Transplantation Induces Profound Changes in the Transcriptional Asset of Hematopoietic Stem Cells: Identification of Specific Signatures Using Machine Learning Techniques

 ,
,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Cells Sources
2.2. Induced Pluripotent Stem Cell (iPS) Line Maintenance
2.3. CD34+ Cell Enrichment
2.4. Real-Time Quantitative (RQ)-PCR Analysis by Low-Density Array/Microfluidic Card
2.5. Immunofluorescence Staining
2.6. Sparse Multivariate Analysis
2.7. Performance Metrics
2.8. Network Inference and Identification of Gene Function
3. Results
3.1. Gene Expression Analysis
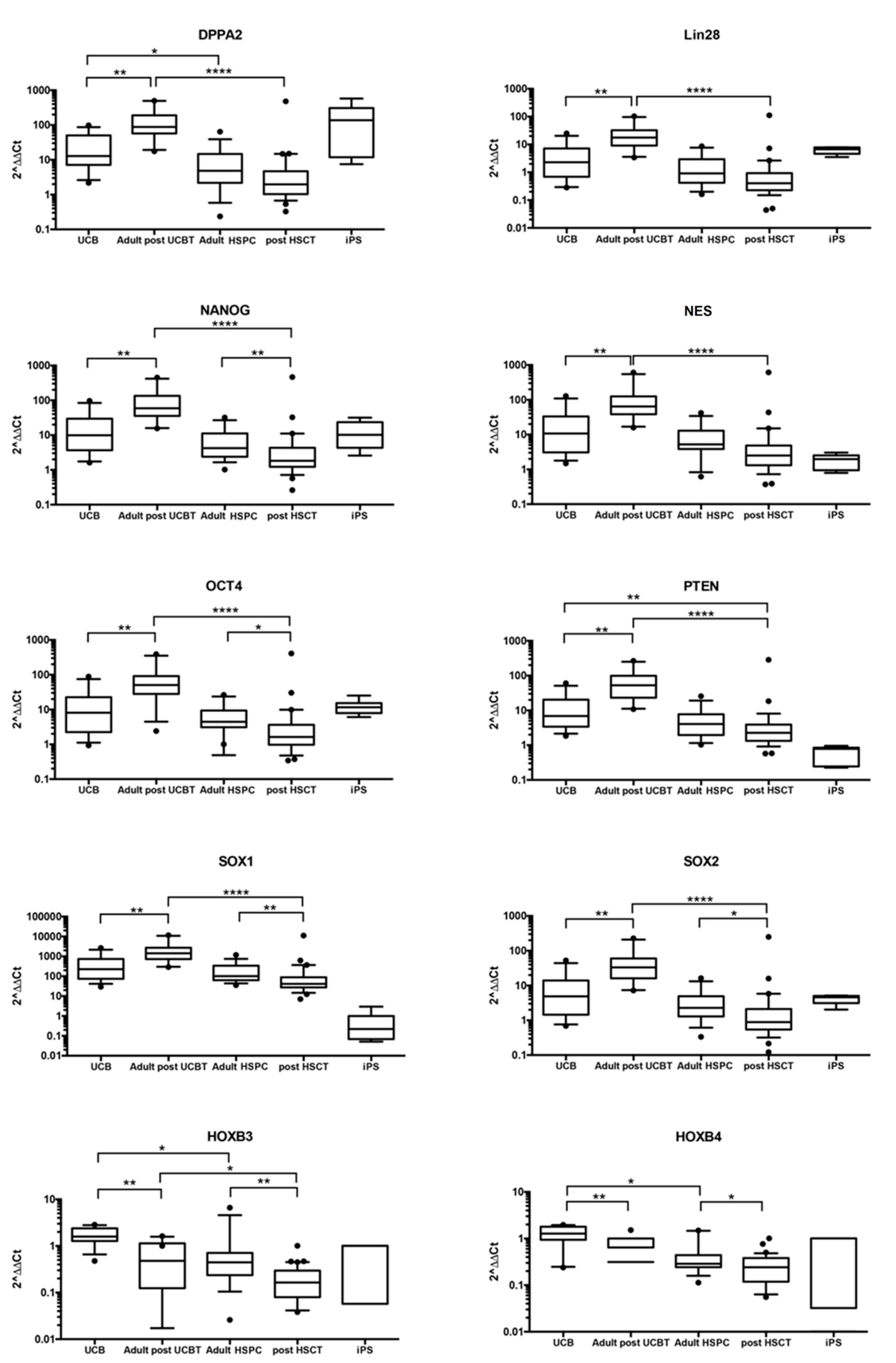
3.1.1. CD34+ Cells after UCBT Express Genes of Self-Renewal, Stem Cell Maintenance, and Reprogramming
3.1.2. Self-Renewal, Stem Cell Maintenance, and Reprogramming Genes Are not Differentially Expressed in CD34+ Cells from UCB vs. Adult CD34+ Cells
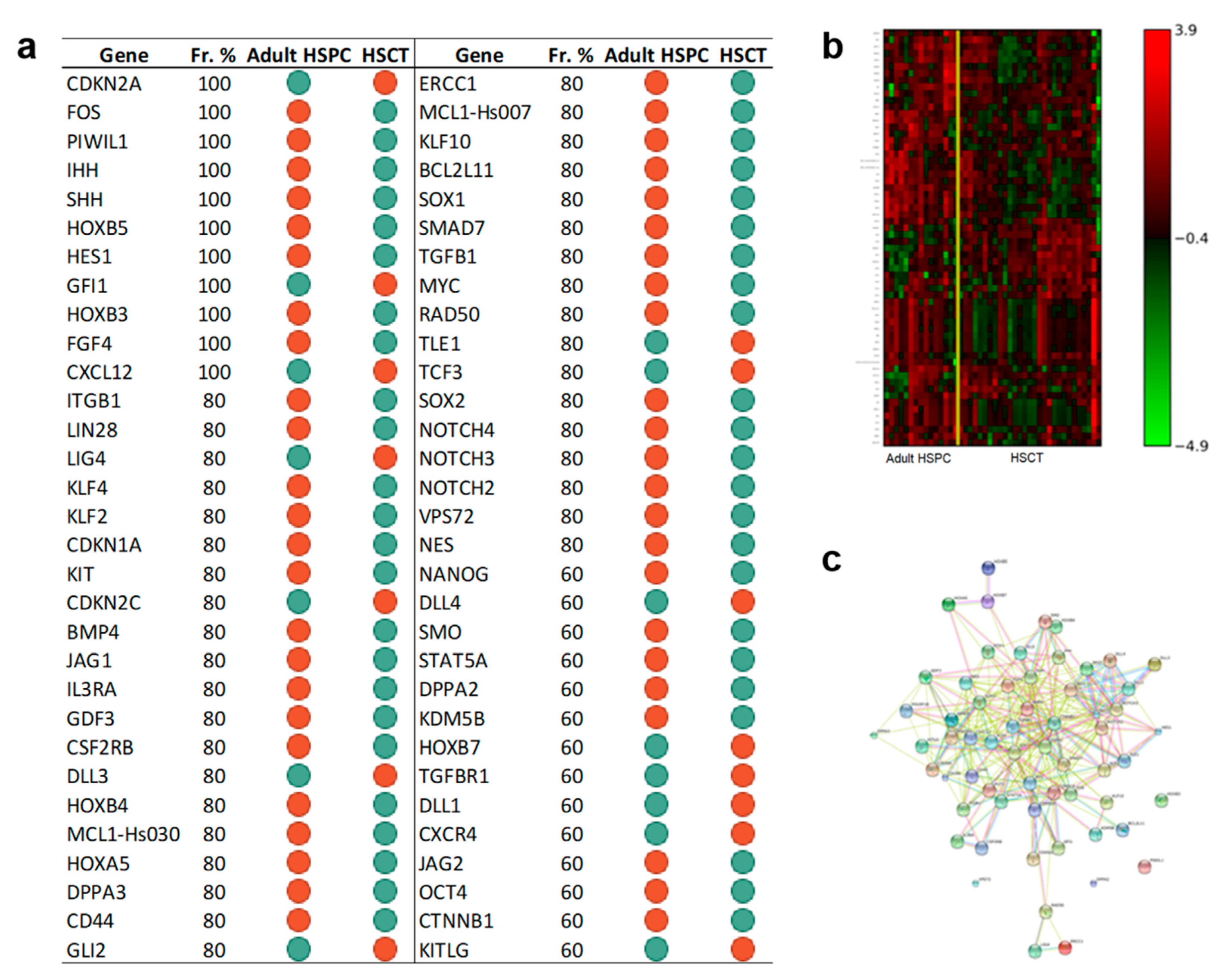
3.1.3. Genes Regulating Self-Renewal, Cell Reprogramming, and Stem Cell Maintenance Are Overexpressed in CD34+ Cells after UCBT but not after Adult HSCT
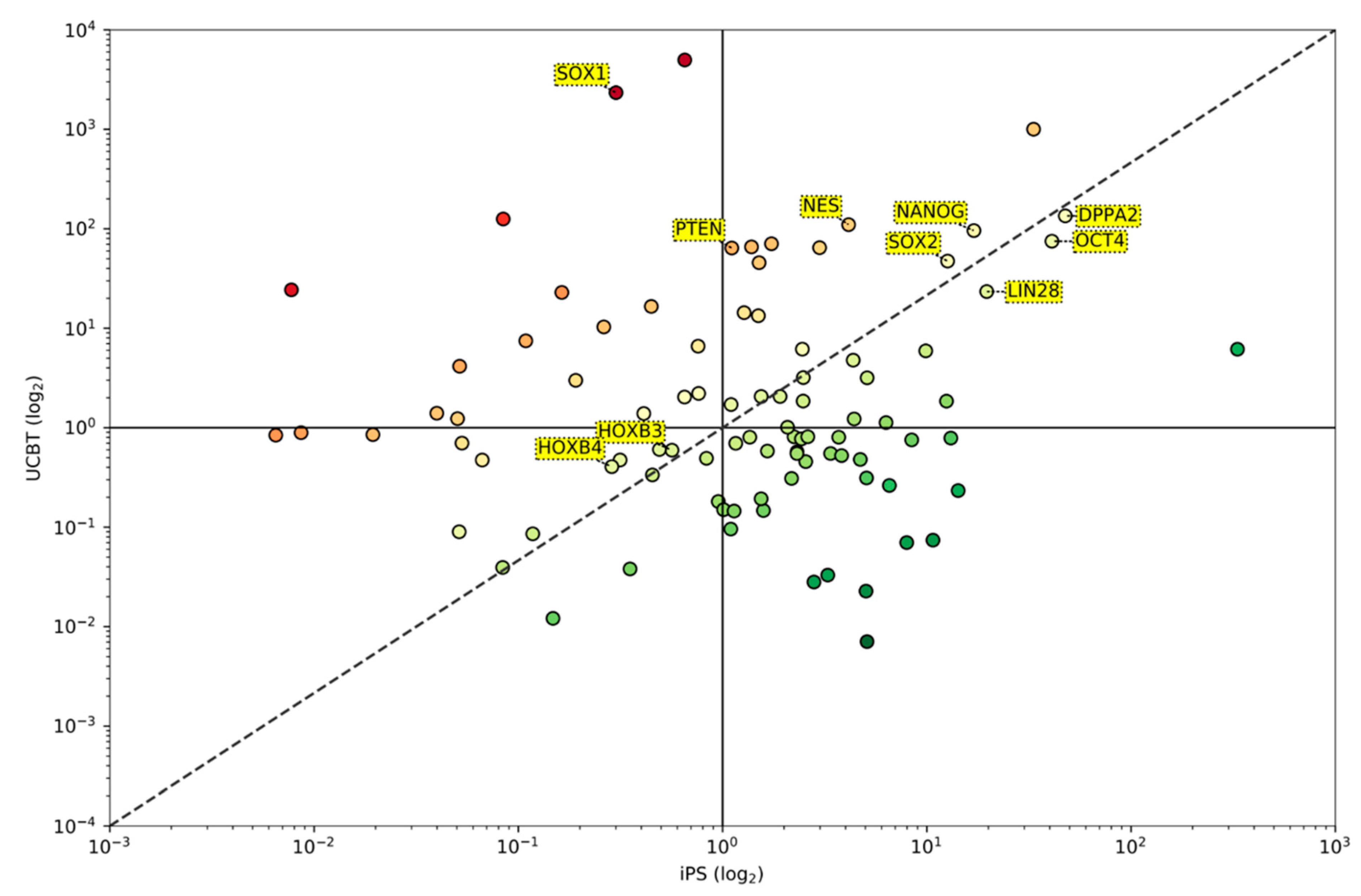
3.1.4. Some Reprogramming Genes Are Similarly Expressed in CD34+ Cells after UCBT and iPS Although Their Overall Picture of Gene Expression Is Divergent
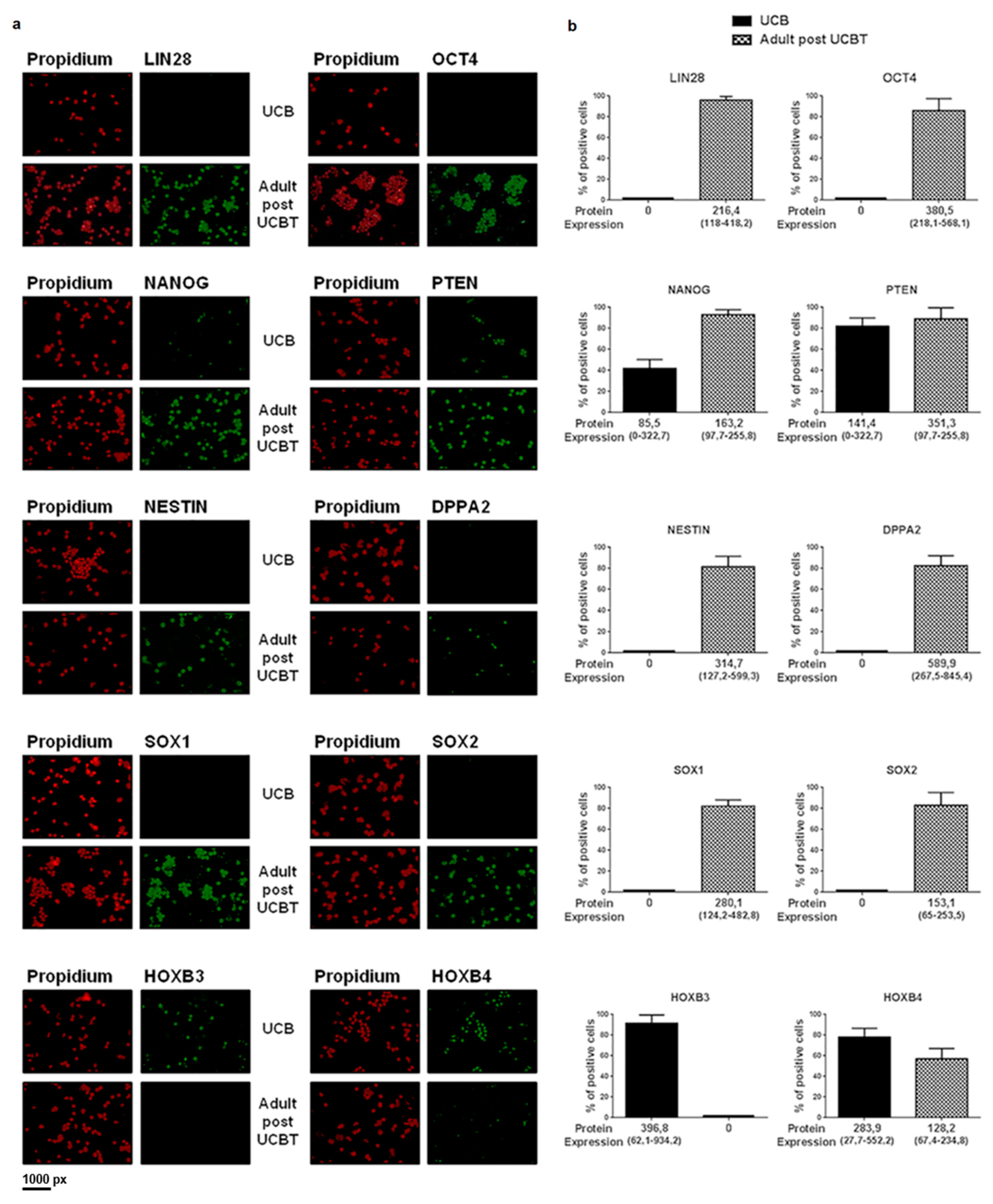
3.2. The Transcription of the Overexpressed Genes Is Confirmed by Protein Analysis
3.3. Sparse Multivariate Regularization Analysis
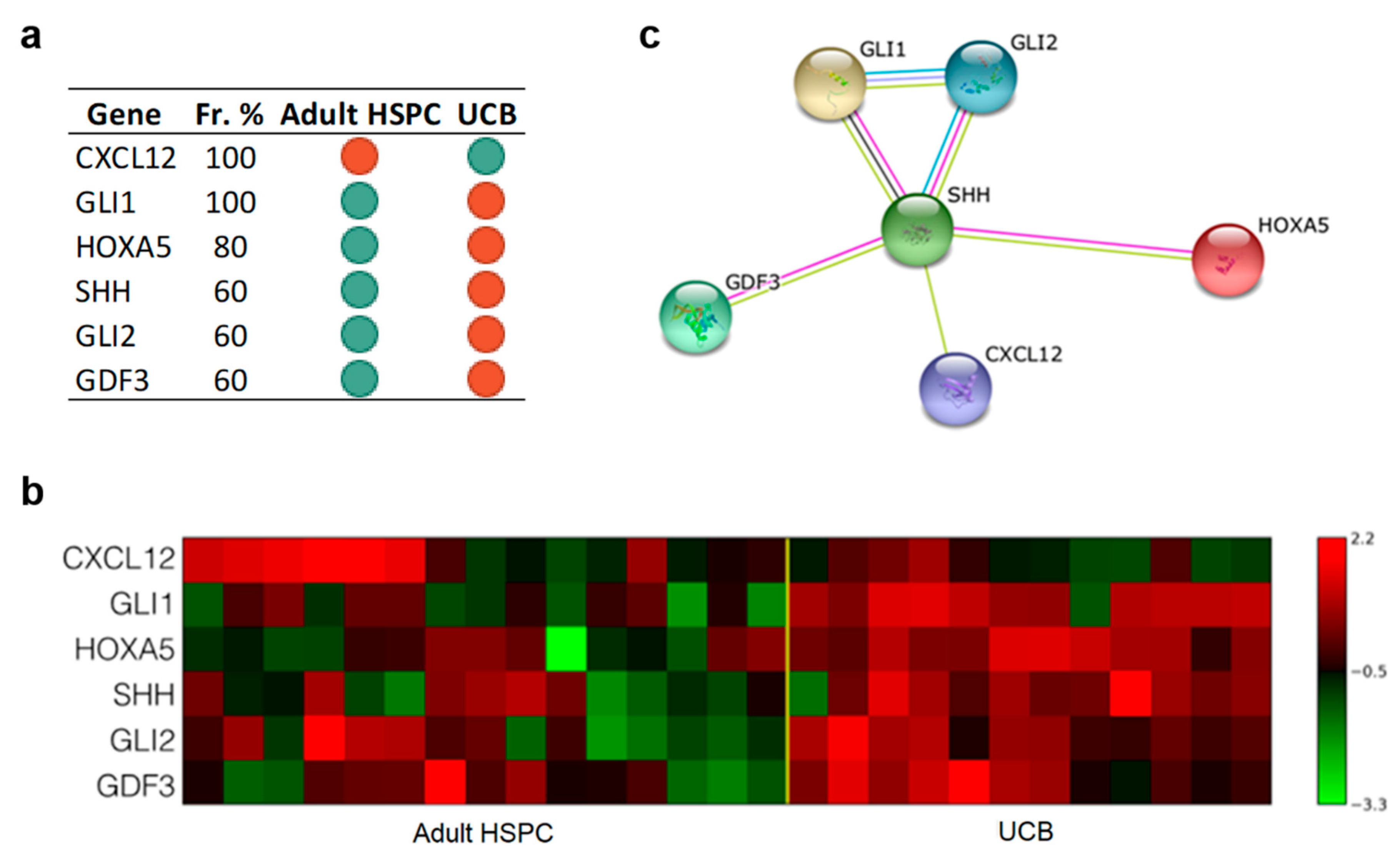
3.3.1. Only Six Genes Are Sufficient to Discriminate UCB from Adult
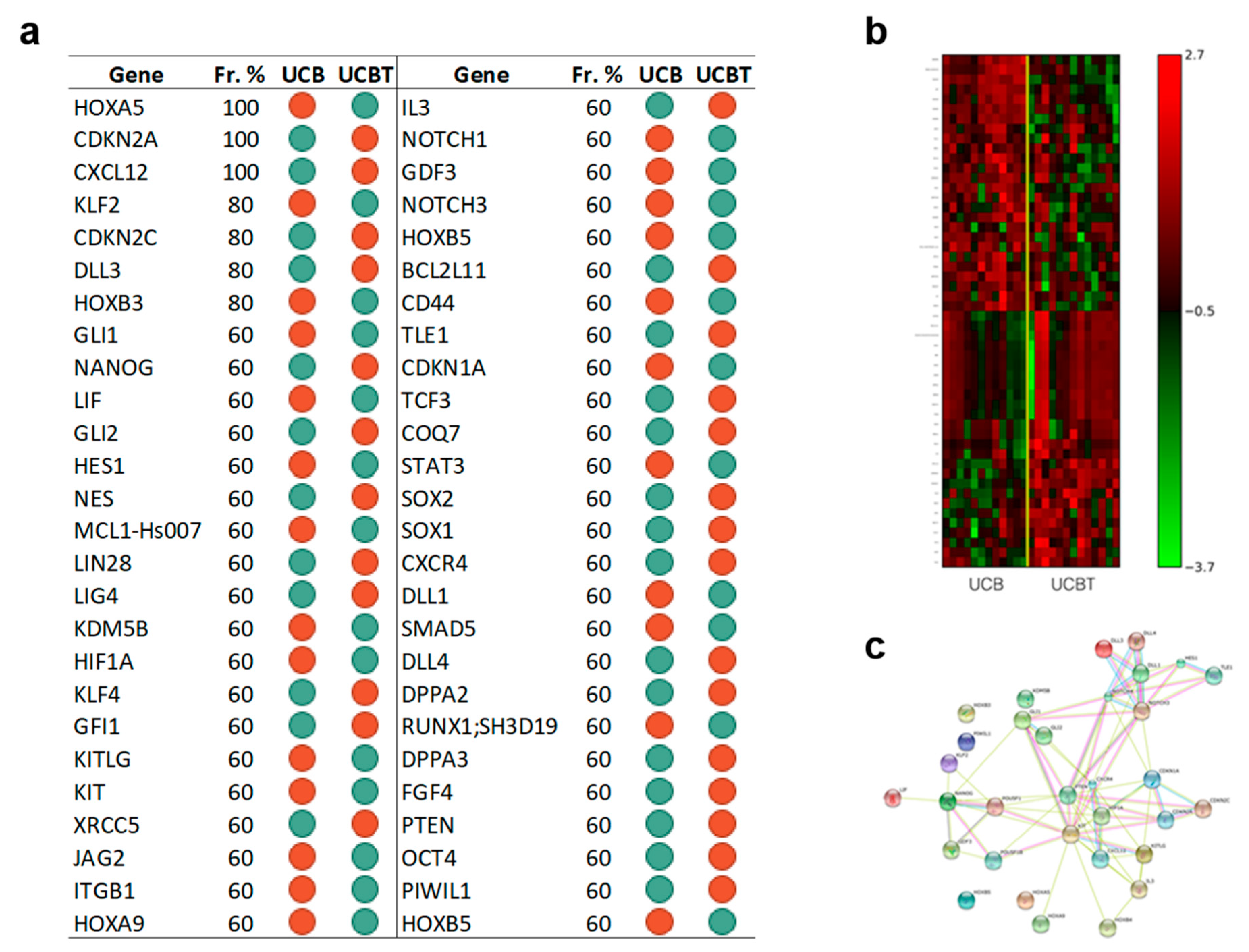
3.3.2. The Set of the Expression Differences of Fifty-Two Genes Discriminates Cord Blood CD34+ Cells before and after Transplantation
3.3.3. The Set of the Expression Differences of Sixty-Two Genes Discriminates Adult CD34+ Cells before and after Transplantation
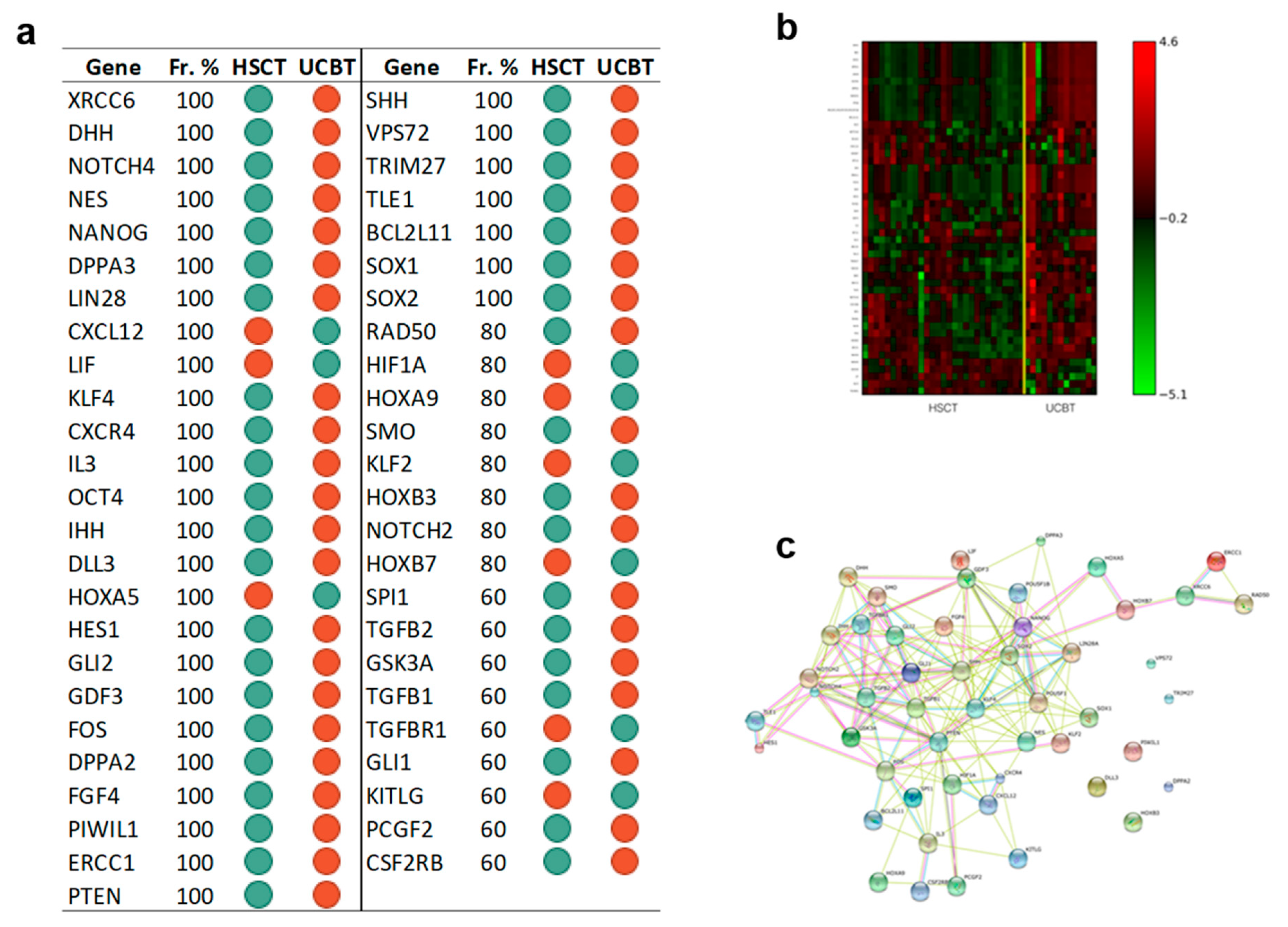
3.3.4. After Transplantation Adult Versus Cord Blood CD34+ Cells Acquire a Divergent Transcriptional Asset; Reprogramming Genes Are a Relevant Part of Such Difference
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gooley, T.A.; Chien, J.W.; Pergam, S.A.; Hingorani, S.; Sorror, M.L.; Boeckh, M.; Martin, P.J.; Sandmaier, B.M.; Marr, K.A.; Appelbaum, F.R.; et al. Reduced mortality after allogeneic hematopoietic-cell transplantation. New Engl. J. Med. 2010, 363, 2091–2101. [Google Scholar] [CrossRef] [Green Version]
- Van Hennik, P.B.; de Koning, A.E.; Ploemacher, R.E. Seeding efficiency of primitive human hematopoietic cells in nonobese diabetic/severe combined immune deficiency mice: Implications for stem cell frequency assessment. Blood 1999, 94, 3055–3061. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.E.; Astle, C.M. Loss of stem cell repopulating ability upon transplantation. Effects of donor age, cell number, and transplantation procedure. J. Exp. Med. 1982, 156, 1767–1779. [Google Scholar] [CrossRef]
- Podestà, M.; Piaggio, G.; Frassoni, F.; Pitto, A.; Zikos, P.; Sessarego, M.; Abate, M.; Van Lint, M.T.; Berisso, G.; Bacigalupo, A. The assessment of the hematopoietic reservoir after immunosuppressive therapy or bone marrow transplantation in severe aplastic anemia. Blood 1998, 91, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Iscove, N.N.; Nawa, K. Hematopoietic stem cells expand during serial transplantation in vivo without apparent exhaustion. Curr. Biol. 1997, 7, 805–808. [Google Scholar] [CrossRef] [Green Version]
- Frassoni, F.; Podesta, M.; Maccario, R.; Giorgiani, G.; Rossi, G.; Zecca, M.; Bacigalupo, A.; Piaggio, G.; Locatelli, F. Cord blood transplantation provides better reconstitution of hematopoietic reservoir compared with bone marrow transplantation. Blood 2003, 102, 1138–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frassoni, F.; Gualandi, F.; Podestà, M.; Raiola, A.M.; Ibatici, A.; Piaggio, G.; Sessarego, M.; Sessarego, N.; Gobbi, M.; Sacchi, N.; et al. Direct intrabone transplant of unrelated cord-blood cells in acute leukaemia: A phase I/II study. Lancet. Oncol. 2008, 9, 831–839. [Google Scholar] [CrossRef]
- De Mol, C.; De Vito, E.; Rosasco, L. Elastic net regularization in learning theory. J. Complex. 2009, 2, 201–230. [Google Scholar] [CrossRef] [Green Version]
- Barla, A.; Mosci, S.; Rosasco, L.; Verri, A. A method for robust variable selection with significance assessment. In Proceedings of the 16th European Symposium on Artificial Neural Networks, Bruges, Belgium, 23–25 April 2008. [Google Scholar]
- Fardin, P.; Barla, A.; Mosci, S.; Rosasco, L.; Verri, A.; Varesio, L. The l1-l2 regularization framework unmasks the hypoxia signature hidden in the transcriptome of a set of heterogeneous neuroblastoma cell lines. BMC Genom. 2009, 10, 474. [Google Scholar] [CrossRef] [Green Version]
- Fardin, P.; Cornero, A.; Barla, A.; Mosci, S.; Acquaviva, M.; Rosasco, L.; Gambini, C.; Verri, A.; Varesio, L. Identification of multiple hypoxia signatures in neuroblastoma cell lines by l1-l2 regularization and data reduction. J. Biomed. Biotechnol. 2010, 878709. [Google Scholar] [CrossRef] [Green Version]
- Squillario, M.; Barla, A. A computational procedure for functional characterization of potential marker genes from m.lecular data: Alzheimer’s as a case study. BMC Med. Genom. 2011, 4, 55. [Google Scholar] [CrossRef] [Green Version]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, A.; Montserrat, N.; Aasen, T.; Gonzalez, F.; Rodríguez-Pizà, I.; Vassena, R.; Raya, A.; Boué, S.; Barrero, M.J.; Corbella, B.A.; et al. Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell 2009, 5, 353–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef]
- Gratwohl, A.; Pasquini, M.C.; Aljurf, M.; Atsuta, Y.; Baldomero, H.; Foeken, L.; Gratwohl, M.; Bouzas, L.F.; Confer, D.; Frauendorfer, K.; et al. One million haemopoietic stem-cell transplants: A retrospective observational study. Lancet. Haematol. 2015, 2, e91–e100. [Google Scholar] [CrossRef]
- Hochedlinger, K.; Jaenisch, R. Induced Pluripotency and Epigenetic Reprogramming. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Thorsteinsdottir, U.; Sauvageau, G.; Humphries, R.K. Enhanced in vivo regenerative potential of HOXB4-transduced hematopoietic stem cells with regulation of their pool size. Blood 1999, 94, 2605–2612. [Google Scholar] [CrossRef]
- Graham, V.; Khudyakov, J.; Ellis, P.; Pevny, L. SOX2 functions to maintain neural progenitor identity. Neuron 2003, 39, 749–765. [Google Scholar] [CrossRef] [Green Version]
- Nichols, J.; Zevnik, B.; Anastassiadis, K.; Niwa, H.; Klewe-Nebenius, D.; Chambers, I.; Schöler, H.; Smith, A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 1998, 95, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Pan, G.; Thomson, J.A. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007, 17, 42–49. [Google Scholar] [CrossRef]
- Young, R.A. Control of the embryonic stem cell state. Cell 2011, 144, 940–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Bernardi, R.; Pandolfi, P.P. A novel signaling network as a critical rheostat for the biology and maintenance of the normal stem cell and the cancer-initiating cell. Curr. Opin. Genet. Dev. 2009, 19, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, Ö.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Grindley, J.C.; Yin, T.; Jayasinghe, S.; He, X.C.; Ross, J.T.; Haug, J.S.; Rupp, D.; Porter-Westpfahl, K.S.; Wiedemann, L.M.; et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006, 441, 518–522. [Google Scholar] [CrossRef]
- Kyba, M.; Perlingeiro, R.C.; Daley, G.Q. HoxB4 confers definitive lymphoid-myeloid engraftment potential on embryonic stem cell and yolk sac hematopoietic progenitors. Cell 2002, 109, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.S.; Kim, B.S.; Sheih, J.H.; Moore, M. Forced expression of HoxB4 enhances hematopoietic differentiation by human embryonic stem cells. Mol. Cells 2008, 25, 487–493. [Google Scholar]
- Wang, L.; Menendez, P.; Shojaei, F.; Li, L.; Mazurier, F.; Dick, J.E.; Cerdan, C.; Levac, K.; Bhatia, M. Generation of hematopoietic repopulating cells from human embryonic stem cells independent of ectopic HOXB4 expression. J. Exp. Med. 2005, 201, 1603–1614. [Google Scholar] [CrossRef]
- Cilloni, D.; Carturan, S.; Bracco, E.; Campia, V.; Rosso, V.; Torti, D.; Calabrese, C.; Gaidano, V.; Niparuck, P.; Favole, A.; et al. Aberrant activation of ROS1 represents a new molecular defect in chronic myelomonocytic leukemia. Leuk. Res. 2013, 37, 520–530. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- De Mol, C.; Mosci, S.; Traskine, M.; Verri, A. A regularized method for selecting nested groups of relevant genes from microarray data. Journal of computational biology. J. Comput. Mol. Cell Biol. 2009, 16, 677–690. [Google Scholar] [CrossRef] [Green Version]
- Mascelli, S.; Barla, A.; Raso, A.; Mosci, S.; Nozza, P.; Biassoni, R.; Morana, G.; Huber, M.; Mircean, C.; Fasulo, D.; et al. Molecular fingerprinting reflects different histotypes and brain region in low grade gliomas. BMC Cancer 2013, 13, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013, 41, W77–W83. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cilloni, D.; Petiti, J.; Campia, V.; Podestà, M.; Squillario, M.; Montserrat, N.; Bertaina, A.; Sabatini, F.; Carturan, S.; Berger, M.; et al. Transplantation Induces Profound Changes in the Transcriptional Asset of Hematopoietic Stem Cells: Identification of Specific Signatures Using Machine Learning Techniques. J. Clin. Med. 2020, 9, 1670. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9061670
Cilloni D, Petiti J, Campia V, Podestà M, Squillario M, Montserrat N, Bertaina A, Sabatini F, Carturan S, Berger M, et al. Transplantation Induces Profound Changes in the Transcriptional Asset of Hematopoietic Stem Cells: Identification of Specific Signatures Using Machine Learning Techniques. Journal of Clinical Medicine. 2020; 9(6):1670. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9061670
Chicago/Turabian StyleCilloni, Daniela, Jessica Petiti, Valentina Campia, Marina Podestà, Margherita Squillario, Nuria Montserrat, Alice Bertaina, Federica Sabatini, Sonia Carturan, Massimo Berger, and et al. 2020. "Transplantation Induces Profound Changes in the Transcriptional Asset of Hematopoietic Stem Cells: Identification of Specific Signatures Using Machine Learning Techniques" Journal of Clinical Medicine 9, no. 6: 1670. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9061670