Landscape of Tumor Suppressor Mutations in Acute Myeloid Leukemia


 , and
, and
Abstract
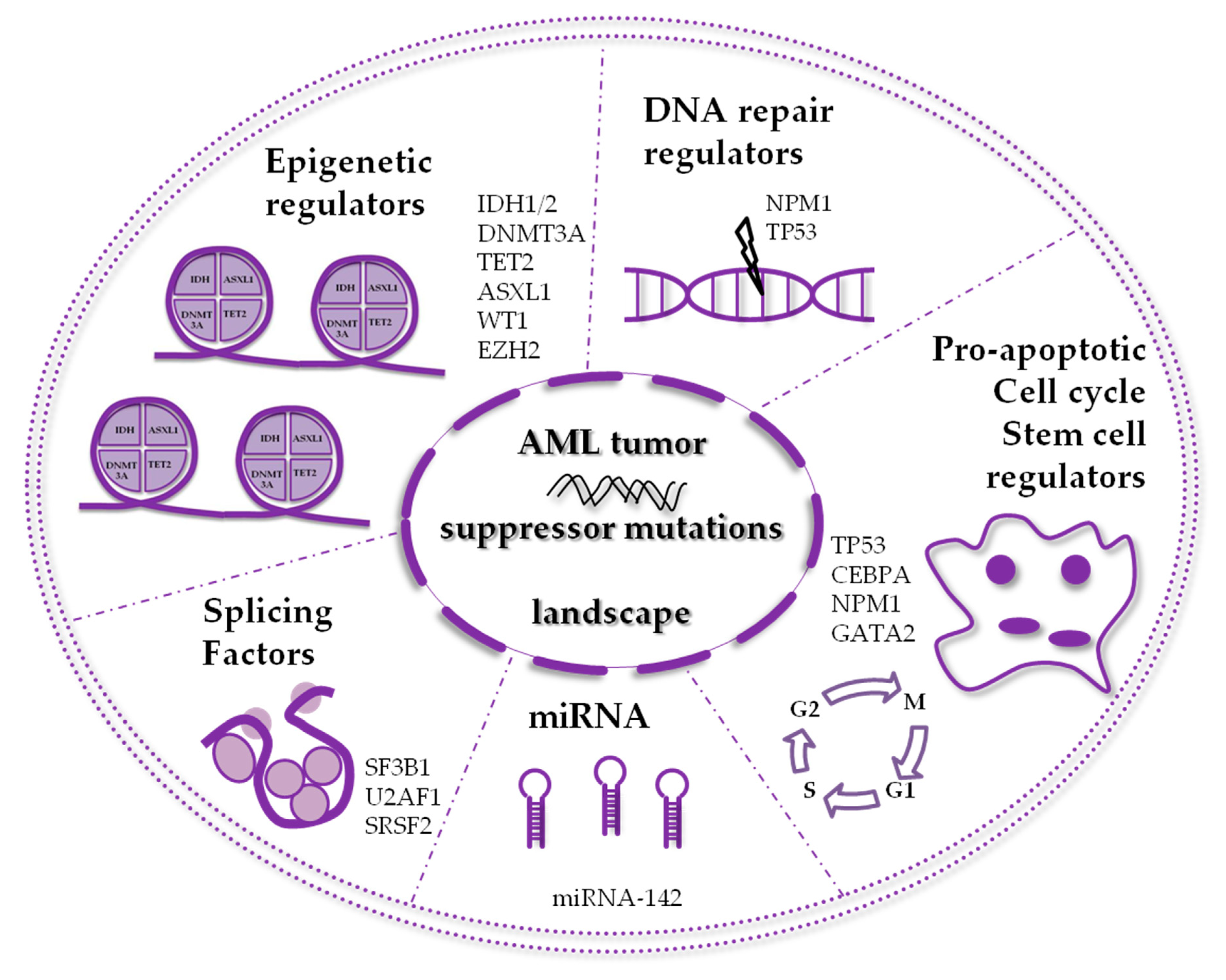
:1. Introduction
2. AML Mutated Tumor Suppressors Involved in Epigenetic Mechanisms
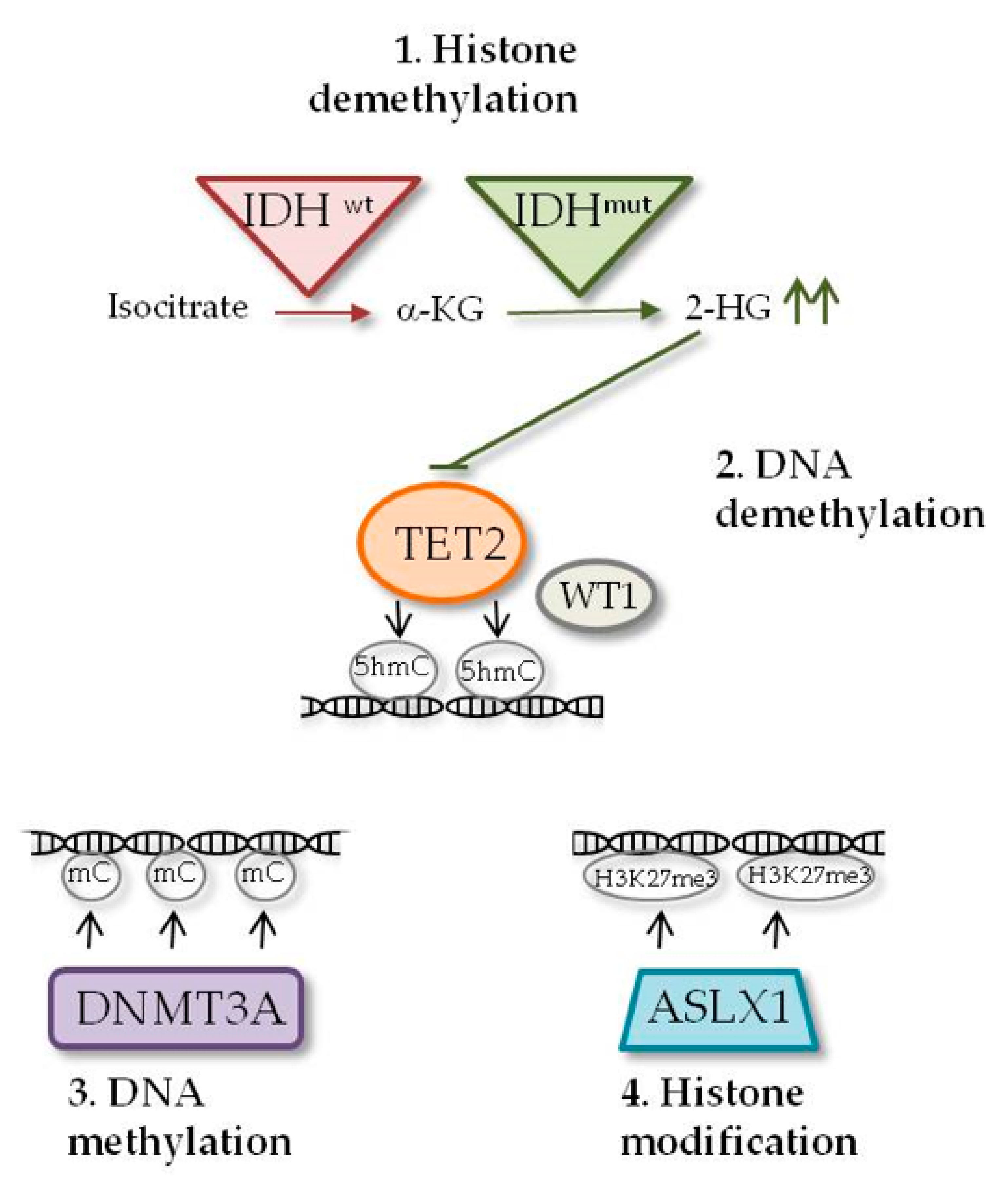
2.1. IDH1 and IDH2 Mutations
2.2. DNMT3A Mutations
2.3. TET2 Mutations
2.4. WT1 Mutations
2.5. ASXL1 Mutations
3. AML Mutated Tumor Suppressors Involved in Non-epigenetic Mechanisms
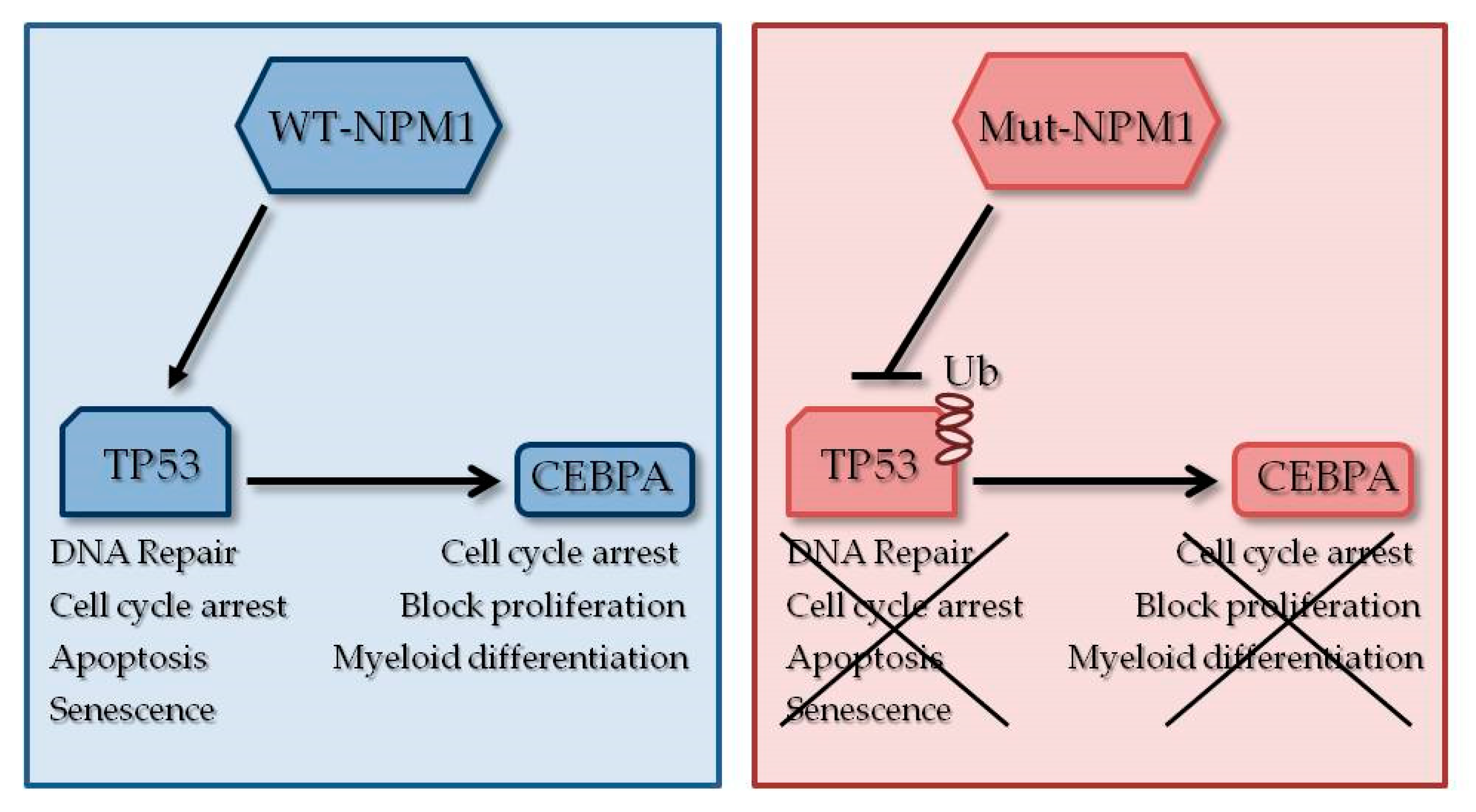
3.1. NPM1 Mutations
3.2. CEBPA Mutations
3.3. TP53 Mutations
4. Other Relevant Mutated Tumor Suppressors
4.1. EZH2 Mutations
4.2. Splicing Factors Mutations
4.3. miRNA Mutations
4.4. GATA2 Mutations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- Grimwade, D. The changing paradigm of prognostic factors in acute myeloid leukaemia. Best Pract. Research. Clin. Haematol. 2012, 25, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, L.; Metzeler, K.H. Clonal hematopoiesis and preleukemia-Genetics, biology, and clinical implications. Geneschromosomes Cancer 2019, 58, 828–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corces, M.R.; Chang, H.Y.; Majeti, R. Preleukemic Hematopoietic Stem Cells in Human Acute Myeloid Leukemia. Front. Oncol. 2017, 7, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.; Andersson, T.M.; Rachet, B.; Bjorkholm, M.; Lambert, P.C. Survival and cure of acute myeloid leukaemia in England, 1971–2006: A population-based study. Br. J. Haematol. 2013, 162, 509–516. [Google Scholar] [CrossRef]
- Sill, H.; Olipitz, W.; Zebisch, A.; Schulz, E.; Wolfler, A. Therapy-related myeloid neoplasms: Pathobiology and clinical characteristics. Br. J. Pharmacol. 2011, 162, 792–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavianpour, M.; Ahmadzadeh, A.; Shahrabi, S.; Saki, N. Significance of oncogenes and tumor suppressor genes in AML prognosis. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 10041–10052. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Rangel, F.A.; Chavez-Valencia, V.; Gomez-Guijosa, M.A.; Cortes-Penagos, C. Acute Myeloid Leukemia-Genetic Alterations and Their Clinical Prognosis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 328–339. [Google Scholar]
- de Jonge, H.J.; Huls, G.; de Bont, E.S. Gene expression profiling in acute myeloid leukaemia. Neth. J. Med. 2011, 69, 167–176. [Google Scholar]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.P.; Gonen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N.; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivarov, V.; Bullinger, L. Expression profiling of leukemia patients: Key lessons and future directions. Exp. Hematol. 2014, 42, 651–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Lee, S.; Urman, A.; Desai, P. Emerging drug profile: Krebs cycle and cancer: IDH mutations and therapeutic implications. Leuk. Lymphoma 2019, 60, 2635–2645. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakheja, D.; Medeiros, L.J.; Bevan, S.; Chen, W. The emerging role of d-2-hydroxyglutarate as an oncometabolite in hematolymphoid and central nervous system neoplasms. Front. Oncol. 2013, 3, 169. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Gong, Y. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomark. Res. 2019, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaidzik, V.I.; Paschka, P.; Spath, D.; Habdank, M.; Kohne, C.H.; Germing, U.; von Lilienfeld-Toal, M.; Held, G.; Horst, H.A.; Haase, D.; et al. TET2 mutations in acute myeloid leukemia (AML): Results from a comprehensive genetic and clinical analysis of the AML study group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 1350–1357. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Kronke, J.; Bullinger, L.; Spath, D.; Kayser, S.; Zucknick, M.; Gotze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef]
- Ok, C.Y.; Loghavi, S.; Sui, D.; Wei, P.; Kanagal-Shamanna, R.; Yin, C.C.; Zuo, Z.; Routbort, M.J.; Tang, G.; Tang, Z.; et al. Persistent IDH1/2 mutations in remission can predict relapse in patients with acute myeloid leukemia. Haematologica 2019, 104, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Nassereddine, S.; Lap, C.J.; Haroun, F.; Tabbara, I. The role of mutant IDH1 and IDH2 inhibitors in the treatment of acute myeloid leukemia. Ann. Hematol. 2017, 96, 1983–1991. [Google Scholar] [CrossRef]
- Ye, D.; Xiong, Y.; Guan, K.L. The mechanisms of IDH mutations in tumorigenesis. Cell Res. 2012, 22, 1102–1104. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.; Wang, Z.; Okano, M.; Nogami, M.; Li, Y.; He, W.W.; Okumura, K.; Li, E. Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene 1999, 236, 87–95. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef]
- Yuan, X.Q.; Chen, P.; Du, Y.X.; Zhu, K.W.; Zhang, D.Y.; Yan, H.; Liu, H.; Liu, Y.L.; Cao, S.; Zhou, G.; et al. Influence of DNMT3A R882 mutations on AML prognosis determined by the allele ratio in Chinese patients. J. Transl. Med. 2019, 17, 220. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627, quiz 3699. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Roller, A.; Grossmann, V.; Bacher, U.; Poetzinger, F.; Weissmann, S.; Nadarajah, N.; Boeck, L.; Kern, W.; Haferlach, C.; Schnittger, S.; et al. Landmark analysis of DNMT3A mutations in hematological malignancies. Leukemia 2013, 27, 1573–1578. [Google Scholar] [CrossRef]
- Gale, R.E.; Lamb, K.; Allen, C.; El-Sharkawi, D.; Stowe, C.; Jenkinson, S.; Tinsley, S.; Dickson, G.; Burnett, A.K.; Hills, R.K.; et al. Simpson’s Paradox and the Impact of Different DNMT3A Mutations on Outcome in Younger Adults With Acute Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2072–2083. [Google Scholar] [CrossRef] [Green Version]
- Gaidzik, V.I.; Schlenk, R.F.; Paschka, P.; Stolzle, A.; Spath, D.; Kuendgen, A.; von Lilienfeld-Toal, M.; Brugger, W.; Derigs, H.G.; Kremers, S.; et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: Results of the AML Study Group (AMLSG). Blood 2013, 121, 4769–4777. [Google Scholar] [CrossRef] [Green Version]
- Russler-Germain, D.A.; Spencer, D.H.; Young, M.A.; Lamprecht, T.L.; Miller, C.A.; Fulton, R.; Meyer, M.R.; Erdmann-Gilmore, P.; Townsend, R.R.; Wilson, R.K.; et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 2014, 25, 442–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Zhao, H.; Hardikar, S.; Singh, A.K.; Goodell, M.A.; Chen, T. A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood 2013, 122, 4086–4089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holz-Schietinger, C.; Matje, D.M.; Reich, N.O. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J. Biol. Chem. 2012, 287, 30941–30951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Rodriguez, B.; Mayle, A.; Park, H.J.; Lin, X.; Luo, M.; Jeong, M.; Curry, C.V.; Kim, S.B.; Ruau, D.; et al. DNMT3A Loss Drives Enhancer Hypomethylation in FLT3-ITD-Associated Leukemias. Cancer Cell 2016, 30, 363–365. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.E.; Qin, T.; Muench, D.E.; Masuda, K.; Venkatasubramanian, M.; Orr, E.; Suarez, L.; Gore, S.D.; Delwel, R.; Paietta, E.; et al. DNMT3A Haploinsufficiency Transforms FLT3ITD Myeloproliferative Disease into a Rapid, Spontaneous, and Fully Penetrant Acute Myeloid Leukemia. Cancer Discov. 2016, 6, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.; Lennartsson, A.; Gaidzik, V.I.; Deneberg, S.; Karimi, M.; Bengtzen, S.; Hoglund, M.; Bullinger, L.; Dohner, K.; Lehmann, S. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics 2014, 9, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Fried, I.; Bodner, C.; Pichler, M.M.; Lind, K.; Beham-Schmid, C.; Quehenberger, F.; Sperr, W.R.; Linkesch, W.; Sill, H.; Wolfler, A. Frequency, onset and clinical impact of somatic DNMT3A mutations in therapy-related and secondary acute myeloid leukemia. Haematologica 2012, 97, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Thol, F.; Damm, F.; Ludeking, A.; Winschel, C.; Wagner, K.; Morgan, M.; Yun, H.; Gohring, G.; Schlegelberger, B.; Hoelzer, D.; et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 2889–2896. [Google Scholar] [CrossRef]
- Marcucci, G.; Metzeler, K.H.; Schwind, S.; Becker, H.; Maharry, K.; Mrozek, K.; Radmacher, M.D.; Kohlschmidt, J.; Nicolet, D.; Whitman, S.P.; et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 742–750. [Google Scholar] [CrossRef] [Green Version]
- Corces-Zimmerman, M.R.; Hong, W.J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [Green Version]
- Ploen, G.G.; Nederby, L.; Guldberg, P.; Hansen, M.; Ebbesen, L.H.; Jensen, U.B.; Hokland, P.; Aggerholm, A. Persistence of DNMT3A mutations at long-term remission in adult patients with AML. Br. J. Haematol. 2014, 167, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, L.; Gundry, M.C.; Goodell, M.A. DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Li, X.; Cassady, K.; Zou, Z.; Zhang, X. TET2 Function in Hematopoietic Malignancies, Immune Regulation, and DNA Repair. Front. Oncol. 2019, 9, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solary, E.; Bernard, O.A.; Tefferi, A.; Fuks, F.; Vainchenker, W. The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia 2014, 28, 485–496. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009, 113, 6403–6410. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.S.; Kim, H.J.; Kim, Y.K.; Jung, S.H.; Yang, D.H.; Lee, J.J.; Lee, I.K.; Kim, N.Y.; Minden, M.D.; Jung, C.W.; et al. Adverse prognostic effect of homozygous TET2 mutation on the relapse risk of acute myeloid leukemia in patients of normal karyotype. Haematologica 2015, 100, e351–e353. [Google Scholar] [CrossRef] [Green Version]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Haferlach, T.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, L.; Abdel-Wahab, O.; Levine, R.L.; Aifantis, I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell 2011, 9, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Han, Y.; Suarez Saiz, F.; Minden, M.D. A tumor suppressor and oncogene: The WT1 story. Leukemia 2007, 21, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Figueroa, M.E. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica 2016, 101, 672–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.F.; Madden, S.L.; Tournay, O.E.; Cook, D.M.; Sukhatme, V.P.; Rauscher, F.J., 3rd. Characterization of the zinc finger protein encoded by the WT1 Wilms’ tumor locus. Oncogene 1991, 6, 2339–2348. [Google Scholar] [PubMed]
- Ariyaratana, S.; Loeb, D.M. The role of the Wilms tumour gene (WT1) in normal and malignant haematopoiesis. Expert Rev. Mol. Med. 2007, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.C.; Romaniuk, P.J. Contribution of individual amino acids to the RNA binding activity of the Wilms’ tumor suppressor protein WT1. Biochemistry 2009, 48, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Haber, D.A.; Sohn, R.L.; Buckler, A.J.; Pelletier, J.; Call, K.M.; Housman, D.E. Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc. Natl. Acad. Sci. USA 1991, 88, 9618–9622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna, I.; Such, E.; Cervera, J.; Barragan, E.; Ibanez, M.; Gomez-Segui, I.; Lopez-Pavia, M.; Llop, M.; Fuster, O.; Dolz, S.; et al. WT1 isoform expression pattern in acute myeloid leukemia. Leuk. Res. 2013, 37, 1744–1749. [Google Scholar] [CrossRef]
- Ellisen, L.W.; Carlesso, N.; Cheng, T.; Scadden, D.T.; Haber, D.A. The Wilms tumor suppressor WT1 directs stage-specific quiescence and differentiation of human hematopoietic progenitor cells. EMBO J. 2001, 20, 1897–1909. [Google Scholar] [CrossRef] [Green Version]
- Baird, P.N.; Simmons, P.J. Expression of the Wilms’ tumor gene (WT1) in normal hemopoiesis. Exp. Hematol. 1997, 25, 312–320. [Google Scholar]
- Miwa, H.; Beran, M.; Saunders, G.F. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia 1992, 6, 405–409. [Google Scholar]
- Cilloni, D.; Renneville, A.; Hermitte, F.; Hills, R.K.; Daly, S.; Jovanovic, J.V.; Gottardi, E.; Fava, M.; Schnittger, S.; Weiss, T.; et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: A European LeukemiaNet study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5195–5201. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.A.; Huang, T.C.; Lin, L.I.; Liu, C.Y.; Chen, C.Y.; Chou, W.C.; Tang, J.L.; Tseng, M.H.; Huang, C.F.; Chiang, Y.C.; et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: Stability during disease evolution and implication of its incorporation into a survival scoring system. Blood 2010, 115, 5222–5231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, S.; Erpelinck-Verschueren, C.A.; Goudswaard, C.S.; Lowenberg, B.; Valk, P.J. Mutant Wilms’ tumor 1 (WT1) mRNA with premature termination codons in acute myeloid leukemia (AML) is sensitive to nonsense-mediated RNA decay (NMD). Leukemia 2010, 24, 660–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaidzik, V.I.; Schlenk, R.F.; Moschny, S.; Becker, A.; Bullinger, L.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; Dohner, H.; et al. Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: A study of the German-Austrian AML Study Group. Blood 2009, 113, 4505–4511. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, H.F.; Sun, Z.; Yao, X.; Litzow, M.R.; Luger, S.M.; Paietta, E.M.; Racevskis, J.; Dewald, G.W.; Ketterling, R.P.; Bennett, J.M.; et al. Anthracycline dose intensification in acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Rampal, R.; Alkalin, A.; Madzo, J.; Vasanthakumar, A.; Pronier, E.; Patel, J.; Li, Y.; Ahn, J.; Abdel-Wahab, O.; Shih, A.; et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014, 9, 1841–1855. [Google Scholar] [CrossRef]
- Pronier, E.; Bowman, R.L.; Ahn, J.; Glass, J.; Kandoth, C.; Merlinsky, T.R.; Whitfield, J.T.; Durham, B.H.; Gruet, A.; Hanasoge Somasundara, A.V.; et al. Genetic and epigenetic evolution as a contributor to WT1-mutant leukemogenesis. Blood 2018, 132, 1265–1278. [Google Scholar] [CrossRef]
- Aref, S.; Sharawy, S.E.; Sabry, M.; Azmy, E.; Raouf, D.A.; Menshawy, N.E. Wilms tumor 1 gene mutations in patients with cytogenetically normal acute myeloid leukemia. Turk. J. Haematol. Off. J. Turk. Soc. Haematol. 2014, 31, 143–148. [Google Scholar] [CrossRef]
- Petiti, J.; Rosso, V.; Lo Iacono, M.; Calabrese, C.; Signorino, E.; Gaidano, V.; Berger, M.; Saglio, G.; Cilloni, D. Prognostic significance of The Wilms’ Tumor-1 (WT1) rs16754 polymorphism in acute myeloid leukemia. Leuk. Res. 2018, 67, 6–11. [Google Scholar] [CrossRef]
- Toogeh, G.; Ramzi, M.; Faranoush, M.; Amirizadeh, N.; Haghpanah, S.; Moghadam, M.; Cohan, N. Prevalence and Prognostic Impact of Wilms’ Tumor 1 (WT1) Gene, Including SNP rs16754 in Cytogenetically Normal Acute Myeloblastic Leukemia (CN-AML): An Iranian Experience. Clin. Lymphomamyeloma Leuk. 2016, 16, e21–e26. [Google Scholar] [CrossRef]
- Fisher, C.L.; Pineault, N.; Brookes, C.; Helgason, C.D.; Ohta, H.; Bodner, C.; Hess, J.L.; Humphries, R.K.; Brock, H.W. Loss-of-function Additional sex combs like 1 mutations disrupt hematopoiesis but do not cause severe myelodysplasia or leukemia. Blood 2010, 115, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, C.L.; Randazzo, F.; Humphries, R.K.; Brock, H.W. Characterization of Asxl1, a murine homolog of Additional sex combs, and analysis of the Asx-like gene family. Gene 2006, 369, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Dey, A. The ASXL-BAP1 axis: New factors in myelopoiesis, cancer and epigenetics. Leukemia 2013, 27, 10–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, R.; Zhou, M.M. The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 2011, 36, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Inoue, D.; Fujino, T.; Kitamura, T. ASXL1 as a critical regulator of epigenetic marks and therapeutic potential of mutated cells. Oncotarget 2018, 9, 35203–35204. [Google Scholar] [CrossRef]
- Alvarez Argote, J.; Dasanu, C.A. ASXL1 mutations in myeloid neoplasms: Pathogenetic considerations, impact on clinical outcomes and survival. Curr. Med. Res. Opin. 2018, 34, 757–763. [Google Scholar] [CrossRef]
- Sahtoe, D.D.; van Dijk, W.J.; Ekkebus, R.; Ovaa, H.; Sixma, T.K. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat. Commun. 2016, 7, 10292. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.C.; Chiu, Y.C.; Lin, C.C.; Kuo, Y.Y.; Hou, H.A.; Tzeng, Y.S.; Kao, C.J.; Chuang, P.H.; Tseng, M.H.; Hsiao, T.H.; et al. The distinct biological implications of Asxl1 mutation and its roles in leukemogenesis revealed by a knock-in mouse model. J. Hematol. Oncol. 2017, 10, 139. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Brecqueville, M.; Devillier, R.; Murati, A.; Mozziconacci, M.J.; Birnbaum, D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J. Hematol. Oncol. 2012, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Herzig, J.K.; Aulitzky, T.; Bullinger, L.; Spath, D.; Teleanu, V.; Kundgen, A.; Kohne, C.H.; et al. ASXL1 mutations in younger adult patients with acute myeloid leukemia: A study by the German-Austrian Acute Myeloid Leukemia Study Group. Haematologica 2015, 100, 324–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Zheng, Y.; Wang, Z.C.; Wang, S.Y. Prognostic significance of ASXL1 mutations in myelodysplastic syndromes and chronic myelomonocytic leukemia: A meta-analysis. Hematology 2016, 21, 454–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnittger, S.; Eder, C.; Jeromin, S.; Alpermann, T.; Fasan, A.; Grossmann, V.; Kohlmann, A.; Illig, T.; Klopp, N.; Wichmann, H.E.; et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia 2013, 27, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Kakosaiou, K.; Panitsas, F.; Daraki, A.; Pagoni, M.; Apostolou, P.; Ioannidou, A.; Vlachadami, I.; Marinakis, T.; Giatra, C.; Vasilatou, D.; et al. ASXL1 mutations in AML are associated with specific clinical and cytogenetic characteristics. Leuk. Lymphoma 2018, 59, 2439–2446. [Google Scholar] [CrossRef]
- Rocquain, J.; Carbuccia, N.; Trouplin, V.; Raynaud, S.; Murati, A.; Nezri, M.; Tadrist, Z.; Olschwang, S.; Vey, N.; Birnbaum, D.; et al. Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer 2010, 10, 401. [Google Scholar] [CrossRef] [Green Version]
- Carbuccia, N.; Trouplin, V.; Gelsi-Boyer, V.; Murati, A.; Rocquain, J.; Adelaide, J.; Olschwang, S.; Xerri, L.; Vey, N.; Chaffanet, M.; et al. Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute myeloid leukemias. Leukemia 2010, 24, 469–473. [Google Scholar] [CrossRef]
- Yang, H.; Kurtenbach, S.; Guo, Y.; Lohse, I.; Durante, M.A.; Li, J.; Li, Z.; Al-Ali, H.; Li, L.; Chen, Z.; et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 2018, 131, 328–341. [Google Scholar] [CrossRef]
- An, Q.; Wright, S.L.; Moorman, A.V.; Parker, H.; Griffiths, M.; Ross, F.M.; Davies, T.; Harrison, C.J.; Strefford, J.C. Heterogeneous breakpoints in patients with acute lymphoblastic leukemia and the dic(9;20)(p11-13;q11) show recurrent involvement of genes at 20q11.21. Haematologica 2009, 94, 1164–1169. [Google Scholar] [CrossRef] [Green Version]
- Eirin-Lopez, J.M.; Frehlick, L.J.; Ausio, J. Long-term evolution and functional diversification in the members of the nucleophosmin/nucleoplasmin family of nuclear chaperones. Genetics 2006, 173, 1835–1850. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M. The role of nucleophosmin in centrosome duplication. Oncogene 2002, 21, 6170–6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjerset, R.A. DNA damage, p14ARF, nucleophosmin (NPM/B23), and cancer. J. Mol. Histol. 2006, 37, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Marine, J.C.; Danovi, D.; Falini, B.; Pelicci, P.G. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 2002, 4, 529–533. [Google Scholar] [CrossRef]
- Ye, M.; Zhang, H.; Amabile, G.; Yang, H.; Staber, P.B.; Zhang, P.; Levantini, E.; Alberich-Jorda, M.; Zhang, J.; Kawasaki, A.; et al. C/EBPa controls acquisition and maintenance of adult haematopoietic stem cell quiescence. Nat. Cell Biol. 2013, 15, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Martelli, M.P.; Bolli, N.; Sportoletti, P.; Liso, A.; Tiacci, E.; Haferlach, T. Acute myeloid leukemia with mutated nucleophosmin (NPM1): Is it a distinct entity? Blood 2011, 117, 1109–1120. [Google Scholar] [CrossRef]
- Heath, E.M.; Chan, S.M.; Minden, M.D.; Murphy, T.; Shlush, L.I.; Schimmer, A.D. Biological and clinical consequences of NPM1 mutations in AML. Leukemia 2017, 31, 798–807. [Google Scholar] [CrossRef]
- Kunchala, P.; Kuravi, S.; Jensen, R.; McGuirk, J.; Balusu, R. When the good go bad: Mutant NPM1 in acute myeloid leukemia. Blood Rev. 2018, 32, 167–183. [Google Scholar] [CrossRef]
- Vassiliou, G.S.; Cooper, J.L.; Rad, R.; Li, J.; Rice, S.; Uren, A.; Rad, L.; Ellis, P.; Andrews, R.; Banerjee, R.; et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat. Genet. 2011, 43, 470–475. [Google Scholar] [CrossRef]
- Falini, B.; Nicoletti, I.; Bolli, N.; Martelli, M.P.; Liso, A.; Gorello, P.; Mandelli, F.; Mecucci, C.; Martelli, M.F. Translocations and mutations involving the nucleophosmin (NPM1) gene in lymphomas and leukemias. Haematologica 2007, 92, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Schnittger, S.; Schoch, C.; Kern, W.; Mecucci, C.; Tschulik, C.; Martelli, M.F.; Haferlach, T.; Hiddemann, W.; Falini, B. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 2005, 106, 3733–3739. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; He, P.; Liu, F.; Shi, L.; Zhu, H.; Zhao, J.; Wang, Y.; Cheng, X.; Zhang, M. Prognostic significance of NPM1 mutations in acute myeloid leukemia: A meta-analysis. Mol. Clin. Oncol. 2014, 2, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratcorona, M.; Brunet, S.; Nomdedeu, J.; Ribera, J.M.; Tormo, M.; Duarte, R.; Escoda, L.; Guardia, R.; Queipo de Llano, M.P.; Salamero, O.; et al. Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: Relevance to post-remission therapy. Blood 2013, 121, 2734–2738. [Google Scholar] [CrossRef] [Green Version]
- Forghieri, F.; Comoli, P.; Marasca, R.; Potenza, L.; Luppi, M. Minimal/Measurable Residual Disease Monitoring in NPM1-Mutated Acute Myeloid Leukemia: A Clinical Viewpoint and Perspectives. Int. J. Mol. Sci. 2018, 19, 3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosna, F.; Capelli, D.; Gottardi, M. Minimal Residual Disease in Acute Myeloid Leukemia: Still a Work in Progress? J. Clin. Med. 2017, 6, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boddu, P.; Kantarjian, H.; Borthakur, G.; Kadia, T.; Daver, N.; Pierce, S.; Andreeff, M.; Ravandi, F.; Cortes, J.; Kornblau, S.M. Co-occurrence of FLT3-TKD and NPM1 mutations defines a highly favorable prognostic AML group. Blood Adv. 2017, 1, 1546–1550. [Google Scholar] [CrossRef] [Green Version]
- Rose, D.; Haferlach, T.; Schnittger, S.; Perglerova, K.; Kern, W.; Haferlach, C. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia 2017, 31, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Redner, R.L.; Rush, E.A.; Faas, S.; Rudert, W.A.; Corey, S.J. The t(5;17) variant of acute promyelocytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood 1996, 87, 882–886. [Google Scholar] [CrossRef] [Green Version]
- Birkenmeier, E.H.; Gwynn, B.; Howard, S.; Jerry, J.; Gordon, J.I.; Landschulz, W.H.; McKnight, S.L. Tissue-specific expression, developmental regulation, and genetic mapping of the gene encoding CCAAT/enhancer binding protein. Genes Dev. 1989, 3, 1146–1156. [Google Scholar] [CrossRef] [Green Version]
- Pundhir, S.; Bratt Lauridsen, F.K.; Schuster, M.B.; Jakobsen, J.S.; Ge, Y.; Schoof, E.M.; Rapin, N.; Waage, J.; Hasemann, M.S.; Porse, B.T. Enhancer and Transcription Factor Dynamics during Myeloid Differentiation Reveal an Early Differentiation Block in Cebpa null Progenitors. Cell Rep. 2018, 23, 2744–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, V.A.; Iwama, A.; Iotzova, G.; Schulz, M.; Elsasser, A.; Vangala, R.K.; Tenen, D.G.; Hiddemann, W.; Behre, G. Granulocyte inducer C/EBPalpha inactivates the myeloid master regulator PU.1: Possible role in lineage commitment decisions. Blood 2002, 100, 483–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuston, E.F. The potential of a single enhancer. Blood 2016, 127, 2943–2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porse, B.T.; Bryder, D.; Theilgaard-Monch, K.; Hasemann, M.S.; Anderson, K.; Damgaard, I.; Jacobsen, S.E.; Nerlov, C. Loss of C/EBP alpha cell cycle control increases myeloid progenitor proliferation and transforms the neutrophil granulocyte lineage. J. Exp. Med. 2005, 202, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Pabst, T.; Mueller, B.U.; Zhang, P.; Radomska, H.S.; Narravula, S.; Schnittger, S.; Behre, G.; Hiddemann, W.; Tenen, D.G. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat. Genet. 2001, 27, 263–270. [Google Scholar] [CrossRef]
- Leroy, H.; Roumier, C.; Huyghe, P.; Biggio, V.; Fenaux, P.; Preudhomme, C. CEBPA point mutations in hematological malignancies. Leukemia 2005, 19, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Pabst, T.; Mueller, B.U. Complexity of CEBPA dysregulation in human acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5303–5307. [Google Scholar] [CrossRef] [Green Version]
- Dufour, A.; Schneider, F.; Metzeler, K.H.; Hoster, E.; Schneider, S.; Zellmeier, E.; Benthaus, T.; Sauerland, M.C.; Berdel, W.E.; Buchner, T.; et al. Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 570–577. [Google Scholar] [CrossRef]
- Mannelli, F.; Ponziani, V.; Bencini, S.; Bonetti, M.I.; Benelli, M.; Cutini, I.; Gianfaldoni, G.; Scappini, B.; Pancani, F.; Piccini, M.; et al. CEBPA-double-mutated acute myeloid leukemia displays a unique phenotypic profile: A reliable screening method and insight into biological features. Haematologica 2017, 102, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Tan, Y.; Lin, H.; Liu, X.; Yu, L.; Yang, Y.; Liu, S.; Bai, O.; Yang, Y.; Jin, F.; et al. Mutational spectrum of acute myeloid leukemia patients with double CEBPA mutations based on next-generation sequencing and its prognostic significance. Oncotarget 2018, 9, 24970–24979. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, V.; Haferlach, C.; Nadarajah, N.; Fasan, A.; Weissmann, S.; Roller, A.; Eder, C.; Stopp, E.; Kern, W.; Haferlach, T.; et al. CEBPA double-mutated acute myeloid leukaemia harbours concomitant molecular mutations in 76.8% of cases with TET2 and GATA2 alterations impacting prognosis. Br. J. Haematol. 2013, 161, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Green, C.L.; Koo, K.K.; Hills, R.K.; Burnett, A.K.; Linch, D.C.; Gale, R.E. Prognostic significance of CEBPA mutations in a large cohort of younger adult patients with acute myeloid leukemia: Impact of double CEBPA mutations and the interaction with FLT3 and NPM1 mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2739–2747. [Google Scholar] [CrossRef] [PubMed]
- Fasan, A.; Haferlach, C.; Alpermann, T.; Jeromin, S.; Grossmann, V.; Eder, C.; Weissmann, S.; Dicker, F.; Kohlmann, A.; Schindela, S.; et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia 2014, 28, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, F.; Chen, X.; Zhang, Y.; Wang, M.; Liu, H.; Cao, P.; Ma, X.; Wang, T.; Zhang, J.; et al. CSF3R Mutations are frequently associated with abnormalities of RUNX1, CBFB, CEBPA, and NPM1 genes in acute myeloid leukemia. Cancer 2018, 124, 3329–3338. [Google Scholar] [CrossRef] [Green Version]
- Lavallee, V.P.; Krosl, J.; Lemieux, S.; Boucher, G.; Gendron, P.; Pabst, C.; Boivin, I.; Marinier, A.; Guidos, C.J.; Meloche, S.; et al. Chemo-genomic interrogation of CEBPA mutated AML reveals recurrent CSF3R mutations and subgroup sensitivity to JAK inhibitors. Blood 2016, 127, 3054–3061. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Gao, S.; Tan, Y.; Lin, H.; Liu, X.; Liu, S.; Yang, Y.; Sun, J.; Li, W. CSF3R mutations were associated with an unfavorable prognosis in patients with acute myeloid leukemia with CEBPA double mutations. Ann. Hematol. 2019, 98, 1641–1646. [Google Scholar] [CrossRef]
- Braun, T.P.; Okhovat, M.; Coblentz, C.; Carratt, S.A.; Foley, A.; Schonrock, Z.; Smith, B.M.; Nevonen, K.; Davis, B.; Garcia, B.; et al. Myeloid lineage enhancers drive oncogene synergy in CEBPA/CSF3R mutant acute myeloid leukemia. Nat. Commun. 2019, 10, 5455. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Lee, S.; Cavallo, L.; Griffith, J. Human p53 binds Holliday junctions strongly and facilitates their cleavage. J. Biol. Chem. 1997, 272, 7532–7539. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef] [Green Version]
- Seifert, H.; Mohr, B.; Thiede, C.; Oelschlagel, U.; Schakel, U.; Illmer, T.; Soucek, S.; Ehninger, G.; Schaich, M.; Study Alliance, L. The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia 2009, 23, 656–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.; et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 2016, 122, 3484–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochazka, K.T.; Pregartner, G.; Rucker, F.G.; Heitzer, E.; Pabst, G.; Wolfler, A.; Zebisch, A.; Berghold, A.; Dohner, K.; Sill, H. Clinical implications of subclonal TP53 mutations in acute myeloid leukemia. Haematologica 2019, 104, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rucker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef]
- Haferlach, C.; Dicker, F.; Herholz, H.; Schnittger, S.; Kern, W.; Haferlach, T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 2008, 22, 1539–1541. [Google Scholar] [CrossRef]
- Roake, C.M.; Artandi, S.E. Control of Cellular Aging, Tissue Function, and Cancer by p53 Downstream of Telomeres. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Christiansen, D.H.; Andersen, M.K.; Pedersen-Bjergaard, J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2001, 19, 1405–1413. [Google Scholar] [CrossRef]
- Prokocimer, M.; Molchadsky, A.; Rotter, V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: Projections on diagnostic workup and therapy. Blood 2017, 130, 699–712. [Google Scholar] [CrossRef]
- Carvajal, L.A.; Neriah, D.B.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Sasca, D.; Hahnel, P.S.; Szybinski, J.; Khawaja, K.; Kriege, O.; Pante, S.V.; Bullinger, L.; Strand, S.; Strand, D.; Theobald, M.; et al. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood 2014, 124, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, M.T.; Teh, C.; Shyh-Chang, N.; Xie, H.; Zhou, B.; Korzh, V.; Lodish, H.F.; Lim, B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009, 23, 862–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Yoon, J.B.; Gu, W. Reactivating the ARF-p53 axis in AML cells by targeting ULF. Cell Cycle 2010, 9, 2946–2951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Lund, K.; Adams, P.D.; Copland, M. EZH2 in normal and malignant hematopoiesis. Leukemia 2014, 28, 44–49. [Google Scholar] [CrossRef]
- Maes, T.; Mascaro, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018, 33, 495–511 e412. [Google Scholar] [CrossRef] [Green Version]
- Visconte, V.; O Nakashima, M.; J Rogers, H. Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features. Cancers 2019, 11, 1844. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Hou, H.A.; Liu, C.Y.; Kuo, Y.Y.; Chou, W.C.; Tsai, C.H.; Lin, C.C.; Lin, L.I.; Tseng, M.H.; Chiang, Y.C.; Liu, M.C.; et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget 2016, 7, 9084–9101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Peng, R.; Wang, J.; Qin, Z.; Xue, L. Circulating microRNAs as potential cancer biomarkers: The advantage and disadvantage. Clin. Epigenetics 2018, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.A.; O’Connell, R.M. MicroRNAs and acute myeloid leukemia: Therapeutic implications and emerging concepts. Blood 2017, 130, 1290–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasedieck, S.; Sorrentino, A.; Langer, C.; Buske, C.; Dohner, H.; Mertens, D.; Kuchenbauer, F. Circulating microRNAs in hematological diseases: Principles, challenges, and perspectives. Blood 2013, 121, 4977–4984. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Su, C.; Deng, T. miR-223 decreases cell proliferation and enhances cell apoptosis in acute myeloid leukemia via targeting FBXW7. Oncol. Lett. 2016, 12, 3531–3536. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. Cr 2019, 38, 53. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Chen, M.T.; Zhang, X.H.; Yin, X.L.; Ning, H.M.; Su, R.; Lin, H.S.; Song, L.; Wang, F.; Ma, Y.N.; et al. The PU.1-Modulated MicroRNA-22 Is a Regulator of Monocyte/Macrophage Differentiation and Acute Myeloid Leukemia. PLoS Genet. 2016, 12, e1006259. [Google Scholar] [CrossRef]
- Senyuk, V.; Zhang, Y.; Liu, Y.; Ming, M.; Premanand, K.; Zhou, L.; Chen, P.; Chen, J.; Rowley, J.D.; Nucifora, G.; et al. Critical role of miR-9 in myelopoiesis and EVI1-induced leukemogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 5594–5599. [Google Scholar] [CrossRef] [Green Version]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009, 113, 6411–6418. [Google Scholar] [CrossRef] [Green Version]
- Trissal, M.C.; Wong, T.N.; Yao, J.C.; Ramaswamy, R.; Kuo, I.; Baty, J.; Sun, Y.; Jih, G.; Parikh, N.; Berrien-Elliott, M.M.; et al. MIR142 Loss-of-Function Mutations Derepress ASH1L to Increase HOXA Gene Expression and Promote Leukemogenesis. Cancer Res. 2018, 78, 3510–3521. [Google Scholar] [CrossRef] [Green Version]
- Ling, K.W.; Ottersbach, K.; van Hamburg, J.P.; Oziemlak, A.; Tsai, F.Y.; Orkin, S.H.; Ploemacher, R.; Hendriks, R.W.; Dzierzak, E. GATA-2 plays two functionally distinct roles during the ontogeny of hematopoietic stem cells. J. Exp. Med. 2004, 200, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, N.P.; Boyd, A.S.; Fugazza, C.; May, G.E.; Guo, Y.; Tipping, A.J.; Scadden, D.T.; Vyas, P.; Enver, T. GATA-2 regulates granulocyte-macrophage progenitor cell function. Blood 2008, 112, 4862–4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.J.; Ma, L.Y.; Huang, Q.H.; Li, G.; Gu, B.W.; Gao, X.D.; Shi, J.Y.; Wang, Y.Y.; Gao, L.; Cai, X.; et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 2076–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crispino, J.D.; Horwitz, M.S. GATA factor mutations in hematologic disease. Blood 2017, 129, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Tien, F.M.; Hou, H.A.; Tsai, C.H.; Tang, J.L.; Chiu, Y.C.; Chen, C.Y.; Kuo, Y.Y.; Tseng, M.H.; Peng, Y.L.; Liu, M.C.; et al. GATA2 zinc finger 1 mutations are associated with distinct clinico-biological features and outcomes different from GATA2 zinc finger 2 mutations in adult acute myeloid leukemia. Blood Cancer J. 2018, 8, 87. [Google Scholar] [CrossRef]
- Hou, H.A.; Lin, Y.C.; Kuo, Y.Y.; Chou, W.C.; Lin, C.C.; Liu, C.Y.; Chen, C.Y.; Lin, L.I.; Tseng, M.H.; Huang, C.F.; et al. GATA2 mutations in patients with acute myeloid leukemia-paired samples analyses show that the mutation is unstable during disease evolution. Ann. Hematol. 2015, 94, 211–221. [Google Scholar] [CrossRef]
- Fasan, A.; Eder, C.; Haferlach, C.; Grossmann, V.; Kohlmann, A.; Dicker, F.; Kern, W.; Haferlach, T.; Schnittger, S. GATA2 mutations are frequent in intermediate-risk karyotype AML with biallelic CEBPA mutations and are associated with favorable prognosis. Leukemia 2013, 27, 482–485. [Google Scholar] [CrossRef]
- Theis, F.; Corbacioglu, A.; Gaidzik, V.I.; Paschka, P.; Weber, D.; Bullinger, L.; Heuser, M.; Ganser, A.; Thol, F.; Schlegelberger, B.; et al. Clinical impact of GATA2 mutations in acute myeloid leukemia patients harboring CEBPA mutations: A study of the AML study group. Leukemia 2016, 30, 2248–2250. [Google Scholar] [CrossRef]
- Hahn, C.N.; Chong, C.E.; Carmichael, C.L.; Wilkins, E.J.; Brautigan, P.J.; Li, X.C.; Babic, M.; Lin, M.; Carmagnac, A.; Lee, Y.K.; et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat. Genet. 2011, 43, 1012–1017. [Google Scholar] [CrossRef]
- Hsu, A.P.; Sampaio, E.P.; Khan, J.; Calvo, K.R.; Lemieux, J.E.; Patel, S.Y.; Frucht, D.M.; Vinh, D.C.; Auth, R.D.; Freeman, A.F.; et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 2011, 118, 2653–2655. [Google Scholar] [CrossRef]
- Ostergaard, P.; Simpson, M.A.; Connell, F.C.; Steward, C.G.; Brice, G.; Woollard, W.J.; Dafou, D.; Kilo, T.; Smithson, S.; Lunt, P.; et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat. Genet. 2011, 43, 929–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, J.R.; Rucker-Braun, E.; Heidrich, K.; von Bonin, M.; Stolzel, F.; Thiede, C.; Middeke, J.M.; Ehninger, G.; Bornhauser, M.; Schetelig, J.; et al. Pilot Study on Mass Spectrometry-Based Analysis of the Proteome of CD34(+)CD123(+) Progenitor Cells for the Identification of Potential Targets for Immunotherapy in Acute Myeloid Leukemia. Proteomes 2018, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alanazi, B.; Munje, C.R.; Rastogi, N.; Williamson, A.J.K.; Taylor, S.; Hole, P.S.; Hodges, M.; Doyle, M.; Baker, S.; Gilkes, A.F.; et al. Integrated nuclear proteomics and transcriptomics identifies S100A4 as a therapeutic target in acute myeloid leukemia. Leukemia 2020, 34, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Aasebo, E.; Brenner, A.K.; Bartaula-Brevik, S.; Gronningsaeter, I.S.; Forthun, R.B.; Hovland, R.; Bruserud, O. High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype. J. Clin. Med. 2019, 8, 970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, A.K.; Aasebo, E.; Hernandez-Valladares, M.; Selheim, F.; Berven, F.; Gronningsaeter, I.S.; Bartaula-Brevik, S.; Bruserud, O. The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome. Cancers 2019, 11, 73. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Prognostic-Risk Group | Cytogenetic Aberrations and Molecular Abnormalities |
|---|---|
| Favorable | t(8:21)(q22;q22) AML1(RUNX1)-ETO(RUNX1T1) inv(16)(p13;1q22)CBFα-MYH11 t(15;17)(q22;q12)PML-RARα NPM1 mutation without FLT3-ITD or with FLT3-ITDlow * CEBPA biallelic mutations |
| Intermediate | NPM1 mutation with FLT3-ITDhigh * NPM1 wild-type without FLT3-ITD or with FLT3-ITDlow * (in the absence of adverse risk genetic lesions) t(9;11)(p22;q23)MLLT3-KMT2A Other cytogenetic abnormalities not included in the other groups |
| Adverse | t(6;9)(p23;q34)DEK/NUP214 inv(3)(q21;q26.2)GATA2,MECOM(EVI1) t(9;22)(q34.1;q11.2)BCR-ABL1 t(v;11q23.3)KMT2A(MLL) rearranged −5 or del(5q) −7 or del(7q) abn(17p) Complex karyotype Monosomal karyotype NPM1 wild-type and FLT3-ITDhigh * RUNX1 mutations (in the absence of favorable risk genetic lesions) ASXL1 mutations (in the absence of favorable risk genetic lesions) TP53 mutations |
| Mutated Tumor Suppressors Involved in Epigenetic Regulation | ||
|---|---|---|
| Mutated Genes | Frequency in AML (%) | Functions, Associations, Prognostic Impact and Specific Drugs |
| IDH1 | 6–10 | Enzyme involved in TCA cycle Important role in lipid metabolism Involved in cellular defense of oxidative damage Mutations cause D-2-hydroxyglutarate (D2HG) accumulation that inhibits various dioxygenases involved in epigenetic regulation Frequent in CN-AML Associated with NPM1 mutations Associated with FLT3, DNMT3A, ASXL1, RUNX1, NRAS mutations Mutually exclusive with TET2 mutations Associated with clonal hematopoiesis in healthy elderly persons Early event in leukemogenesis Prognostic impact context-dependent IDH1 inhibitor ivosidenib approved by FDA |
| IDH2R140 | 5–15 | Enzyme involved in TCA cycle Involved in cellular defense of oxidative damage Mutations cause D-2-hydroxyglutarate (D2HG) accumulation that inhibits various dioxygenases involved in epigenetic regulation Frequent in CN-AML Frequency increases with age Associated with NPM1 mutations Associated with FLT3, DNMT3A, ASXL1, RUNX1, NRAS mutations Mutually exclusive with TET2 mutations Associated with clonal hematopoiesis in healthy elderly persons Early event in leukemogenesis Prognostic impact could be more favorable than other IDH mutations IDH2 inhibitor enasidenib approved by FDA |
| IDH2R172 | 1–4 | Enzyme involved in TCA cycle Involved in cellular defense of oxidative damage Mutations cause D-2-hydroxyglutarate (D2HG) accumulation that inhibits various dioxygenases involved in epigenetic regulation Frequent in CN-AML AML with IDH2R172 mutation (in the absence of other lesions) may represent a separate disease class, associated with a distinct microarray gene expression and microRNA expression profile Mutually exclusive with NPM1 mutations Associated with FLT3, DNMT3A, ASXL1, RUNX1, NRAS mutations Mutually exclusive with TET2 mutations No consistent data on prognostic impact Associated with clonal hematopoiesis in healthy elderly persons Early event in leukemogenesis IDH2 inhibitor enasidenib approved by FDA |
| DNMT3A | 15–30 | Catalyzes the addition of a methyl group to the cytosine residue of CpG dinucleotides Essential for de novo DNA methylation and regulation of gene expression Frequent in CN-AML Frequency increases with age Associated with NPM1, FLT3-ITD, IDH1, IDH2R140 and IDH2R172 mutation Prognostic impact not consistent and context-dependent Particularly poor prognosis in DNMT3Amut/NPM1mut/FLT3-ITD Persistent DNMT3A transcript levels in hematologic CR Associated with clonal hematopoiesis in healthy elderly persons Early event in leukemogenesis |
| TET2 | 12–34 | Regulates differentiation or proliferation and epigenetic modifications Key family of enzymes for DNA demethylation Frequent in CN-AML Frequency increases with age Associated with NPM1 mutation Mutually exclusive with IDH1 and IDH2 mutations Prognostic impact associated with inferior OS in CN-AML Associated with clonal hematopoiesis in healthy elderly persons Early event in leukemogenesis Mutations in TET2 may respond to hypomethylating agents (HMAs) therapy |
| WT1 | 6–15 | Zinc finger transcription factor Multiple isoforms from two splicing events Involved in regulation of cell survival, proliferation, and differentiation Overexpressed in AML where it is used as a diagnostic molecular marker and for MRD monitoring Overexpression correlate with chemotherapy resistance, decreased OS and higher relapse rate Mutations in exons 1, 7 and 9 in AML Frequent in younger patients Associated with FLT3-ITD and CEBPA biallelic mutation Associated with worse prognosis and resistance to chemotherapy Possible role in the same epigenetic pathway of TET2 and IDH1/2 Anticorrelated with TET2, IDH1 and IDH2 mutations Use of HMAs such azacitidine as a potential strategy of therapy in WT1 mutated patients Polymorphism SNP rs16754 positive prognostic factor in patients with AML |
| ASXL1 | 5–18 | Chromatin-binding protein, epigenetic scaffold protein Enhancer of the trithorax and polycomb genes Mutations in the ASXL1 described in many subtypes of myeloid malignances Associated with adverse prognosis, shorter OS and higher risk of progression Frequent in CMML Frequency increases significantly with age Correlate with t(8; 21), +8 and − 7 chromosomal aberrations Associated with RUNX1 and IDH2 mutations Associated with clonal hematopoiesis in healthy elderly persons Early event in leukemogenesis |
| Mutated Tumor Suppressors Involved in Non-Epigenetic Mechanisms | ||
|---|---|---|
| Mutated Genes | Frequency in AML (%) | Functions, Associations, Prognostic Impact and Specific Drugs |
| NPM1 | 25–30 | Nucleus-cytoplasm shuttling protein Involved in the regulation of centrosome duplication, DNA repair, ribosomal protein assembly and apoptotic response to oncogenic stimuli Key regulator of tumor suppressors TP53 and p19ARF Frequent in adult CN-AML Mutations mostly located into exon 12 Correlates with good response to conventional therapy Classified as favorable risk, high complete remission rates, EFS and OS Co-occurrence with FLT3 mutation associated with an intermediate prognosis Associated with DNMT3A, IDH1, IDH2 and TET2 mutations Used for monitoring of MRD |
| CEBPA (biallelic) | 5–20 | Zinc finger transcription factor Regulates differentiation of multipotent precursor cells to myeloid progenitors Directs granulocyte and monocyte differentiation Controls self-renewal properties of hematopoietic stem and progenitor cells Frequent in de novo AML Frequently biallelic Biallelic mutations are associated with favorable prognosis if compared to single allele mutation AML subgroup with CEBPA mutations recognized as a distinct diagnostic entity by the 2016 WHO classification of myeloid neoplasms Direct transcriptional repression by AML1-ETO, RARα-PLZF and FLT-ITD Associated with TET2, GATA2, WT1, DNMT3A and ASXL1 mutations Associated with a more favorable prognosis |
| TP53 | 5–20 | Guardian of the genome Regulates cell cycle arrest, apoptosis, senescence and DNA repair Mutation frequency rises in therapy-related and complex karyotype AML (approximately 70%) Mutations associated with absence of clinical remission, poor OS and DFS Majority of mutations in the region encoding the DNA-binding domain Mutations typically heterozygous followed by a rapid loss of heterozygosity Mutually exclusive with NPM1, FLT3, MDM2 and ARF Associated with -5, -7, -17 cytogenetic abnormalities In presence of wild-type form, several inactivating processes including MDM2 and MDMX overexpression, miRNA overexpression, FLT3-ITD mutations and impact on TP53 pathway Targeted therapy influenced by low frequency mutations Therapy focused on reactivate the wild-type TP53 Dual inhibitors of MDM2 and MDMX in clinical trials in AML Combination therapies with BCL2 inhibitors (venetoclax) |
| Mutated Gene | Prognosis | Current Diagnostic Practice 1 | |
|---|---|---|---|
| ASXL1 | Poor | Worse OS Correlation with age > 60 years and higher WBC counts | Recommended by 2017 ELN guidelines |
| CEBPA | Variable | Positive in CN-AML Biallelic mutations have better EFS, DFS and OS Single mutations with NPM1mut/FLT3-ITDlow cases have worse OS compared with CEBPA wild-type NPM1mut/FLT3-ITDlow cases Impaired outcome with concurrent TET2 mutation Better OS with concurrent GATA2 mutation | Recommended by 2017 ELN guidelines |
| DNMT3A | Poor | Linked to adverse outcomes | Recommended: pre-leukemic event, could indicate higher probability of relapse |
| IDH1 | Not consistent data | Impaired outcome in R132 mut/FLT3 wild-type patients | Recommended: new specific inhibitor (ivosidenib) in clinical trials |
| IDH2 | Not consistent data | R172 showed no correlation to outcome or response R140 improved OS and decreased response rates | Recommended: new specific inhibitor (enasidenib) in clinical trials |
| NPM1 | Good | Improved OS, DFS, and relapse-free survival (RFS) | Recommended by 2017 ELN guidelines |
| TET2 | Not consistent data | Impaired OS in multivariate analysis Impaired DFS | Recommended: could respond to HMAs treatment |
| WT1 | Poor | Often concurrent with FLT3 mutations Impaired OS and RFS | Recommended: could respond to HMAs treatment |
| TP53 | Poor | Associated with resistance to chemotherapy Impaired OS and DFS Association with complex karyotype | Recommended by 2017 ELN guidelines |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panuzzo, C.; Signorino, E.; Calabrese, C.; Ali, M.S.; Petiti, J.; Bracco, E.; Cilloni, D. Landscape of Tumor Suppressor Mutations in Acute Myeloid Leukemia. J. Clin. Med. 2020, 9, 802. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9030802
Panuzzo C, Signorino E, Calabrese C, Ali MS, Petiti J, Bracco E, Cilloni D. Landscape of Tumor Suppressor Mutations in Acute Myeloid Leukemia. Journal of Clinical Medicine. 2020; 9(3):802. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9030802
Chicago/Turabian StylePanuzzo, Cristina, Elisabetta Signorino, Chiara Calabrese, Muhammad Shahzad Ali, Jessica Petiti, Enrico Bracco, and Daniela Cilloni. 2020. "Landscape of Tumor Suppressor Mutations in Acute Myeloid Leukemia" Journal of Clinical Medicine 9, no. 3: 802. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9030802