Mechanotransduction in Wound Healing and Fibrosis
by
 , , and
, , and
Britta Kuehlmann
1,2,† ,
,
Clark A. Bonham
1,†,
Isabel Zucal
2,
Lukas Prantl
2 and
Geoffrey C. Gurtner
1,* 1
Division of Plastic and Reconstructive Surgery, Department of Surgery, Stanford University, Stanford, CA 94305, USA
2
University Center for Plastic, Reconstructive, Aesthetic and Hand Surgery, University Hospital Regensburg and Caritas Hospital St. Josef, 93053 Regensburg, Germany
*
Author to whom correspondence should be addressed.
†
Both authors contributed equally to this manuscript.
J. Clin. Med. 2020, 9(5), 1423; https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9051423
Submission received: 19 April 2020
/
Revised: 6 May 2020
/
Accepted: 7 May 2020
/
Published: 11 May 2020
(This article belongs to the Special Issue Clinical Trends in Regenerative Medicine)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Skin injury is a common occurrence and mechanical forces are known to significantly impact the biological processes of skin regeneration and wound healing. Immediately following the disruption of the skin, the process of wound healing begins, bringing together numerous cell types to collaborate in several sequential phases. These cells produce a multitude of molecules and initiate multiple signaling pathways that are associated with skin disorders and abnormal wound healing, including hypertrophic scars, keloids, and chronic wounds. Studies have shown that mechanical forces can alter the microenvironment of a healing wound, causing changes in cellular function, motility, and signaling. A better understanding of the mechanobiology of cells in the skin is essential in the development of efficacious therapeutics to reduce skin disorders, normalize abnormal wound healing, and minimize scar formation.
1. Introduction
Wound healing is a complex process of overlapping, consecutive phases that restores the structural integrity of the skin following injury. Recent studies have identified mechanical signaling networks in the skin that influence wound healing. These pathways elicit changes in the physiology and structure of the skin, which can delay regeneration and lead to fibrosis. In recent years, the effects of force and mechanical stress on wound healing have gained significant clinical attention and the importance of these pathways has been confirmed in clinical trials. Cells have been shown to respond to mechanical stress by altering their functional, migratory, and signaling capabilities through a process called mechanotransduction. All such events have a profound effect on wound healing, leading to changes in the final wound phenotype, including overhealing (fibrosis, keloids) and underhealing (chronic wounds).
2. Wound Healing and Fibrosis in the Skin
Wound healing undergoes several distinct phases following injury to the skin: hemostasis, inflammation, proliferation, and tissue remodeling. Immediately after tissue disruption, platelets converge and adhere to severed blood vessels and prevent excessive bleeding. These fibrin clots provide a variety of beneficial proteins and signaling molecules to progress healing into the inflammatory phase [1].
Circulating monocytes undergo chemotaxis as a result of inflammatory cell signaling, migrating to the wound bed where other signaling molecules induce their differentiation into macrophages. Along with newly recruited neutrophils and tissue-resident macrophages, these migratory macrophages attempt to clean the wound bed of harmful and foreign substances. In chronic wounds, this phase is drawn out, and it often does not come to a successful conclusion, inhibiting the proper, successful progression of wound healing.
In acute wounds, following the successful cellular debridement of the wound, macrophages and other cells begin to secrete signaling molecules in order to recruit fibroblasts to the wound. These fibroblasts undergo differentiation into myofibroblasts, characterized by their alpha smooth muscle actin (α-SMA) bundles that give them a contractile capability [2]. These myofibroblasts synthesize collagen and other extracellular matrix (ECM) components that serve as the foundation for the healing wound [3]. The myofibroblasts work to contract the wound following sufficient collagen and ECM deposition.
As the wound begins to close, cell signaling causes phenotypical changes within the cells and the focus shifts to restructuring the newly deposited components to produce the final healed scar. Matrix metalloproteinases (MMPs) and their respective inhibitors, tissue inhibitors of metalloproteinases (TIMPs), are produced to break down and remodel the collagen and ECM bundles [4]. This final phase concludes wound healing but yields a scar that differs in structure and tensile strength when compared to regular, healthy skin.
Scarring can differ among individuals, with some being more susceptible to keloids and hypertrophic scars than others. Most scars cause distress to the individual and they can lead to functional deficiencies, with billions of dollars being spent on scar treatment each year [5]. Thus, a better understanding of scar formation might lead to greater prevention of undesirable outcomes.
3. Mechanotransduction in Skin and Wounds
Human skin constantly deals with intrinsic and extrinsic forces throughout life. The effect of mechanical force is dependent upon the stiffness and biomechanical properties of the skin, which vary between anatomical locations [6]. Mechanical stimuli contribute to alterations in the wound healing process and underlie the increased susceptibility to excessive scar formation found in particular regions of the body [7,8]. Modern plastic surgery already employs several mechanomodulatory procedures in order to counter these effects, including z-plasty and the use of steristrips [9,10]. Both relax skin tension at the site of the wound, effectively reducing scar development, although there remains room for improvement [11,12,13]. As our understanding of mechanotransduction grows, we are focusing on “next generation” biomolecular approaches to further reduce scarring and fibrosis.
It is important to first grasp how mechanotransduction works at the cellular and tissue levels to better understand how the signaling pathways involved in wound healing and skin fibrosis are affected by mechanical force. As physical force is applied to the skin, mechanical signals are conveyed into chemical information by molecules that transmit this information to the cell (model of cellular tensegrity) [14,15,16,17]. The extracellular matrix (ECM) and the extracellular fluid (ECF) are essential to the transduction of mechanical forces into cells and control the constant remodeling of the skin. Transmembrane structures play an integral role in this process, as the membrane itself is fluid and requires specialized structures to transduce forces. Components of the cell membrane and cytoskeleton (e.g., actin and RhoA), ion channels, catenin complexes, cell adhesion molecules (e.g., focal adhesions and integrins), and several signaling pathways (e.g., Wnt, FAK-ERK, MAPK/ERK) are known to act as mechanosensors [18,19,20]. These sensors transmit mechanical signals to cells that bind to the extracellular matrix (ECM) and trigger a further signaling cascade of responses [21,22,23] (Figure 1). For instance, the destruction of the ECM in aged skin leads to the further breakdown of fibroblasts within the dermis, as these cells thereby cease to receive mechanical information [24]. As a result, collagen production is decreased, while that of collagen-degrading enzymes, such as matrix metalloproteinases (MMPs), is increased [25,26].
Previous studies have demonstrated the significance of mechanical force in modulating hypertrophic scar (HTS) formation in a mechanically stress-induced murine scar model [27,28]. Sustained mechanical load applied to incisions resulted in fibrotic responses and HTS formation, demonstrating that mechanomodulation is linked to scarring through the stimulation of an inflammatory response [29]. Clinical evidence also supports the importance of mechanical stress on the development of scarring, as the use of a compression device on human patients postoperatively led to reductions in scar size through mechanical offloading in multiple human randomized clinical trials (RCTs) [30,31]. Specifically, a contracting elastomeric silicone dressing device has been shown to significantly improve scars through application of compressive forces to the incision by minimizing impact on the wound through the off-loading of tension [30,32]. Similarly, multiple RCTs of skin taping to improve scar appearance have shown clinical benefit in the appearance of scars [33,34,35,36].
In the following paragraphs, we will review the pathways that mediate mechanotransduction and underlie these clinical findings.
4. Tensegrity and CTF
The skeletal and membranous components of cells both serve as mediators between physical force and downstream cellular signaling and activity. Tensegrity describes the alignment of structural components of the cytoskeleton in response to mechanical forces in order to preserve the cell’s tensional integrity. The cells are exposed to mechanical prestress as the cytoskeletal components are connected with each other and to the ECM, leading to a continuous balance between opposing forces (e.g., actomyosin contraction is resisted by microtubules intracellularly and by the stiffness of the ECM outside the cell). This ongoing balance of tensional forces provides the cell with stability: when mechanical stress comes from the ECM or surrounding cells, the cytoskeleton’s arrangement changes to counteract this stress. The cytoskeleton is further subdivided into hierarchically subordered structural components that ensure stability themselves, for example the nucleus, the submembranous cytoskeleton, and the actomyosin filament bundles work independently [16,17]. These structural components ensure a connection between the ECM, as well as the cell surface membrane and nuclear chromatin, generating epigenetic modulation of intranuclear programs when the mechanical forces of the surrounding tissue act upon the cell surface [37]. Conversely, cell traction force (CTF) is generated by intracellular actin/myosin interactions that are regulated by α-SMA and TGF-β. Information is then passed on to the ECM via focal adhesions, regulating cell migration and ECM organization and modulating mechanical signal generation [38]. In summary, mechanical stimuli from the outside are transduced to the nucleus and vice versa via tensegrity and CTF, respectively. Tensegrity strongly determines wound healing processes, scar formation and cellular activity, shape, and motility alike [39,40].
5. TGF-β
TGF-β represents a crucial soluble factor promoting fibrosis, with TGF-β1 inducing myofibroblast stimulation. The activated myofibroblasts secrete TGF-β, creating a positive feedback loop that further exacerbates fibrosis. As a growth factor, it is involved in structural changes of the cell and the secretion of pro-fibrotic factors, such as α-SMA, which interact with myosin to increase tension [41,42,43].
In the ECM, TGF-β1 can bind to the latency-associated peptide (LAP) and the latent TGF-β-binding-protein-1 (LTBP-1). Via integrins, LTBP-1 can be activated by intracellular forces and TGF-β1 is released. As cell tension and force propagation increase, so does TGF-β1 liberation and biomechanical tissue stiffness in fibrosis therefore increases TGF-β1 activation [44]. TGF-β1 itself promotes the expression of genes that are integral to the deposition of the ECM, such as collagens [45], fibronectin [46], and plasminogen activator inhibitor type-1 (PAI-1) [47] and they accumulate in fibrosis.
TGF-β2 is known to have pro-fibrotic properties and accumulation has been found in human fibrotic liver disease [48]. However, TGF-β3 is characterized by the regulation of epidermal and dermal cell motility, which plays a key role in wound repair and promotes wound healing without fibrotic scar formation [49]. In this regard, Ferguson et al. assessed the intradermal administration of avotermin, recombinant, active, human TGF-β3 for scar prevention. Visual assessment of scar formation was performed after six and twelve months on a visual analogue scale (VAS) with the intervention group displaying significant improvement. However, only phase I and II of human clinical trials were passed, whereas phase III not [50].
Recently, several studies have investigated pharmacologic therapies targeting TGF-β signaling in fibrotic diseases throughout the body, affecting the kidney [51], lungs [52], and heart, among others [53]. P144, a peptide inhibitor of TGF-β1, was tested by Santiago et al. in mice that received daily injections of bleomycin representing a model of human scleroderma. Their study displayed that there was a significant decrease in skin fibrosis and soluble collagen content after application of P144 lipogel for two weeks. TGF-β and α-SMA levels were lower, resulting in reduced wound scarring and fibrosis [54]. Nevertheless, few of these studies showed positive outcomes in patients [41]. Creating a systemic therapy without impeding otherwise unrelated homeostatic processes remains complicated due to the versatility of TGF-β.
6. FAK-ERK-MCP1 Pathway
Focal adhesion kinase (FAK), which is a non-receptor cytoplasmic tyrosine kinase, is one of the key mediators of skin mechanobiology and it is activated after cutaneous injury [27,55]. Mechanical forces potentiate the activation of FAK through phosphorylation following injury of the skin [56,57,58]. FAK contributes to cell signaling through its linking of mechanical stress from the ECM to the cytoplasmic cytoskeleton [59], activating inflammatory pathways. Fibroblasts are recruited to the wound by inflammatory signaling, where their secretion of profibrotic cytokines brings about increased collagen synthesis.
Interestingly, various pathologies that are associated with poor wound healing have been shown to have atypical levels of FAK. Wong et al. demonstrated that mechanical force regulates pathologic scarring through inflammatory FAK-ERK-MCP1 pathways, and that molecular strategies targeting focal adhesion kinase (FAK) can effectively uncouple mechanical force from fibrosis [27] (Figure 2). Thus, FAK remains a potentially promising target for drug approaches to minimize scar formation [27]. Further, FAK is found to be downregulated in diabetes under high glucose conditions as a result of increased calpain 1 activation [60]. Liu et al. have shown that fibroblasts are prone to high glucose stimuli, unlike keratinocytes, which are resistant to high glucose-induced FAK degradation by calpain 1 [60]. Further studies have shown that mice with keratinocyte FAK knockout display delayed wound closure with reduced collagen density and dermal thickness. FAK knockout keratinocytes exhibited hyperactive MMP9 and p38 signaling when cultured in vitro, highlighting FAK as an upstream regulator that is essential to wound healing [61]. The aberrant secretion of MMP9 is a result of blocked FAK in suspended human keratinocytes and the loss of epithelial FAK has been shown to impede normal repair pathways [61]. These findings highlight the significance of properly functioning mechanotransduction pathways during the regeneration processes and that some level of their activity is crucial in proper wound closure.
Pharmacological approaches to locally inhibit FAK have previously shown anti-scarring effects in in vitro and preclinical animal studies [62,63]. In 2018, Ma et al. were able to show that the application of FAK inhibitor (FAKI) pullulan-collagen-based hydrogel scaffolds promotes wound healing with reduced scar formation [64]. FAKI hydrogels inhibited the phosphorylation of FAK in a sustained manner that was mediated by slow release. As a result, collagen deposition and myofibroblast counts were both reduced in FAKI treated wounds. Further, FAKI treated healed skin displayed improved mechanical integrity, as seen by the quantification of the Young’s modulus [64]. Such findings point to the therapeutic effects that targeting mechanotransduction pathways may bring.
7. Canonical Wnt Pathway and β-catenin Pathway
Like the FAK-ERK-MCP1 pathway, Wnt/β-catenin signaling has been shown to play an essential role in the self-renewing ability of the skin [65,66]. The canonical Wnt pathway is essential to passing extracellular mechanical signals into the cell through its surface receptors. β-catenin is a structural component of adherens junctions in epithelial cells, regulating cell-cell interactions (Figure 3). After binding to the cytoplasmic domain of E-cadherin, β-catenin binds to α-catenin and it acts as a component of intercellular adhesive junctions [67], which mechanically link cadherins to actin [68]. α-catenin is thought to be essential for linking cadherin to actin filaments.
In response to physical force and the upregulation of Wnt signaling, β-catenin accumulates within the nucleus [69]. Interestingly, fibroblasts expressing increased amounts of β-catenin are found in hypertrophic scars and keloids, implicating β-catenin in cutaneous fibroproliferative diseases [70]. Further, skin biopsies from patients with systemic sclerosis display increased expression of several Wnt molecules, implicating Wnt signaling in fibrotic diseases of the skin [71,72]. As such, the Wnt/β-catenin pathway affects scar formation in the dermis through the upregulation of pro-fibrotic function [73,74], and prolonged activation of Wnt/β-catenin signaling has been observed in human hyperplastic wounds [75].
Ray et al. showed that β-catenin provides stability to the epidermis under stress, while the loss of β-catenin results in a loss of response to mechanical stimuli in vitro [76]. The loss of β-catenin weakened tight junctions’ associations with the cytoskeleton and in turn cellular responsiveness to mechanical stress. Increased Wnt signaling was also shown to increase the production of reactive oxygen species (ROS), leading to DNA damage and, thus, increased senescence [77]. Furthermore, the Wnt/β-catenin pathway is connected to TGF-β signaling as β-catenin induces TGF-β to induce fibroblast activity in human skin [78,79]. The manipulation of Wnt/ β-catenin signaling, through both up- and down-regulation, significantly alters the wound healing and scarring responses via the manipulation of mechanosignaling. Regardless, the specific role and changes of Wnt/β-catenin in wounded skin still need to be further elucidated, though manipulation of the pathway could prove to be a promising therapeutic.
8. YAP/TAZ
The Hippo pathway is a highly conserved network that moderates tissue growth in adults [80]. Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) are two major downstream regulators that serve as mechanotransducers in response to the stiffness and arrangement of the cytoskeleton [39]. Because the cytoskeleton’s organization and tensional state are influenced by the cell’s physical position in three-dimensional space, mechanical strains are applied to the cell that alter its rigidity and structure [81,82]. While the cell is free from outside stress, YAP/TAZ persists in an inactive state within the cytoplasm that is modulated by mechanical stress upstream. Focal adhesion formation through structures, such as integrin, FAK, and SRC, among others, causes YAP/TAZ to dephosphorylate from their inactive complex and subsequently relocate to the nucleus, where they drive transcription in concert with several coactivators [81,82,83]. Such activity impacts the metabolic function of cells and influences myriad processes throughout the body, from myofibroblast differentiation in fibrosis, to the activity of endothelial cells in angiogenesis [82,84,85]. As such, YAP/TAZ are essential regulators of collective migration and proliferation and they are likely increasingly expressed at sites that are susceptible to higher levels of tension, making them particularly important to scar development [81].
The unspecific nature of YAP/TAZ activity and its regulatory role within various profibrotic pathways makes it a promising therapeutic target, although much remains to be elucidated. Accordingly, contemporary research has attempted to identify the essentials of YAP/TAZ signaling in the progression of wound regeneration. Lee et al. showed, in an siRNA-mediated knockdown of YAP/TAZ model, that YAP/TAZ localization to the nucleus is essential for proper wound healing in full-thickness wounds [86]. Another interesting study suggests that YAP functions as a molecular switch of stem/progenitor cell activation in the epidermis and it is essential for epidermal differentiation and proliferation [87,88]. Additionally, YAP is known to influence skin size and tissue overgrowth [89] (Figure 4).
9. ILK-PI3K/Akt Pathway
Mechanical strain activates Integrin-linked kinase (ILK)-Phosphoinositide 3-kinase (PI3K)/Akt signaling [90]. Generally, PI3K/Akt activity influences cell proliferation, motility, growth, survival, and apoptosis. There are three classes of PI3Ks, of which Class I is activated by receptor tyrosine kinases (RTKs) or G protein-coupled receptors (GPCRs). Akt is a serine/threonine protein kinase that is activated following recruitment to the plasma membrane and it acts downstream from PI3K [91]. The ILK-PI3K/Akt pathway is activated through transmembrane integrin receptors and it initiates signaling of Akt following PI3K-dependent phosphorylation (Figure 5). Pathway activation in fibroblasts increases their motility, as well as α-SMA expression and collagen I production. Li et al. demonstrated that ILK-overexpression leads to greater scar contracture and scar hypertrophy. Conversely, ILK and PI3K/AKT inhibitors inhibited wound contraction and re-epithelialization, consequently delaying wound healing in vivo. Similar effects were observed following the application of Akt inhibitors in vitro [92].
Moreover, the PI3K/Akt-pathway is a crucial pathway in human keratinocyte differentiation. In fact, the PI3K/Akt-pathway is activated early in the keratinocyte differentiation. Activation is contingent on the expression of epidermal growth factor receptor (EGFR), E-cadherin-mediated adhesion, and Src families of tyrosine kinases, respectively. Blockade of PI3K was shown to cause cell death in keratinocytes, underscoring the importance of balanced PI3K/Akt signaling to proper keratinocyte functionality [93].
Investigating the effect of stretch force in vitro, Yano et al. observed that the mechanical stretching of keratinocytes led to the activation of the Akt-pathway, generating proliferative and antiapoptotic signals. Thus, the degradation of keratinocytes is inhibited and skin preservation is promoted [94]. Moreover, previous studies of our lab have shown that Akt activation fluctuates with both the frequency and degree of strain imposed, and that wound tension in mouse models promotes dermal fibroblasts to express the higher activation of Akt [95]. Further, Gao et al. evaluated peaks of PI3K- and p-Akt-expression during reconstruction in the wound healing process and suggested western blot-quantification as a marker for wound time estimation [96]. Akt also serves to activate Mammalian Target of Rapamycine (mTOR), which mediates the fibroblast response to TGF-β [97] and MMP1-inhibition, preventing its proteolytic collagenase activity [98].
The aforementioned studies implicate the ILK-PI3K/Akt pathway as an essential player in wound healing, with both over- and under-expression hindering the regenerative process. As such, the pharmacological modulation of the ILK-PI3K/Akt pathway in fibroblasts and keratinocytes might serve to ameliorate hypertrophic scarring.
10. Rho-GTPases
Rho-GTPases are intracellular proteins that mediate between integrins and the cytoskeleton, regulating cell tension, intracellular force, motility, and adherence in response to mechanical force [99]. Rho-GTPases are activated by tension on integrins in focal adhesions as well as by soluble factors, such as growth factors [100] (Figure 6). RhoA, Rac1, and Cdc42 generate contractile forces by coordinating myosin II activity by Rho kinase (ROCK) stimulation [101] and promoting actin filament assembly [102]. As a consequence, wound contracture and fibrosis are increased. Previous studies examining fibroblasts in cardiac and pulmonary fibrosis have determined that the inhibition of Rho-associated signaling reduces collagen synthesis [103]. Bond et al. used Fasudil, a Rho-associated kinase inhibitor, to hinder fibroblast contractility and prevent excessive scarring in rat models. As a result of Fasudil application, wound contracture was reduced when compared to control wounds [104]. Finally, Rho-GTPases represent key mediators for cell proliferation, survival, and motility, not only in fibroblasts, but also in keratinocytes [105]. Hence, future investigations on Rho signaling pathway inhibitors may lead to an effective therapeutic option to prevent fibrosis and contracture in wounds, as well as to treat diseases that are characterized by excessive fibrosis, such as idiopathic pulmonary fibrosis (IPF).
11. Calcium-Dependent ion Channels
Mechanosensitive calcium-dependent ion channels serve as integral mechanotransducers, as incidental stretching can force open channels, allowing calcium (Ca2+) influx and calcium-dependent pro-fibrotic pathway initiation. The mechanosensitive cation channel TRPV4 (transient receptor potential vanilloid 4) and the voltage-gated L-type channels, among others, influence myofibroblast differentiation through extracellular calcium influx. Active TRPV4 was further shown to enhance actomyosin remodeling and promote nuclear translocation of the α-SMA transcription coactivator, another factor contributing to fibrogenesis [106,107].
Calcium levels alter nitric oxide (NO)-pathways that are involved in promoting angiogenesis, proliferation of fibroblasts, epithelial cells, and keratinocytes [108].
Ca2+-dependent ion channels have proven to be effective therapeutic targets across various diseases. Mukherjee et al. tested several types of calcium channel blockers (CCBs), namely 1 μM of Nifedipine, 1 μM of Verapamil (both L-type blockers), 2.7 μM of Mibefradil (a mixed L-/T-type blocker), 40 μM of NiCl2 (selective T-type blocker at this concentration), 30 mM of KCl (depolarizes the cell membrane and inactivates T-type current), and 1 mM of NiCl2 (L- and T-type current blocker) in human fibroblasts, showing that intracellular Ca2+ oscillations evoked by exogenously applied transforming growth factor-β (TGF-β) were significantly reduced [109]. Such inhibitors are believed to interfere with fibroblast Ca2+ influx, disrupting Ca2+ oscillations and, in turn, fibroblasts’ abilities to synthesize collagen, thereby protecting from pulmonary fibrosis in humans. Nifedipine was further shown to reduce bleomycin-induced lung fibrosis in rat models, impeding Ca2+ influx [109]. The utilization of CCBs has also been demonstrated to encourage wound healing in rat models, with studies assessing the administration of verapamil, diltiazem, difedipine, amlodipine, and azelnidipine. Accelerated wound healing and enhanced tensile strength of the skin were observed in treatment groups when compared with the untreated controls [110,111,112,113].
12. GPCRs
G protein-coupled receptors (GPCRs) are characterized by the presence of seven transmembrane domain proteins and they are activated by focal adhesion complexes [114]. GPCRs can promote both pro-fibrotic and anti-fibrotic phenotypes in fibroblasts. Receptor class and downstream signaling pathways determine whether fibrosis is supported or not [115] (Figure 7).
When pro-fibrotic pathways are activated, GPCRs take part in collagen synthesis, as well as fibroblast contraction [116]. Conversely, Roberts et al. explored the ability of GPCRs to inhibit fibrotic development. GPCRs can increase the cAMP levels in human lung fibroblasts, leading to the inhibition of fibroblast proliferation and differentiation. Formoterol, prostaglandin E2, treprostinil, and forskolin were shown to reach maximal cAMP responses, whereas partial GPCR-agonists achieved full inhibition of fibroblasts, providing a promising set of targets for novel IPF treatments [117]. Interestingly, it was demonstrated that maximal cAMP response does not correlate with maximal inhibition of proliferation and differentiation in fibroblasts. Moreover, Dooling et al. described the inhibition of tumor fibrosis and decreased force generation of the actomyosin cytoskeleton by the application of GPCR-agonists [118].
13. Future Perspectives
In conclusion, mechanotransduction contributes to cellular proliferation, survival, differentiation, mobility, shape, and apoptosis, whether by first-hand effect or downstream paracrine signaling. Thus, both external and internal mechanical forces have the ability to alter phenotypical responses to disease and dysfunction. As the skin serves as the body’s barrier to external stimuli, it is almost always subjected to such mechanical forces that impede wound repair processes and intensify scar production. The development of target molecules to prevent excessive scarring and fibrosis might prove difficult, as target molecules are often characterized by non-specific target cells. Nevertheless, a better understanding and identification of mechanotransduction pathways will contribute to the development of suitable pharmaceutical agents.
14. Conclusions
Scars, keloids, and fibrosis affect a relevant fraction of the population and they often require corrective treatment. However, modern therapies are typically ineffective in the case of scar reduction and patients with large, obvious scars suffer from both a physical and mental perspective. In fact, the exact pathogenesis of many fibrotic diseases, such as keloids, is yet poorly understood, in part because of lacking adequate animal models. The detection of further key players in fibrosis and scar formation is still required [119,120]. Extending beyond experiences of poor wound healing, all surgical procedures inevitably produce a scar, superficially visible or not. From lifesaving cases requiring major incisions to common aesthetic procedures, the resulting scar formation remains an everyday problem in plastic surgery, bringing about a need for improved therapeutics. With an understanding of the factors that contribute to increased scarring and worsened wound healing, such improvements are being made.
Pathways, including the FAK-ERK-MCP1 and YAP/TAZ, have been shown to impact different stages of regeneration through mechanotransduction, and specific targeting of these pathways has improved scarring. Device approaches have shown promise in mitigating scar formation in humans by countering the scarring effects of stretching with compression, but the ultimate solution will require drug appeal. Understanding mechanotransduction pathways will unravel the mechanisms that regulate scar formation and wound healing and lead to the development of effective therapeutics to reduce scar formation in a number of fibrotic diseases.
Funding
Gurtner is a founder of Neodyne Biosciences, the manufacturer of the embrace device mentioned in this manuscript. All comments regarding Neodyne and embrace are derived from peer reviewed, published literature.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| α-SMA | Alpha Smooth Muscle Actin |
| Ca2+ | calcium |
| CaSR | calcium-sensing receptors |
| CCB | calcium channel-blocker |
| CTF | cell traction force |
| DNA | deoxyribonucleic acid |
| ECF | extracellular fluid |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular signal-related kinase |
| FAK | focal adhesion kinase |
| FAKI | focal adhesion kinase inhibitor |
| GPCR | G protein-coupled receptor |
| HCN | hyperpolarization-activated Cyclic Nucleotide-gated |
| HTS | hypertrophic scar |
| ILK | integrin-linked kinase |
| IPF | idiopathic pulmonary fibrosis |
| K+ | kalium |
| LAP | latency-associated peptide |
| LTBP-1 | latent TGF-β-binding-protein-1 |
| miRNA | microRNA |
| MMP | matrix metalloproteinase |
| mRNA | messenger RNAs |
| MS | mechanosensitive |
| MSCs | mechanosensitive ion channels |
| mTOR | mammalian target of rapamycine |
| MYLK | myosin light chain kinase |
| Na+ | natrium |
| NO | nitric oxide |
| PAI-1 | plasminogen activator inhibitor type-1 |
| PI3K | phosphoinositide 3-kinase |
| RAN | ribonucleic acid |
| RTK | receptor tyrosine kinase |
| ROCK | rho-associated coiled-coil kinase |
| ROS | reactive oxygen species |
| siRNA | small interfering RNA |
| TAZ | tafazzin |
| TGFβ | transforming growth factor beta |
| TIMP | tissue inhibitors of metalloproteinase |
| TRP | transient receptor potential |
| TRPV4 | transient receptor potential vanilloid 4 |
| VAS | visual analogue scale |
| YAP | yes-associated protein |
References
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Masur, S.K.; Dewal, H.S.; Dinh, T.T.; Erenburg, I.; Petridou, S. Myofibroblasts differentiate from fibroblasts when plated at low density. Proc. Natl. Acad. Sci. USA 1996, 93, 4219–4223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosain, A.; DiPietro, L.A. Aging and wound healing. World J. Surg. 2004, 28, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Sen, C.K.; Gordillo, G.M.; Roy, S.; Kirsner, R.; Lambert, L.; Hunt, T.K.; Gottrup, F.; Gurtner, G.C.; Longaker, M.T. Human skin wounds: A major and snowballing threat to public health and the economy. Wound Repair Regen. 2009, 17, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.; Levi, K.; Akaishi, S.; Schultz, G.; Dauskardt, R.H. Scar zones: Region-specific differences in skin tension may determine incisional scar formation. Plast Reconstr. Surg. 2012, 129, 1272–1276. [Google Scholar] [CrossRef]
- Bayat, A.; McGrouther, D.A.; Ferguson, M.W. Skin scarring. BMJ 2003, 326, 88–92. [Google Scholar] [CrossRef]
- Barnes, L.A.; Marshall, C.D.; Leavitt, T.; Hu, M.S.; Moore, A.L.; Gonzalez, J.G.; Longaker, M.T.; Gurtner, G.C. Mechanical Forces in Cutaneous Wound Healing: Emerging Therapies to Minimize Scar Formation. Adv. Wound Care 2018, 7, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Wakure, A. Scar revision. Indian J. Plast Surg. 2013, 46, 408–418. [Google Scholar] [CrossRef]
- Block, L.; Gosain, A.; King, T.W. Emerging Therapies for Scar Prevention. Adv. Wound Care (New Rochelle) 2015, 4, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, E.A.; Liang, H.; Mahadevan, L. Topology, Geometry, and Mechanics of Z-Plasty. Phys. Rev. Lett. 2018, 120, 068101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerrigan, C.L.; Homa, K. Evaluation of a new wound closure device for linear surgical incisions: 3M Steri-Strip S Surgical Skin Closure versus subcuticular closure. Plast Reconstr. Surg. 2010, 125, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nast, A.; Eming, S.; Fluhr, J.; Fritz, K.; Gauglitz, G.; Hohenleutner, S.; Panizzon, R.G.; Sebastian, G.; Sporbeck, B.; Koller, J. German S2k guidelines for the therapy of pathological scars (hypertrophic scars and keloids). J. Dtsch. Dermatol. Ges. 2012, 10, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Control of capillary growth and differentiation by extracellular matrix. Use of a tensegrity (tensional integrity) mechanism for signal processing. Chest 1991, 99, 34–40. [Google Scholar] [CrossRef]
- Ingber, D.E. Cellular mechanotransduction: Putting all the pieces together again. FASEB J. 2006, 20, 811–827. [Google Scholar] [CrossRef]
- Ingber, D.E. Tensegrity I. Cell structure and hierarchical systems biology. J. Cell Sci. 2003, 116, 1157–1173. [Google Scholar] [CrossRef] [Green Version]
- Ingber, D.E. Tensegrity II. How structural networks influence cellular information processing networks. J. Cell Sci. 2003, 116, 1397–1408. [Google Scholar] [CrossRef] [Green Version]
- Knapik, D.M.; Perera, P.; Nam, J.; Blazek, A.D.; Rath, B.; Leblebicioglu, B.; Das, H.; ChuWu, L.; Hewett, T.E.; Agarwal, S.K.J.; et al. Mechanosignaling in bone health, trauma and inflammation. Antioxid. Redox Signal. 2014, 20, 970–985. [Google Scholar] [CrossRef] [Green Version]
- Young, S.R.; Gerard-O’Riley, R.; Kim, J.B.; Pavalko, F.M. Focal adhesion kinase is important for fluid shear stress-induced mechanotransduction in osteoblasts. J. Bone Miner. Res. 2009, 24, 411–424. [Google Scholar] [CrossRef]
- Marjoram, R.J.; Lessey, E.C.; Burridge, K. Regulation of RhoA activity by adhesion molecules and mechanotransduction. Curr. Mol. Med. 2014, 14, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Alenghat, F.J.; Ingber, D.E. Mechanotransduction: All signals point to cytoskeleton, matrix, and integrins. Sci. STKE 2002, 119, pe6. [Google Scholar] [CrossRef] [PubMed]
- Sukharev, S.; Betanzos, M.; Chiang, C.S.; Guy, H.R. The gating mechanism of the large mechanosensitive channel MscL. Nature 2001, 409, 720–724. [Google Scholar] [CrossRef]
- Geiger, B.; Spatz, J.P.; Bershadsky, A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009, 10, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Varani, J.; Dame, M.K.; Rittie, L.; Fligiel, S.E.G.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Decreased collagen production in chronologically aged skin: Roles of age-dependent alteration in fibroblast function and defective mechanical stimulation. Am. J. Pathol. 2006, 168, 1861–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, R.; Hsu, C.K. Mechanobiological dysregulation of the epidermis and dermis in skin disorders and in degeneration. J. Cell. Mol. Med. 2013, 17, 817–822. [Google Scholar] [CrossRef]
- Fligiel, S.E.; Varani, J.; Datta, S.C.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Collagen degradation in aged/photodamaged skin in vivo and after exposure to matrix metalloproteinase-1 in vitro. J. Investig. Dermatol. 2003, 120, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.; Rustad, K.C.; Akaishi, S.; Sorkin, M.; Glotzbach, J.P.; Januszyk, M.; Nelson, E.R.; Levi, K.; Paterno, J.; Vial, I.N.; et al. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat. Med. 2011, 18, 148–152. [Google Scholar] [CrossRef] [Green Version]
- Aarabi, S.; Bhatt, K.A.; Shi, Y.; Paterno, J.; Chang, E.I.; Loh, S.A.; Holmes, J.W.; Longaker, M.T.; Yee, H.; Gurtner, G.C. Mechanical load initiates hypertrophic scar formation through decreased cellular apoptosis. FASEB J. 2007, 21, 3250–3261. [Google Scholar] [CrossRef] [Green Version]
- Longaker, M.T.; Rohrich, R.J.; Greenberg, L.; Furnas, H.; Wald, R.; Bansal, V.; Seify, H.; Tran, A.; Weston, J.; Korman, J.M.; et al. A randomized controlled trial of the embrace advanced scar therapy device to reduce incisional scar formation. Plast Reconstr. Surg. 2014, 134, 536–546. [Google Scholar] [CrossRef] [Green Version]
- Lim, A.F.; Weintraub, J.; Kaplan, E.N.; Januszyk, M.; Cowley, C.; McLaughlin, P.; Beasley, B.; Gurtner, G.C.; Longaker, M.T. The embrace device significantly decreases scarring following scar revision surgery in a randomized controlled trial. Plast Reconstr. Surg. 2014, 133, 398–405. [Google Scholar] [CrossRef] [Green Version]
- Orgill, D.P.; Ogawa, R. Discussion: The embrace device significantly decreases scarring following scar revision surgery in a randomized controlled trial. Plast Reconstr. Surg. 2014, 133, 406–407. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Dauskardt, R.H.; Wong, V.W.; Bhatt, K.A.; Wu, K.; Vial, I.N.; Padois, K.; Korman, J.M.; Longaker, M.T. Improving cutaneous scar formation by controlling the mechanical environment: Large animal and phase I studies. Ann. Surg. 2011, 254, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Lionelli, G.T.; Lawrence, W.T. Wound dressings. Surg. Clin. N. Am. 2003, 83, 617–638. [Google Scholar] [CrossRef]
- Reiffel, R.S. Prevention of hypertrophic scars by long-term paper tape application. Plast Reconstr. Surg. 1995, 96, 1715–1718. [Google Scholar] [CrossRef]
- Rosengren, H.; Askew, D.A.; Heal, C.; Buettner, P.G.; Humphreys, W.O.; Semmens, L.A. Does taping torso scars following dermatologic surgery improve scar appearance? Derm. Pr. Concept. 2013, 3, 75–83. [Google Scholar] [CrossRef]
- Atkinson, J.A.; McKenna, K.T.; Barnett, A.G.; McGrath, D.J.; Rudd, M. A randomized, controlled trial to determine the efficacy of paper tape in preventing hypertrophic scar formation in surgical incisions that traverse Langer’s skin tension lines. Plast Reconstr. Surg. 2005, 116, 1648–1656. [Google Scholar] [CrossRef]
- Gieni, R.S.; Hendzel, M.J. Mechanotransduction from the ECM to the genome: Are the pieces now in place? J. Cell. Biochem. 2008, 104, 1964–1987. [Google Scholar] [CrossRef]
- Wang, J.H.; Lin, J.S. Cell traction force and measurement methods. Biomech. Model. Mechanobiol. 2007, 6, 361–371. [Google Scholar] [CrossRef]
- Duscher, D.; Maan, Z.N.; Wong, V.W.; Rennert, R.C.; Januszyk, M.; Rodrigues, M.; Hu, M.; Whitmore, A.J.; Whittam, A.J.; Longaker, M.T.; et al. Mechanotransduction and fibrosis. J. Biomech. 2014, 47, 1997–2005. [Google Scholar] [CrossRef] [Green Version]
- Noguera, R.; Nieto, O.A.; Tadeo, I.; Fariñas, F.; Alvaro, T. Extracellular matrix, biotensegrity and tumor microenvironment. An update and overview. Histol. Histopathol. 2012, 27, 693–705. [Google Scholar]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharm. 2017, 8, 461. [Google Scholar] [CrossRef] [Green Version]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, R.G. Tissue mechanics and fibrosis. Biochim. Biophys. Acta 2013, 1832, 884–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscemi, L.; Ramonet, D.; Klingberg, F.; Formey, A.; Smith-Clerc, J.; Meister, J.J.; Hinz, B. The single-molecule mechanics of the latent TGF-β1 complex. Curr. Biol. 2011, 21, 2046–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrecchia, F.; Vindevoghel, L.; Lechleider, R.J.; Uitto, J.; Roberts, A.B.; Mauviel, A. Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific manner. Oncogene 2001, 20, 3332–3340. [Google Scholar] [CrossRef] [Green Version]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef] [Green Version]
- Milani, S.; Herbst, H.; Schuppan, D.; Stein, H.; Surrenti, C. Transforming growth factors beta 1 and beta 2 are differentially expressed in fibrotic liver disease. Am. J. Pathol. 1991, 139, 1221–1229. [Google Scholar]
- Occleston, N.L.; Laverty, H.G.; O’Kane, S.; Ferguson, M.W. Prevention and reduction of scarring in the skin by Transforming Growth Factor beta 3 (TGFbeta3): From laboratory discovery to clinical pharmaceutical. J. Biomater. Sci. Polym. Ed. 2008, 19, 1047–1063. [Google Scholar] [CrossRef]
- Ferguson, M.W.; Duncan, J.; Bond, J.; Bush, J.; Durani, P.; So, K.; Taylor, L.; Chantrey, J.; Mason, T.; James, G.; et al. Prophylactic administration of avotermin for improvement of skin scarring: Three double-blind, placebo-controlled, phase I/II studies. Lancet 2009, 373, 1264–1274. [Google Scholar] [CrossRef]
- Voelker, J.; Berg, P.H.; Sheetz, M. Anti-TGF- β1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, A. Pirfenidone treatment of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2012, 6, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Hermida, N.; López, B.; González, A.; Dotor, J.; Lasarte, J.J.; Sarobe, P.; Borrás-Cuesta, F.; Díez, J. A synthetic peptide from transforming growth factor-beta1 type III receptor prevents myocardial fibrosis in spontaneously hypertensive rats. Cardiovasc. Res. 2009, 81, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Santiago, B.; Gutierrez-Cañas, I.; Dotor, J.; Palao, G.; Lasarte, J.J.; Ruiz, J.; Prieto, J.; Borrás-Cuesta, F.; Pablos, J.L. Topical application of a peptide inhibitor of transforming growth factor-beta1 ameliorates bleomycin-induced skin fibrosis. J. Investig. Dermatol. 2005, 125, 450–455. [Google Scholar] [CrossRef] [Green Version]
- Januszyk, M.; Kwon, S.H.; Wong, V.W.; Padmanabhan, J.; Maan, Z.N.; Whittam, A.J.; Major, M.R.; Gurtner, G.C. The Role of Focal Adhesion Kinase in Keratinocyte Fibrogenic Gene Expression. Int. J. Mol. Sci. 2017, 18, 1915. [Google Scholar] [CrossRef] [Green Version]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Zhao, X.; Guan, J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [Green Version]
- David, F.S.; Zage, P.E.; Marcantonio, E.E. Integrins interact with focal adhesions through multiple distinct pathways. J. Cell. Physiol. 1999, 181, 74–82. [Google Scholar] [CrossRef]
- Liu, W.; Ma, K.; Kwon, S.H.; Garg, R.; Patta, Y.R.; Fujiwara, T.; Gurtner, G.C. The Abnormal Architecture of Healed Diabetic Ulcers Is the Result of FAK Degradation by Calpain 1. J. Investig. Dermatol. 2017, 137, 1155–1165. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.; Garg, R.K.; Sorkin, M.; Rustad, K.C.; Akaishi, S.; Levi, K.; Nelson, E.R.; Tran, M.; Rennert, R.; Liu, W.; et al. Loss of keratinocyte focal adhesion kinase stimulates dermal proteolysis through upregulation of MMP9 in wound healing. Ann. Surg. 2014, 260, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Li, X.; Wu, X.; Hui, B.; Han, S.; Gao, J.; Li, Y.; Shi, J.; Zhu, H.; Zhao, B.; et al. Simultaneous deactivation of FAK and Src improves the pathology of hypertrophic scar. Sci. Rep. 2016, 6, 26023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, V.W.; Sorkin, M.; Glotzbach, J.P.; Longaker, M.T.; Gurtner, G.C. Surgical approaches to create murine models of human wound healing. J. Biomed. Biotechnol. 2011, 2011, 969618. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Kwon, S.H.; Padmanabhan, J.; Duscher, D.; Trotsyuk, A.A.; Dong, Y.; Inayathullah, M.; Rajadas, J.; Gurtner, G.C. Controlled Delivery of a Focal Adhesion Kinase Inhibitor Results in Accelerated Wound Closure with Decreased Scar Formation. J. Investig. Dermatol. 2018, 138, 2452–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widelitz, R.B. Wnt signaling in skin organogenesis. Organogels 2008, 4, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, X.; Nusse, R. Wnt signaling in skin development, homeostasis, and disease. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Yonemura, S. A mechanism of mechanotransduction at the cell-cell interface: Emergence of alpha-catenin as the center of a force-balancing mechanism for morphogenesis in multicellular organisms. BioEssays 2011, 33, 732–736. [Google Scholar] [CrossRef]
- Leckband, D.E.; de Rooij, J. Cadherin adhesion and mechanotransduction. Annu. Rev. Cell Dev. Biol. 2014, 30, 291–315. [Google Scholar] [CrossRef]
- Wu, M.; Fannin, J.; Rice, K.M.; Wang, B.; Blough, E.R. Effect of aging on cellular mechanotransduction. Ageing Res. Rev. 2011, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Cheon, S.S.; Cheah, A.Y.L.; Turley, S.; Nadesan, P.; Poon, R.; Clevers, H.; Alman, B.A. beta-Catenin stabilization dysregulates mesenchymal cell proliferation, motility, and invasiveness and causes aggressive fibromatosis and hyperplastic cutaneous wounds. Proc. Natl. Acad. Sci. USA 2002, 99, 6973–6978. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Melichian, D.; Komura, K.; Hinchcliff, M.E.; Lam, A.P.; Lafyatis, R.; Gottardi, C.; MacDougald, O.A.; Varga, J. Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: A novel mouse model for scleroderma? Arthritis Rheum. 2011, 63, 1707–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaire, R.; Farina, G.; Bayle, J.; Dimarzio, M.; Pendergrass, S.A.; Milano, A.; Whitfield, M.L.; Lafyatis, R. Antagonistic effect of the matricellular signaling protein CCN3 on TGF-beta- and Wnt-mediated fibrillinogenesis in systemic sclerosis and Marfan syndrome. J. Investig. Dermatol. 2010, 130, 1514–1523. [Google Scholar] [CrossRef] [Green Version]
- Bastakoty, D.; Young, P.P. Wnt/beta-catenin pathway in tissue injury: Roles in pathology and therapeutic opportunities for regeneration. FASEB J. 2016, 30, 3271–3284. [Google Scholar] [CrossRef]
- Amini-Nik, S.; Cambridge, E.; Yu, W.; Guo, A.; Whetstone, H.; Nadesan, P.; Poon, R.; Hinz, B.; Alman, B.A. beta-Catenin-regulated myeloid cell adhesion and migration determine wound healing. J. Clin. Investig. 2014, 124, 2599–2610. [Google Scholar] [CrossRef] [Green Version]
- Cheon, S.; Poon, R.; Yu, C.; Khoury, M.; Shenker, R.; Fish, J.; Alman, B.A. Prolonged beta-catenin stabilization and tcf-dependent transcriptional activation in hyperplastic cutaneous wounds. Lab Investig. 2005, 85, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Foote, H.P.; Lechler, T. beta-Catenin protects the epidermis from mechanical stresses. J.Cell Biol. 2013, 202, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.C.; Ng, A.; Kim, B.H.; Bianco, A.; Xavier, R.J.; Elledge, S.J. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev. 2010, 24, 1507–1518. [Google Scholar] [CrossRef] [Green Version]
- Sato, M. Upregulation of the Wnt/beta-catenin pathway induced by transforming growth factor-beta in hypertrophic scars and keloids. Acta Derm.-Venereol. 2006, 86, 300–307. [Google Scholar] [CrossRef] [Green Version]
- Cheon, S.S.; Wei, Q.; Gurung, A.; Youn, A.; Bright, T.; Poon, R.; Whetstone, H.; Guha, A.; Alman, B.A. Beta-catenin regulates wound size and mediates the effect of TGF-beta in cutaneous healing. FASEB J. 2006, 20, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Elbediwy, A.; Vincent-Mistiaen, Z.I.; Spencer-Dene, B.; Stone, R.K.; Boeing, S.; Wculek, S.K.; Cordero, J.; Tan, E.H.; Ridgway, R.; Brunton, V.G.; et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 2016, 143, 1674–1687. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, H.; Yamamoto, K.; Agarwala, S.; Terai, K.; Fukui, H.; Fukuhara, S.; Ando, K.; Miyazaki, T.; Yokota, Y.; Schmelzer, E.; et al. Flow-Dependent Endothelial YAP Regulation Contributes to Vessel Maintenance. Dev. Cell. 2017, 40, 523–536.e6. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Byun, M.R.; Furutani-Seiki, M.; Hong, J.H.; Jung, H.S. YAP and TAZ regulate skin wound healing. J. Investig. Dermatol. 2014, 134, 518–525. [Google Scholar] [CrossRef] [Green Version]
- Beverdam, A.; Claxton, C.; Zhang, X.; James, G.; Harvey, K.F.; Key, B. Yap controls stem/progenitor cell proliferation in the mouse postnatal epidermis. J. Investig. Dermatol. 2013, 133, 1497–1505. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pasolli, H.A.; Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. USA 2011, 108, 2270–2275. [Google Scholar] [CrossRef] [Green Version]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Li, B.S.; Guo, W.J.; Hong, L.; Liu, Y.D.; Liu, C.; Hong, S.S.; Wu, D.B.; Min, J. Role of mechanical strain-activated PI3K/Akt signaling pathway in pelvic organ prolapse. Mol. Med. Rep. 2016, 14, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Li, Y.Y.; Sun, J.E.; Lin, W.H.; Zhou, R.X. ILK-PI3K/AKT pathway participates in cutaneous wound contraction by regulating fibroblast migration and differentiation to myofibroblast. Lab. Investig. 2016, 96, 741–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calautti, E.; Li, J.; Saoncella, S.; Brissette, J.L.; Goetinck, P.F. Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J. Biol. Chem. 2005, 280, 32856–32865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, S.; Komine, M.; Fujimoto, M.; Okochi, H.; Tamaki, K. Activation of Akt by mechanical stretching in human epidermal keratinocytes. Exp. Dermatol. 2006, 15, 356–361. [Google Scholar] [CrossRef]
- Paterno, J.; Vial, I.N.; Wong, V.W.; Rustad, K.C.; Sorkin, M.; Shi, Y.; Bhatt, K.A.; Thangarajah, H.; Glotzbach, J.P.; Gurtner, G.C. Akt-mediated mechanotransduction in murine fibroblasts during hypertrophic scar formation. Wound Repair Regen. 2011, 19, 49–58. [Google Scholar] [CrossRef]
- Gao, Y.L.; Liu, C.S.; Zhao, R.; Wang, L.L.; Li, S.S.; Liu, M.; Zhang, M.; Jiang, S.K.; Tian, Z.L.; Wang, M.; et al. Effects of PI3K/Akt Pathway in Wound Healing Process of Mice Skin. Fa Yi Xue Za Zhi 2016, 32, 7–12. [Google Scholar]
- Rahimi, R.A.; Andrianifahanana, M.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Blenis, J.; Leof, E.B. Distinct roles for mammalian target of rapamycin complexes in the fibroblast response to transforming growth factor-beta. Cancer Res. 2009, 69, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Bujor, A.M.; Pannu, J.; Bu, S.; Smith, E.A.; Muise-Helmericks, R.C.; Trojanowska, M. Akt blockade downregulates collagen and upregulates MMP1 in human dermal fibroblasts. J. Investig. Dermatol. 2008, 128, 1906–1914. [Google Scholar] [CrossRef] [Green Version]
- Lessey, E.C.; Guilluy, C.; Burridge, K. From mechanical force to RhoA activation. Biochemistry 2012, 51, 7420–7432. [Google Scholar] [CrossRef]
- Zhao, X.H.; Laschinger, C.; Arora, P.; Szászi, K.; Kapus, A.; McCulloch, C.A. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J. Cell Sci. 2007, 120, 1801–1809. [Google Scholar] [CrossRef] [Green Version]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, N.; Kato, T.; Fujita, A.; Ishizaki, T.; Narumiya, S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat. Cell Biol. 1999, 1, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, K.X.; Li, Y.J.; Guo, B.Y.; Wang, M. Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, suppresses high glucose-induced proliferation and collagen synthesis in rat cardiac fibroblasts. Clin. Exp. Pharm. Physiol. 2011, 38, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.E.; Kokosis, G.; Ren, L.; Selim, M.A.; Bergeron, A.; Levinson, H. Wound contraction is attenuated by fasudil inhibition of Rho-associated kinase. Plast Reconstr. Surg. 2011, 128, 438–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huveneers, S.; Danen, E.H. Adhesion signaling—Crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Scheraga, R.G.; Olman, M.A. A Focus on “Eye on” Channels in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 132–133. [Google Scholar] [CrossRef]
- Rahaman, S.; Grove, L.; Paruchuri, S.; Southern, B.; Abraham, S.; Niese, K.; Scheraga, R.; Ghosh, S.; Thodeti, C.; Zhang, D.; et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J. Clin. Investig. 2014, 124, 5225–5238. [Google Scholar] [CrossRef]
- Frank, S.; Stallmeyer, B.; Kämpfer, H.; Kolb, N.; Pfeilschifter, J. Nitric oxide triggers enhanced induction of vascular endothelial growth factor expression in cultured keratinocytes (HaCaT) and during cutaneous wound repair. FASEB J. 1999, 13, 2002–2014. [Google Scholar] [CrossRef]
- Mukherjee, S.; Ayaub, E.A.; Murphy, J.; Lu, C.; Kolb, M.; Ask, K.; Janssen, L.J. Disruption of Calcium Signaling in Fibroblasts and Attenuation of Bleomycin-Induced Fibrosis by Nifedipine. Am. J. Respir. Cell Mol. Biol. 2015, 53, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Bagheri, M.; Jahromi, B.M.; Mirkhani, H.; Solhjou, Z.; Noorafshan, A.; Zamani, A.; Amirghofran, Z. Azelnidipine, a new calcium channel blocker, promotes skin wound healing in diabetic rats. J. Surg. Res. 2011, 169, 101–107. [Google Scholar] [CrossRef]
- Bhaskar, H.N.; Udupa, S.L.; Udupa, A.L. Effect of nifedipine and amlodipine on wound healing in rats. Indian J. Physiol. Pharm. 2004, 48, 111–114. [Google Scholar]
- Ashkani-Esfahani, S.; Hosseinabadi, O.K.; Moezzi, P.; Moafpourian, Y.; Kardeh, S.; Rafiee, S.; Fatheazam, R.; Noorafshan, A.; Nadimi, E.; Mehrvarz, S.; et al. Verapamil, a Calcium-Channel Blocker, Improves the Wound Healing Process in Rats with Excisional Full-Thickness Skin Wounds Based on Stereological Parameters. Adv. Skin Wound Care 2016, 29, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, A.A.; Mojiri Forushani, H.; Mohammad Asgari, H. Wound healing potential of topical amlodipine in full thickness wound of rabbit. Jundishapur J. Nat. Pharm. Prod. 2014, 9, e15638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, V.W.; Akaishi, S.; Longaker, M.T.; Gurtner, G.C. Pushing back: Wound mechanotransduction in repair and regeneration. J. Investig. Dermatol. 2011, 131, 2186–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haak, A.J.; Ducharme, M.T.; Diaz Espinosa, A.M.; Tschumperlin, D.J. Targeting GPCR Signaling for Idiopathic Pulmonary Fibrosis Therapies. Trends Pharm. Sci. 2020, 41, 172–182. [Google Scholar] [CrossRef]
- D’Souza, K.M.; Malhotra, R.; Philip, J.L.; Staron, M.L.; Theccanat, T.; Jeevanandam, V.; Akhter, S.A. G protein-coupled receptor kinase-2 is a novel regulator of collagen synthesis in adult human cardiac fibroblasts. J. Biol. Chem. 2011, 286, 15507–15516. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.J.; Broome, R.E.; Kent, T.C.; Charlton, S.J.; Rosethorne, E.M. The inhibition of human lung fibroblast proliferation and differentiation by Gs-coupled receptors is not predicted by the magnitude of cAMP response. Respir. Res. 2018, 19, 56. [Google Scholar] [CrossRef]
- Dooling, L.J.; Discher, D.E. Inhibiting Tumor Fibrosis and Actomyosin through GPCR activation. Trends Cancer 2019, 5, 197–199. [Google Scholar] [CrossRef]
- Bayat, A.; Bock, O.; Mrowietz, U.; Ollier, W.E.; Ferguson, M.W. Genetic susceptibility to keloid disease and hypertrophic scarring: Transforming growth factor beta1 common polymorphisms and plasma levels. Plast Reconstr. Surg. 2003, 111, 535–543. [Google Scholar] [CrossRef]
- Chike-Obi, C.J.; Cole, P.D.; Brissett, A.E. Keloids: Pathogenesis, clinical features, and management. Semin. Plast Surg. 2009, 23, 178–184. [Google Scholar] [CrossRef] [Green Version]
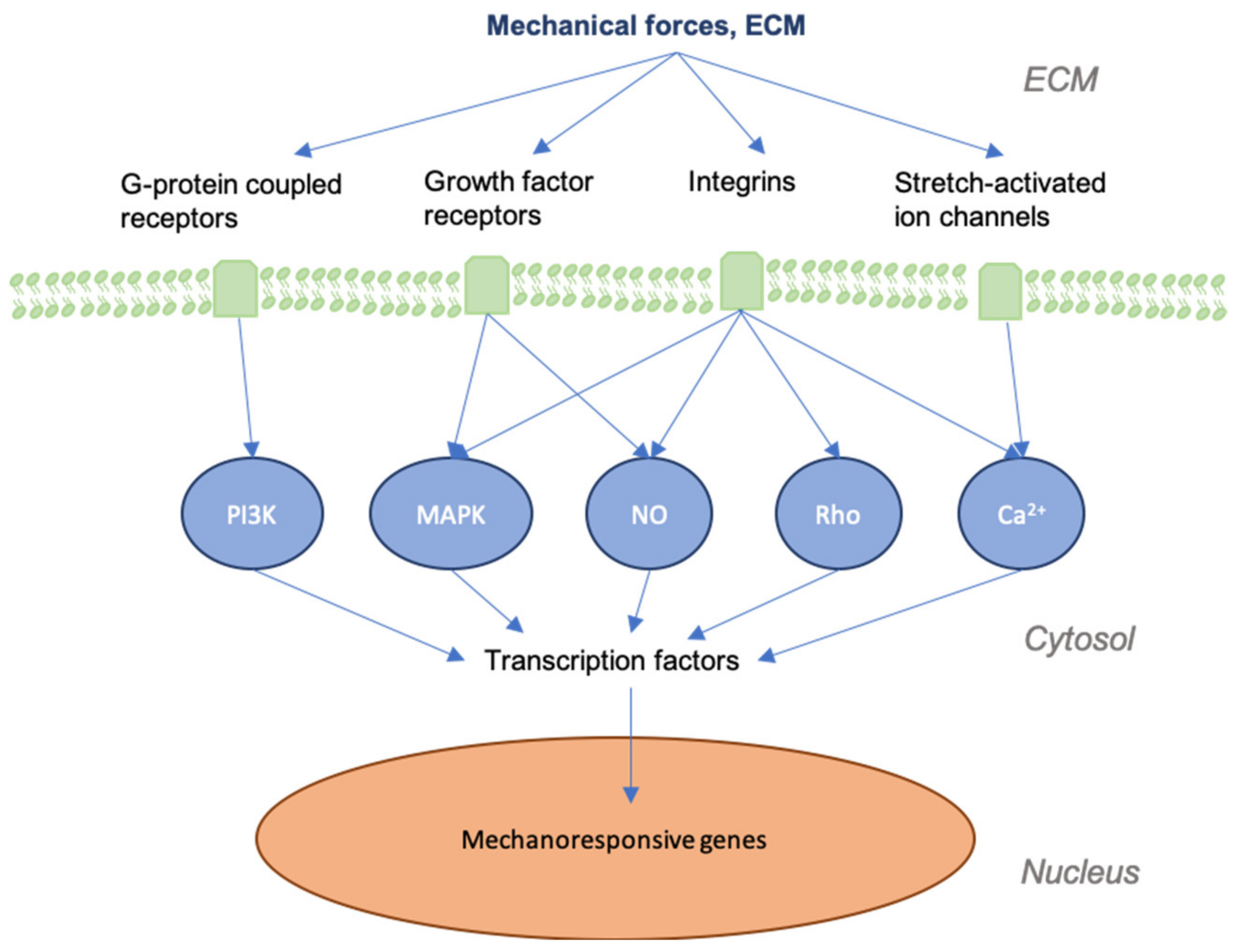
Figure 1.
Graphic depicting mechanical forces applied to skin and the associated effects at cellular levels. As skin is stretched, the dermal matrix expands, opening ion channels, receptors, and other mechanotransductors, altering their accessibility to their respective ligands. This alters many of the signaling cascades that are involved in regeneration and the final state of the repaired wound.
Figure 1.
Graphic depicting mechanical forces applied to skin and the associated effects at cellular levels. As skin is stretched, the dermal matrix expands, opening ion channels, receptors, and other mechanotransductors, altering their accessibility to their respective ligands. This alters many of the signaling cascades that are involved in regeneration and the final state of the repaired wound.

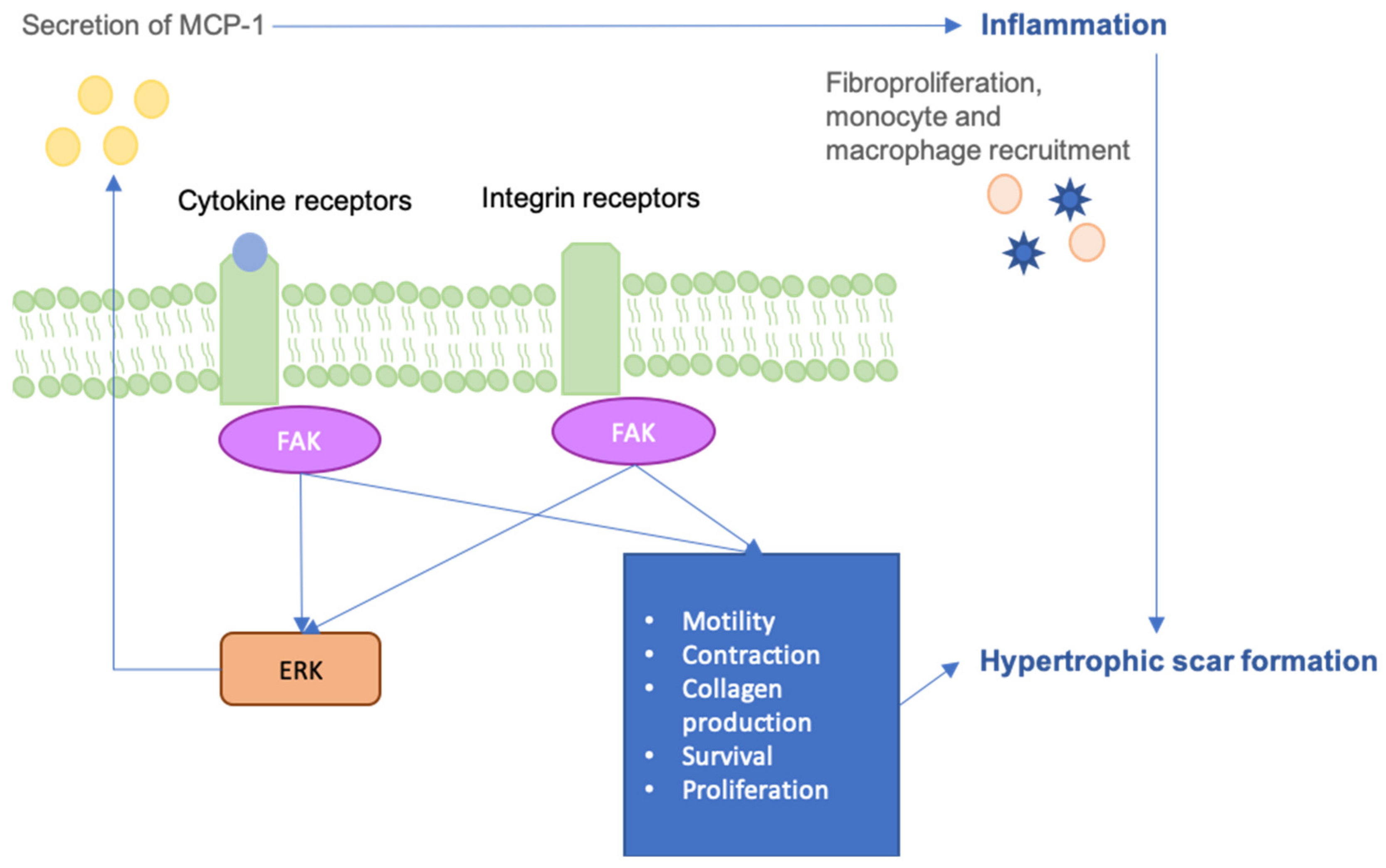
Figure 2.
The Focal Adhesion Kinase (FAK) is a critical upstream mediator of these scar-forming processes, linking mechanical stress to inflammatory pathways.
Figure 2.
The Focal Adhesion Kinase (FAK) is a critical upstream mediator of these scar-forming processes, linking mechanical stress to inflammatory pathways.

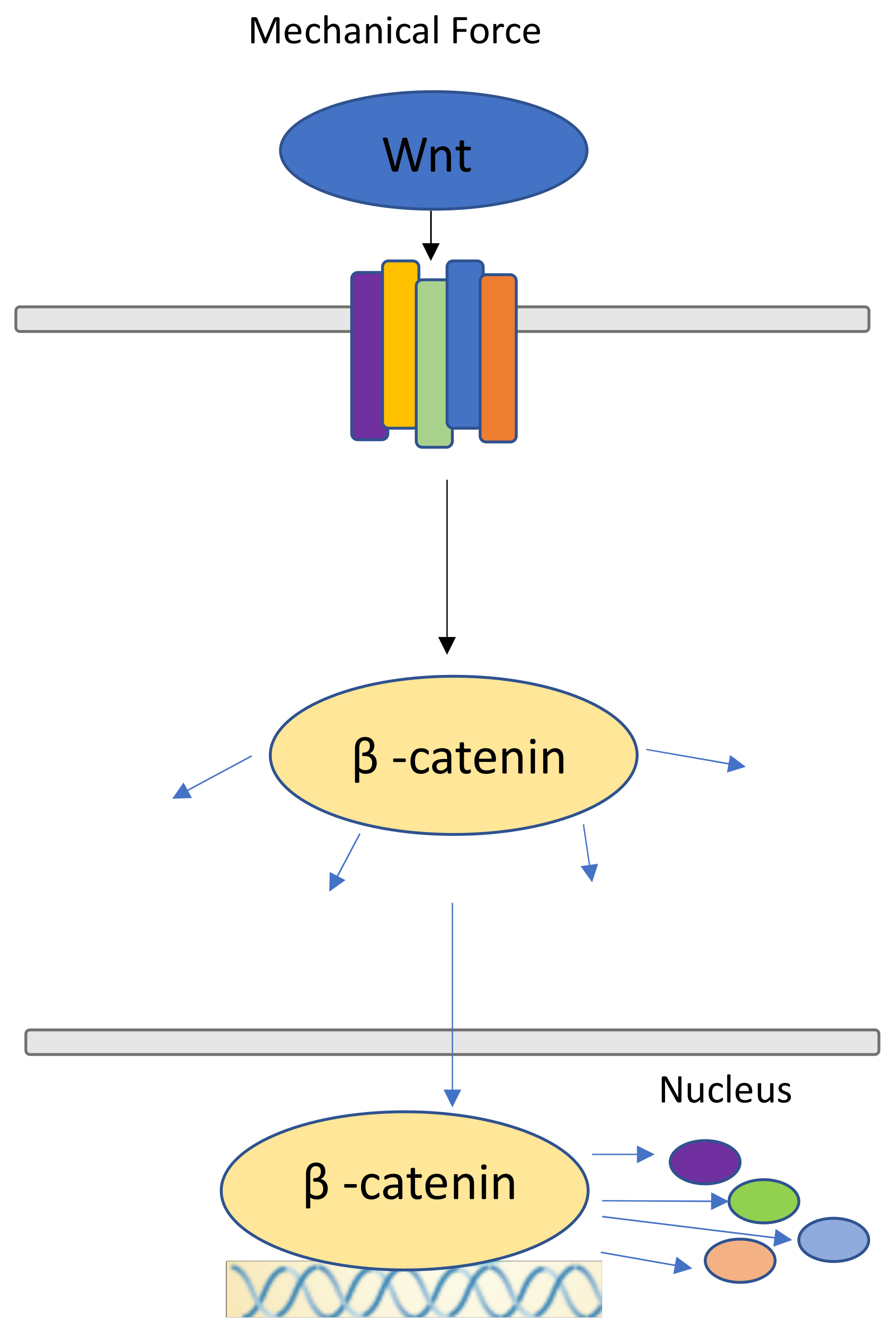
Figure 3.
Wnt/ β-catenin signaling pathway. Wnt molecules attached to the respective frizzled-receptors made more prone by mechanical force, releasing β-catenin molecules into the cytoplasm, where they are free to participate in other signaling pathways and can travel to the nucleus. Here, they serve as upstream regulators for other molecules.
Figure 3.
Wnt/ β-catenin signaling pathway. Wnt molecules attached to the respective frizzled-receptors made more prone by mechanical force, releasing β-catenin molecules into the cytoplasm, where they are free to participate in other signaling pathways and can travel to the nucleus. Here, they serve as upstream regulators for other molecules.

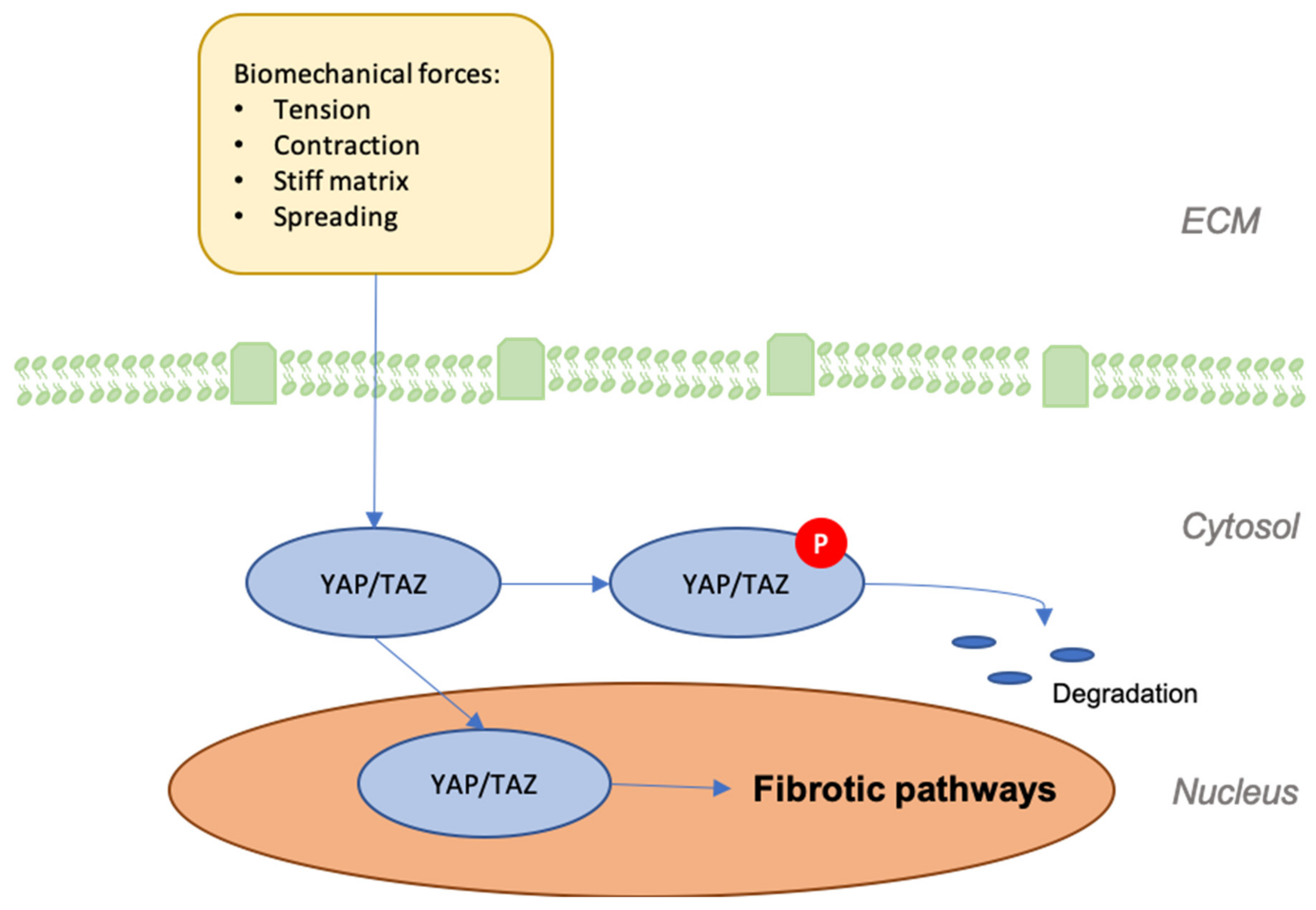
Figure 4.
Graphic depicting the effects of mechanical forces on the expression of Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) and their effects on the development of fibrosis.
Figure 4.
Graphic depicting the effects of mechanical forces on the expression of Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) and their effects on the development of fibrosis.

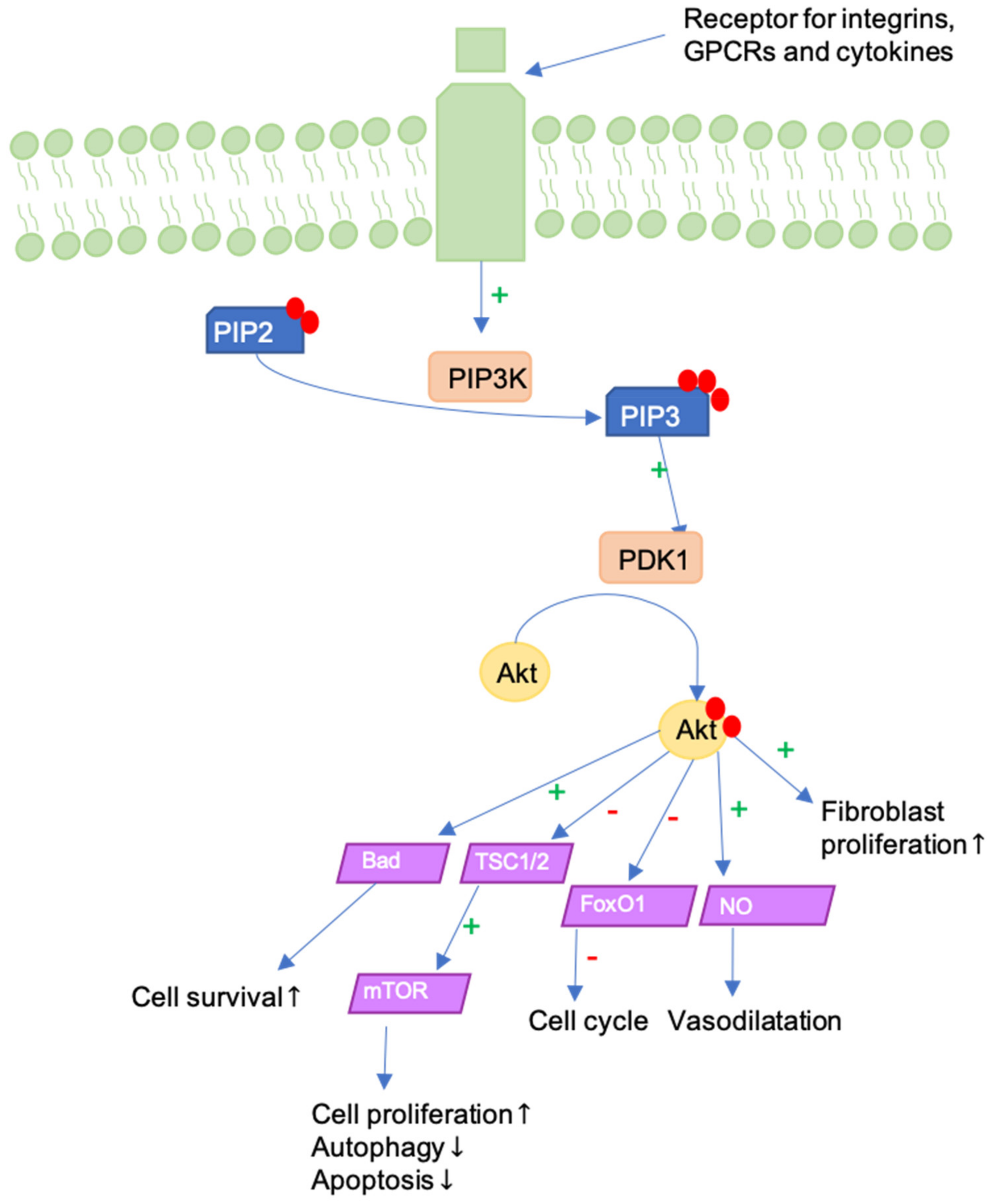
Figure 5.
The PI3K/Akt pathway can be activated in different ways, as for example by G-protein coupled receptors (GPCRs), integrin or cytokine receptors and it represents a mediator between ECM and fibroblasts’ cell cycle. In this figure, PI3K-dependent phosphorylation and activation of Akt is represented, which in turn leads to enhanced proliferation and cell survival. Furthermore, apoptosis is regulated, and vasodilatation is promoted.
Figure 5.
The PI3K/Akt pathway can be activated in different ways, as for example by G-protein coupled receptors (GPCRs), integrin or cytokine receptors and it represents a mediator between ECM and fibroblasts’ cell cycle. In this figure, PI3K-dependent phosphorylation and activation of Akt is represented, which in turn leads to enhanced proliferation and cell survival. Furthermore, apoptosis is regulated, and vasodilatation is promoted.

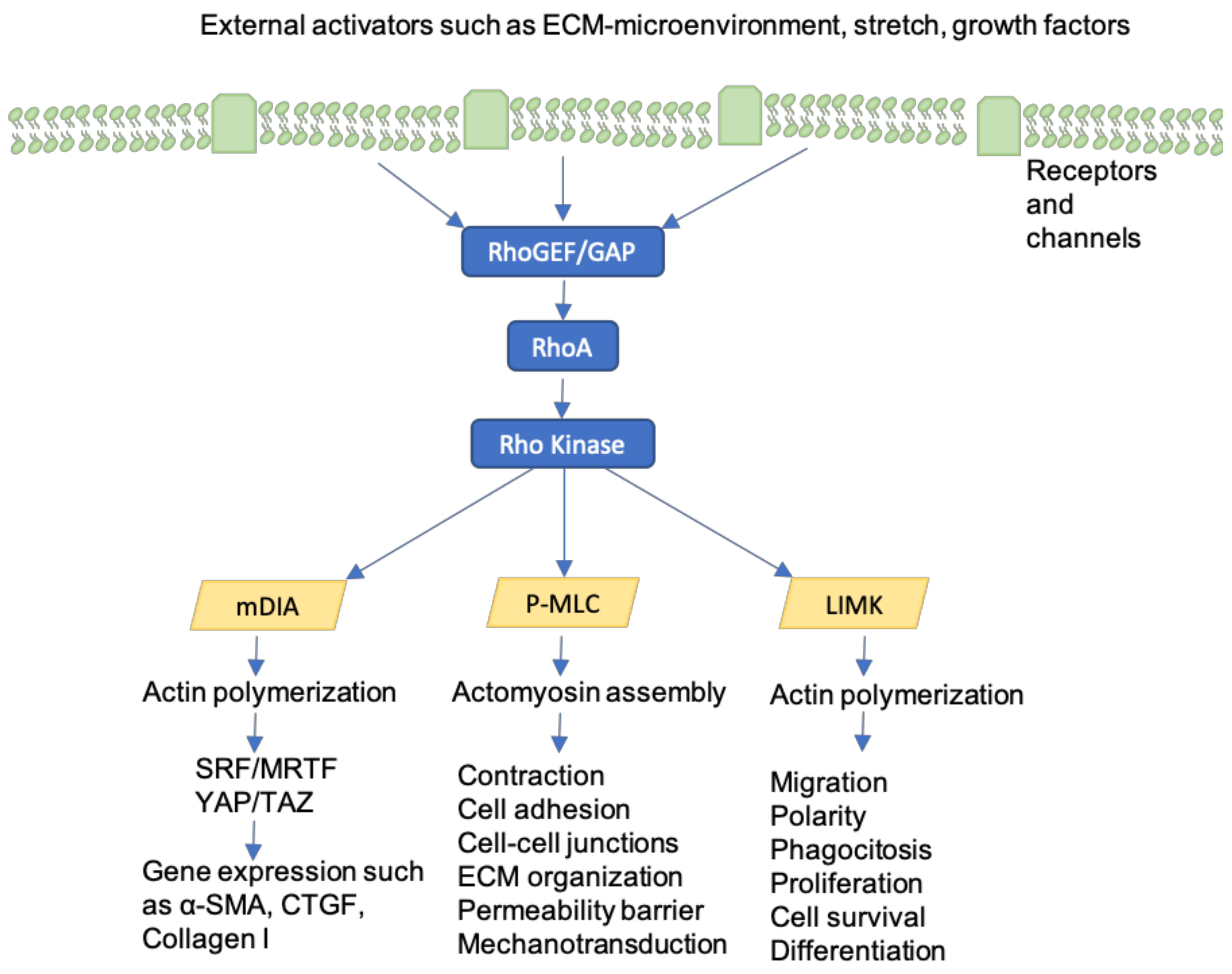
Figure 6.
Activation of Rho-GTPases and its’ implications are represented. External factors such as tension on integrins in focal adhesions, cell stretch, growth factors and changes in the ECM environment can lead to Rho-GTPase activation. Through different pathways, cell contraction, motility and consequently wound contracture are promoted. In fact, on the one hand, actin polymerization, α-SMA and collagen I gene expression are enhanced. On the other hand, cell adhesion, cell contraction, ECM organization, migration and polarity are regulated. Finally, cell proliferation, survival and differentiation are coordinated as well.
Figure 6.
Activation of Rho-GTPases and its’ implications are represented. External factors such as tension on integrins in focal adhesions, cell stretch, growth factors and changes in the ECM environment can lead to Rho-GTPase activation. Through different pathways, cell contraction, motility and consequently wound contracture are promoted. In fact, on the one hand, actin polymerization, α-SMA and collagen I gene expression are enhanced. On the other hand, cell adhesion, cell contraction, ECM organization, migration and polarity are regulated. Finally, cell proliferation, survival and differentiation are coordinated as well.

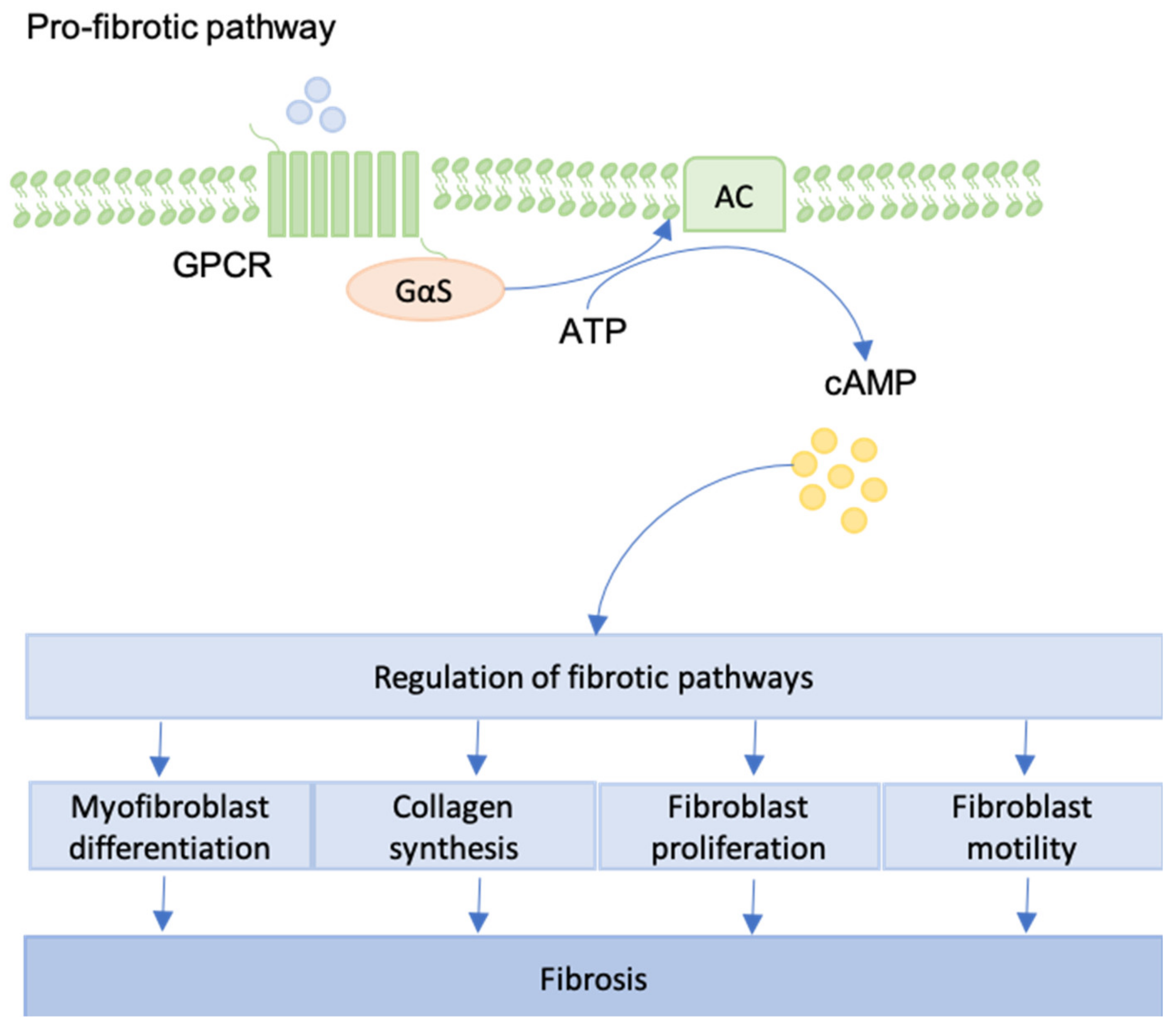
Figure 7.
An overview of the pro-fibrotic pathway generated by GPCR-activation is provided. For example, focal adhesion complexes can lead to pro-fibrotic GPCR-activation. Via cAMP generation, fibroblast differentiation, proliferation and motility are regulated. Collagen synthesis can be enhanced as well leading to genesis and progression of fibrosis.
Figure 7.
An overview of the pro-fibrotic pathway generated by GPCR-activation is provided. For example, focal adhesion complexes can lead to pro-fibrotic GPCR-activation. Via cAMP generation, fibroblast differentiation, proliferation and motility are regulated. Collagen synthesis can be enhanced as well leading to genesis and progression of fibrosis.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kuehlmann, B.; Bonham, C.A.; Zucal, I.; Prantl, L.; Gurtner, G.C. Mechanotransduction in Wound Healing and Fibrosis. J. Clin. Med. 2020, 9, 1423. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9051423
AMA Style
Kuehlmann B, Bonham CA, Zucal I, Prantl L, Gurtner GC. Mechanotransduction in Wound Healing and Fibrosis. Journal of Clinical Medicine. 2020; 9(5):1423. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9051423
Chicago/Turabian StyleKuehlmann, Britta, Clark A. Bonham, Isabel Zucal, Lukas Prantl, and Geoffrey C. Gurtner. 2020. "Mechanotransduction in Wound Healing and Fibrosis" Journal of Clinical Medicine 9, no. 5: 1423. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9051423
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.