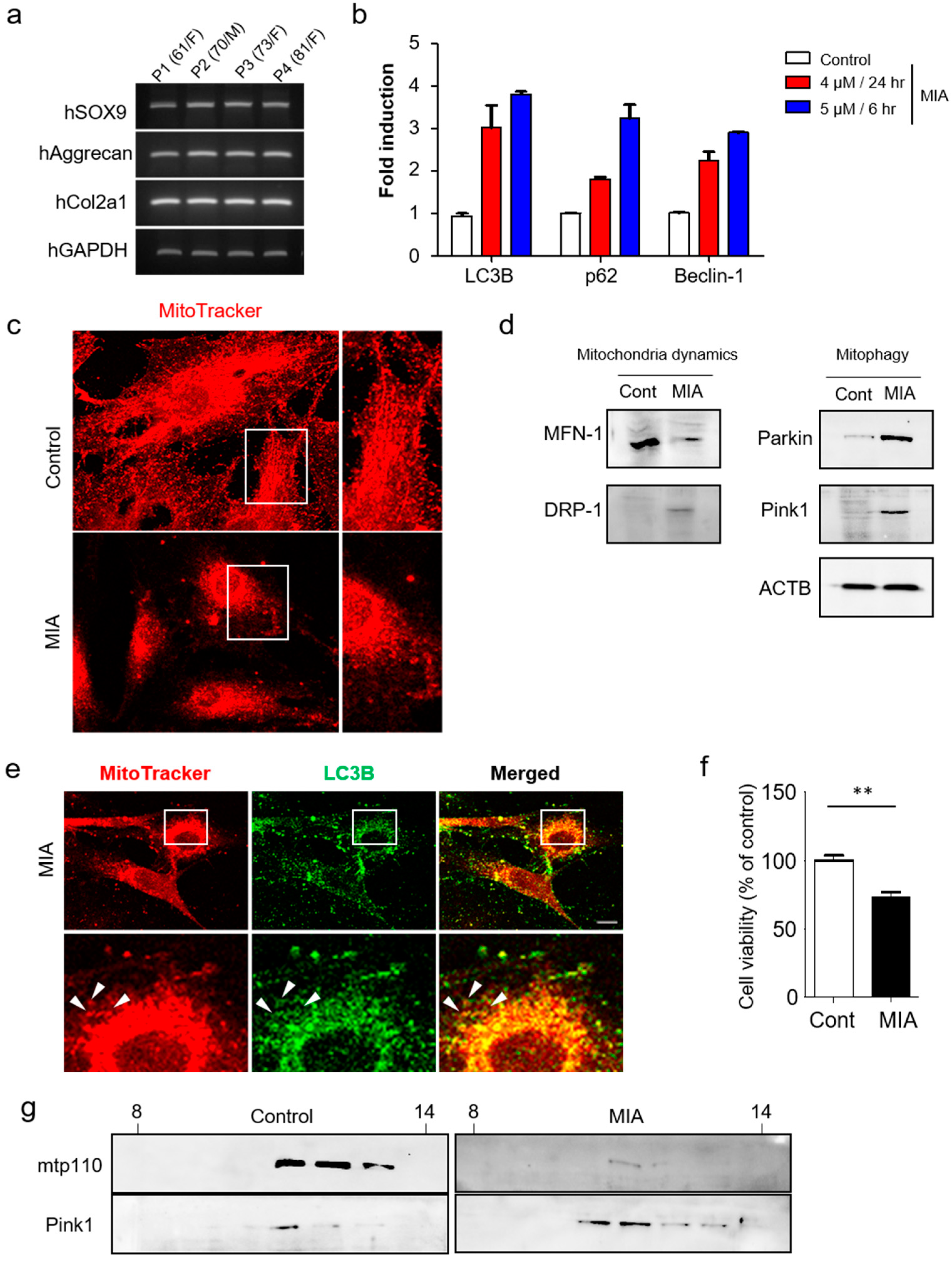
3.1. Pink1 and Parkin Expressions Increase in a MIA-Induced Osteoarthritis Model
3.2. Pink1-Mediated Mitophagy is Involved in Mitochondrial Fragmentation and Cell Death in Human Primary Chondrocytes
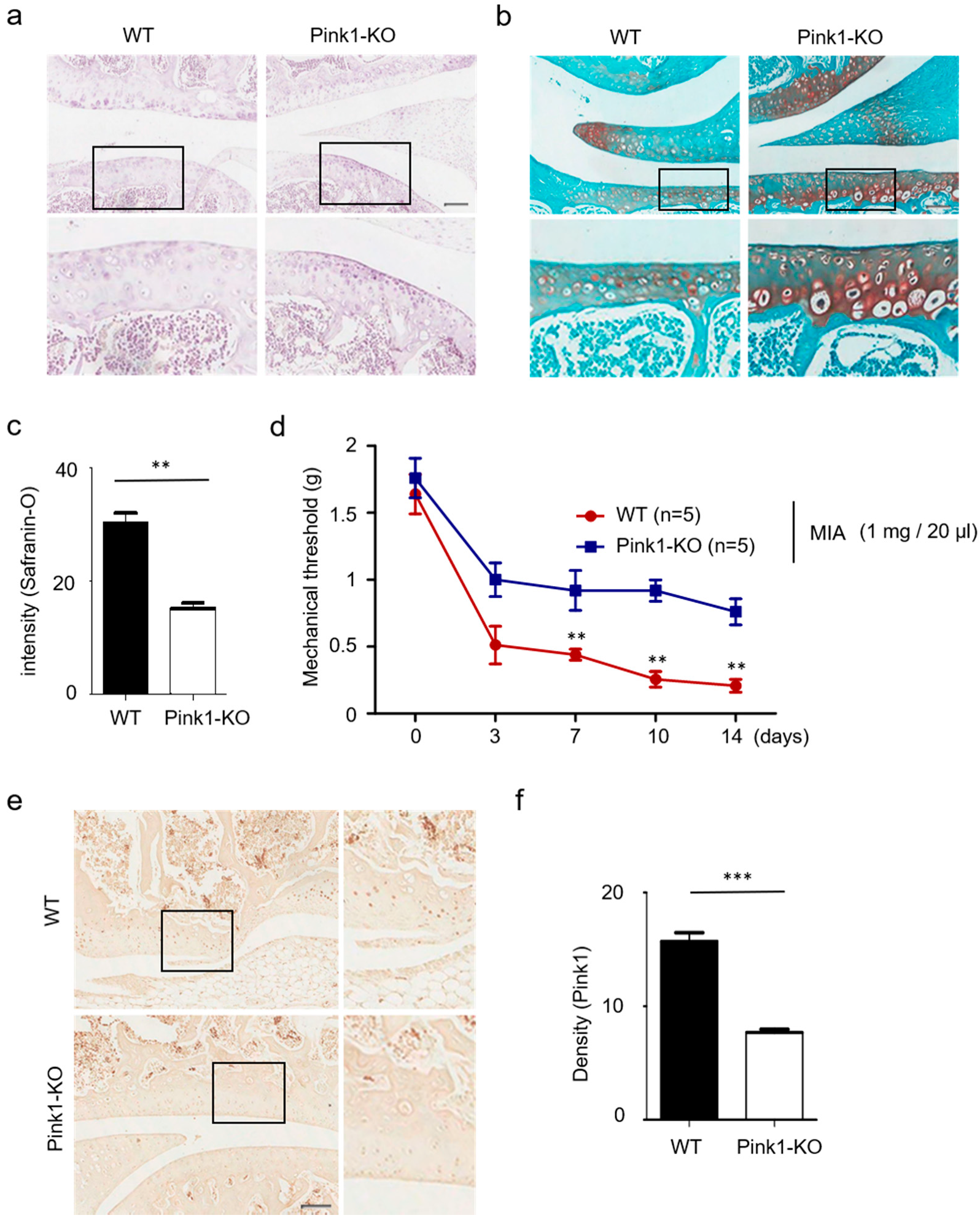
3.3. Pink1 Knock-Out Decreases Cartilage Damage in MIA-Induced Osteoarthritis
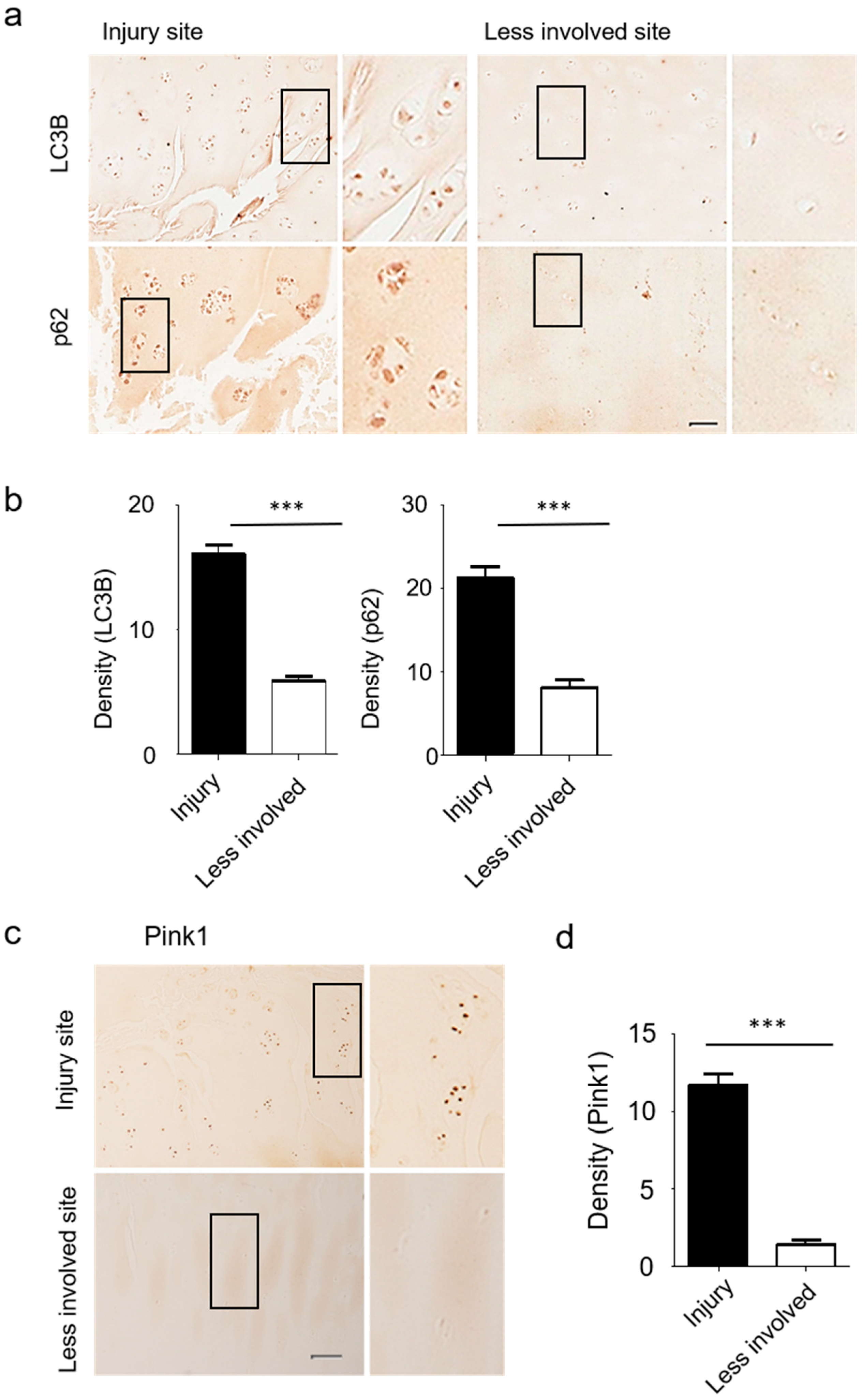
3.4. Autophagy and Mitophagy-Related Genes are Highly Expressed in Human Osteoarthritic Cartilage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Ethics Approval
References
- Choi, H.S.; Im, S.; Park, J.W.; Suh, H.J. Protective Effect of Deer Bone Oil on Cartilage Destruction in Rats with Monosodium Iodoacetate (MIA)-Induced Osteoarthritis. Biol. Pharm. Bull. 2016, 39, 2042–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Zhang, T.; Sun, H.; Wang, S.; Liu, M. Protective effects of dioscin against cartilage destruction in a monosodium iodoacetate (MIA)-indcued osteoarthritis rat model. Biomed. Pharmacother. 2018, 108, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, T.; Sousa-Valente, J.; Malcangio, M. The Monoiodoacetate Model of Osteoarthritis Pain in the Mouse. J. Vis. Exp. 2016, 111. [Google Scholar] [CrossRef] [PubMed]
- Chiu, P.R.; Hu, Y.C.; Huang, T.C.; Hsieh, B.S.; Yeh, J.P.; Cheng, H.L.; Huang, L.W.; Chang, K.L. Vitamin C Protects Chondrocytes against Monosodium Iodoacetate-Induced Osteoarthritis by Multiple Pathways. Int. J. Mol. Sci. 2016, 18, 38. [Google Scholar] [CrossRef]
- Das, G.C.; Hollinger, F.B. Molecular pathways for glucose homeostasis, insulin signaling and autophagy in hepatitis C virus induced insulin resistance in a cellular model. Virology 2012, 434, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Yan, X.; Yu, L. The late stage of autophagy: Cellular events and molecular regulation. Protein Cell 2010, 1, 907–915. [Google Scholar] [CrossRef]
- Ekiz, H.A.; Can, G.; Baran, Y. Role of autophagy in the progression and suppression of leukemias. Crit. Rev. Oncol. Hematol. 2012, 81, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Wang, W.; Zhang, H.; Hu, Y.; Wang, M.; Yin, Z. The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int. J. Mol. Med. 2013, 32, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Cheng, N.T.; Meng, H.; Ma, L.F.; Zhang, L.; Yu, H.M.; Wang, Z.Z.; Guo, A. Role of autophagy in the progression of osteoarthritis: The autophagy inhibitor, 3-methyladenine, aggravates the severity of experimental osteoarthritis. Int. J. Mol. Med. 2017, 39, 1224–1232. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Nieminen, A.L.; Qian, T.; Trost, L.C.; Elmore, S.P.; Nishimura, Y.; Crowe, R.A.; Cascio, W.E.; Bradham, C.A.; Brenner, D.A.; et al. The mitochondrial permeability transition in cell death: A common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta 1998, 1366, 177–196. [Google Scholar] [CrossRef]
- Stolz, A.; Dikic, I. PINK1-PARKIN interplay: Down to ubiquitin phosphorylation. Mol. Cell 2014, 56, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.H.; Shin, J.; Shin, N.; Yin, Y.; Lee, S.Y.; Kim, C.S.; Kim, S.R.; Zhang, E.; Kim, D.W. PINK1 mediates spinal cord mitophagy in neuropathic pain. J. Pain Res. 2019, 12, 1685–1699. [Google Scholar] [CrossRef] [PubMed]
- Cetrullo, S.; D’Adamo, S.; Guidotti, S.; Borzi, R.M.; Flamigni, F. Hydroxytyrosol prevents chondrocyte death under oxidative stress by inducing autophagy through sirtuin 1-dependent and -independent mechanisms. Biochim. Biophys. Acta 2016, 1860, 1181–1191. [Google Scholar] [CrossRef]
- Wang, L.; Cho, Y.L.; Tang, Y.; Wang, J.; Park, J.E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 2018, 28, 787–802. [Google Scholar] [CrossRef]
- Choi, I.; Kim, J.; Jeong, H.K.; Kim, B.; Jou, I.; Park, S.M.; Chen, L.; Kang, U.J.; Zhuang, X.; Joe, E.H. PINK1 deficiency attenuates astrocyte proliferation through mitochondrial dysfunction, reduced AKT and increased p38 MAPK activation, and downregulation of EGFR. Glia 2013, 61, 800–812. [Google Scholar] [CrossRef]
- Bakker, B.; Eijkel, G.B.; Heeren, R.M.; Karperien, M.; Post, J.N.; Cillero-Pastor, B. Oxygen Regulates Lipid Profiles in Human Primary Chondrocyte Cultures. Osteoarthr. Cartil. 2016, 24, S456–S457. [Google Scholar] [CrossRef]
- Clayton, D.A.; Shadel, G.S. Purification of mitochondria by sucrose step density gradient centrifugation. Cold Spring Harb. Protoc. 2014, 2014, pdb prot080028. [Google Scholar] [CrossRef]
- Wattiaux, R.; Wattiaux-De Coninck, S.; Ronveaux-Dupal, M.F. Deterioration of rat-liver mitochondria during centrifugation in a sucrose gradient. Eur. J. Biochem. 1971, 22, 31–39. [Google Scholar] [CrossRef]
- Taylor, S.W.; Warnock, D.E.; Glenn, G.M.; Zhang, B.; Fahy, E.; Gaucher, S.P.; Capaldi, R.A.; Gibson, B.W.; Ghosh, S.S. An alternative strategy to determine the mitochondrial proteome using sucrose gradient fractionation and 1D PAGE on highly purified human heart mitochondria. J. Proteome Res. 2002, 1, 451–458. [Google Scholar] [CrossRef]
- Kim, S.H.; Chung, J.M. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992, 50, 355–363. [Google Scholar] [CrossRef]
- Jiang, L.; Li, L.; Geng, C.; Gong, D.; Jiang, L.; Ishikawa, N.; Kajima, K.; Zhong, L. Monosodium iodoacetate induces apoptosis via the mitochondrial pathway involving ROS production and caspase activation in rat chondrocytes in vitro. J. Orthop. Res. 2013, 31, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; de Crombrugghe, B. Toward understanding SOX9 function in chondrocyte differentiation. Matrix Biol. 1998, 16, 529–540. [Google Scholar] [CrossRef]
- Shi, S.; Mercer, S.; Eckert, G.J.; Trippel, S.B. Regulation of articular chondrocyte aggrecan and collagen gene expression by multiple growth factor gene transfer. J. Orthop. Res. 2012, 30, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Ghayor, C.; Herrouin, J.F.; Chadjichristos, C.; Ala-Kokko, L.; Takigawa, M.; Pujol, J.P.; Galera, P. Regulation of human COL2A1 gene expression in chondrocytes. Identification of C-Krox-responsive elements and modulation by phenotype alteration. J. Biol. Chem. 2000, 275, 27421–27438. [Google Scholar] [CrossRef]
- Santel, A.; Frank, S.; Gaume, B.; Herrier, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003, 116, 2763–2774. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Westrate, L.M.; Wu, H.X.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Uno, M.; Koyano, F.; Go, E.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. A Dimeric PINK1-containing Complex on Depolarized Mitochondria Stimulates Parkin Recruitment. J. Biol. Chem. 2013, 288, 36372–36384. [Google Scholar] [CrossRef] [Green Version]
- Soto-Hermida, A.; Fernández-Moreno, M.; Oreiro, N.; Fernández-López, C.; Pértega, S.; Cortés-Pereira, E.; Rego-Pérez, I.; Blanco, F.J. Mitochondrial DNA (mtDNA) haplogroups influence the progression of knee osteoarthritis. Data from the Osteoarthritis Initiative (OAI). PLoS ONE. 2014, 9, e112735. [Google Scholar] [CrossRef]
- Charlier, E.; Relic, B.; Deroyer, C.; Malaise, O.; Neuville, S.; Collee, J.; Malaise, M.G.; De Seny, D. Insights on Molecular Mechanisms of Chondrocytes Death in Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 2146. [Google Scholar] [CrossRef]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.; El-Sherbeny, A.; Roon, P.; Schoenlein, P.V.; Ganapathy, V.; Smith, S.B. Apoptotic cell death in the mouse retinal ganglion cell layer is induced in vivo by the excitatory amino acid homocysteine. Exp. Eye Res. 2001, 73, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Berenbaum, F.; Griffin, T.M.; Liu-Bryan, R. Metabolic Regulation of Inflammation in Osteoarthritis. Arthritis Rheumatol. 2017, 69, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Rayman, M.P.; Gualillo, O.; Sellam, J.; van der Kraan, P.; Fearon, U. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2017, 13, 302–311. [Google Scholar] [CrossRef]
- Ryter, S.W.; Cloonan, S.M.; Choi, A.M.K. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 2013, 36, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Li, Q.; Wang, H.Y.; Zhang, J.L.; Du, J.; Feng, H.M.; Blumwald, E.; Yu, L.; Xu, G.H. Two NHX-type transporters from Helianthus tuberosus improve the tolerance of rice to salinity and nutrient deficiency stress. Plant Biotechnol. J. 2018, 16, 310–321. [Google Scholar] [CrossRef]
- Kissova, I.; Deffieu, M.; Manon, S.; Camougrand, N. Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem. 2004, 279, 39068–39074. [Google Scholar] [CrossRef]
- Cover, C.; Mansouri, A.; Knight, T.R.; Bajt, M.L.; Lemasters, J.J.; Pessayre, D.; Jaeschke, H. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J. Pharmacol. Exp. Ther. 2005, 315, 879–887. [Google Scholar] [CrossRef]
- Escalera-Rivera, K.; Catheline, S.; Eliseev, R.; Jonason, J. The role of mitochondrial dysfunction in the development of post-traumatic osteoarthritis. J. Bone Miner. Res. 2018, 33, 74. [Google Scholar]
- Weihofen, A.; Thomas, K.J.; Ostaszewski, B.L.; Cookson, M.R.; Selkoe, D.J. Pink1 Forms a Multiprotein Complex with Miro and Milton, Linking Pink1 Function to Mitochondrial Trafficking. Biochemistry-Us 2009, 48, 2045–2052. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.T.; Mi, L.; Wang, T.; Yuan, L.; Li, X.H.; Dong, L.S.; Zhao, P.; Fu, J.L.; Yao, B.Y.; Zhou, Z.C. PINK1/Parkin-mediated mitophagy play a protective role in manganese induced apoptosis in SH-SY5Y cells. Toxicol. In Vitro 2016, 34, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.F.; Ni, H.; Li, L.L. Leptin Maintained Zinc Homeostasis Against Glutamate-Induced Excitotoxicity by Preventing Mitophagy-Mediated Mitochondrial Activation in HT22 Hippocampal Neuronal Cells. Front. Neurol. 2018, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Proïcs, E.; Rubio-Patiño, C.; Obba, S.; Zunino, B.; Bossowski, J.P.; Rozier, R.M.; Chiche, J.; Mondragón, L.; Riley, J.S.; et al. Parkin-Independent Mitophagy Controls Chemotherapeutic Response in Cancer Cells. Cell Rep. 2017, 20, 2846–2859. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, H.J.; Park, H.; Shin, N.; Kwon, H.H.; Yin, Y.; Hwang, J.-A.; Song, H.-J.; Kim, J.; Kim, D.W.; Beom, J. Pink1-Mediated Chondrocytic Mitophagy Contributes to Cartilage Degeneration in Osteoarthritis. J. Clin. Med. 2019, 8, 1849. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8111849
Shin HJ, Park H, Shin N, Kwon HH, Yin Y, Hwang J-A, Song H-J, Kim J, Kim DW, Beom J. Pink1-Mediated Chondrocytic Mitophagy Contributes to Cartilage Degeneration in Osteoarthritis. Journal of Clinical Medicine. 2019; 8(11):1849. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8111849
Chicago/Turabian StyleShin, Hyo Jung, Hyewon Park, Nara Shin, Hyeok Hee Kwon, Yuhua Yin, Jeong-Ah Hwang, Hee-Jung Song, Jinhyun Kim, Dong Woon Kim, and Jaewon Beom. 2019. "Pink1-Mediated Chondrocytic Mitophagy Contributes to Cartilage Degeneration in Osteoarthritis" Journal of Clinical Medicine 8, no. 11: 1849. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8111849