Current and Future Trends in Molecular Biomarkers for Diagnostic, Prognostic, and Predictive Purposes in Non-Melanoma Skin Cancer


 ,
,  and
and
Abstract
:1. Introduction
2. Etiology
2.1. Ultraviolet (UV) Light
2.2. Genetic Background
2.3. Infectious Agents
3. Current Molecular Biomarkers for NMSC
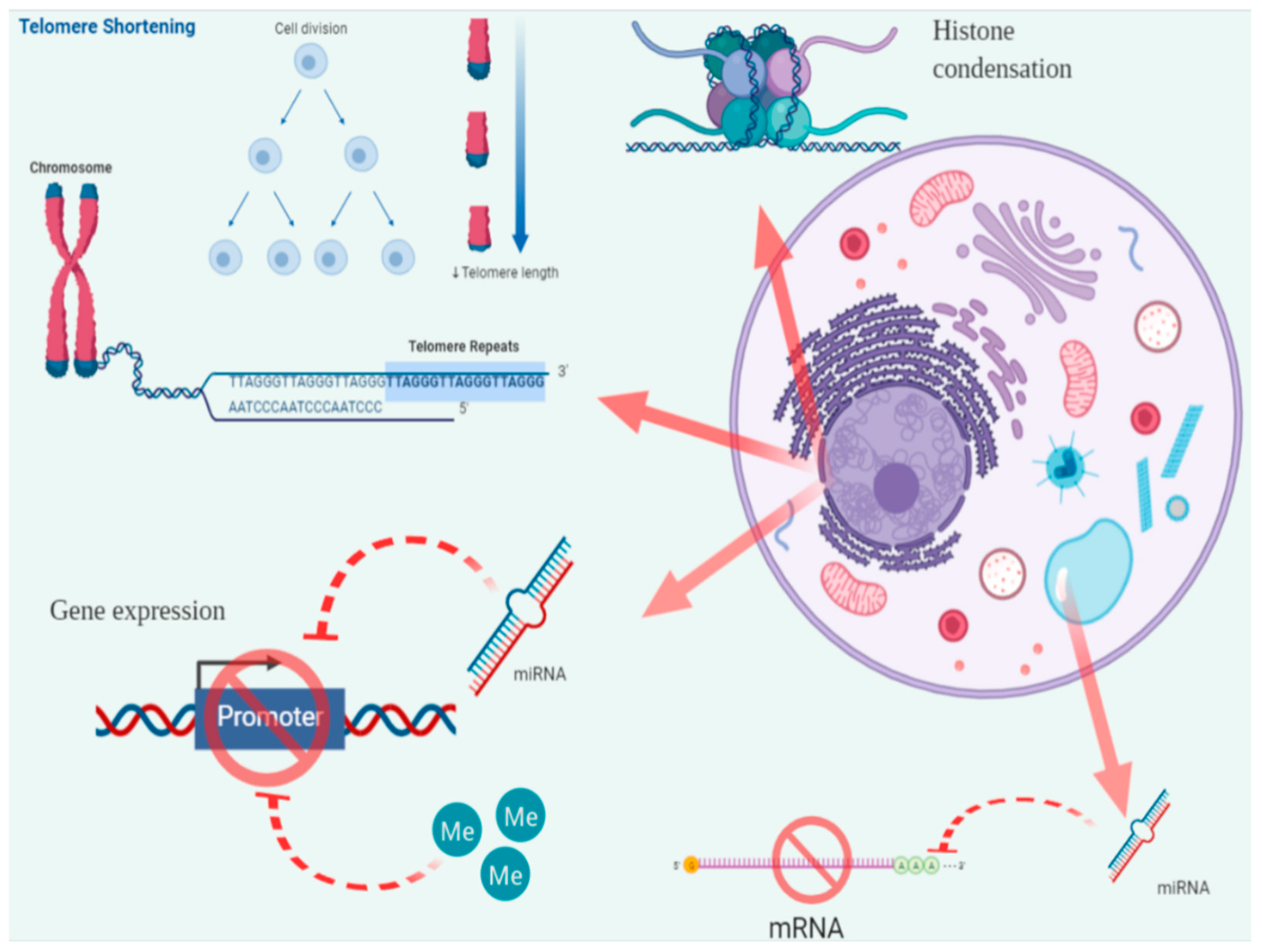
3.1. Telomere Length (TL)
3.2. Telomerase Activity (TA)
3.3. Epigenetic Modifications
3.3.1. CpG Island Methylation (CIM)
3.3.2. Histone Methylation and Acetylation
3.3.3. MicroRNAs (miRNAs or miRs)
4. Biomarkers under Evaluation
Micronuclei Frequency (MNf)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NMSC | non-melanoma skin cancer |
| BCC | basal cell carcinoma |
| SCC | squamous cell carcinoma |
| MCC | Merkel cell carcinoma |
| BD | Bowen’s Disease |
| AK | Actinic Keratosis |
| UV | Ultraviolet |
| β-HPV | β-Human papilloma virus |
| UVB | ultraviolet B |
| UVA | Ultraviolet A |
| EGFR | epidermal growth factor receptor |
| FGFR | fibroblast growth factor receptors |
| EBV | Epstein–Barr virus |
| MCPyV | Merkel Cell Polyomavirus |
| LTAg | large T-antigen |
| STAg | small t-antigen |
| TL | Telomere length |
| hTERT | human telomerase reverse transcriptase |
| hTR | human telomere RNA |
| TERC | telomerase RNA component |
| TA | telomerase activity |
| PBL | peripheral blood lymphocytes |
| CIM | CpG island methylation |
| DNMTs | DNA methyltransferases |
| LAD | lamina-associated domains |
| HATs | acetyltransferases |
| HDACs | histone deacetyltransferases |
| pri-miRNAs | primary miRNAs |
| pre-miRNAs | precursor miRNAs |
| 3′ UTR | 3′-untranslated region |
| 5′ UTR | 5′-untranslated region |
| MN | Micronuclei |
| MNf | Micronuclei frequency |
References
- Leiter, U.; Eigentler, T.; Garbe, C. Epidemiology of skin cancer. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2014; Volume 810, pp. 120–140. [Google Scholar]
- Rigel, D.S.; Friedman, R.J.; Kopf, A.W. Lifetime risk for development of skin cancer in the U.S. population: Current estimate is now 1 in 5. J. Am. Acad. Dermatol. 1996, 35, 1012–1013. [Google Scholar] [CrossRef]
- Apalla, Z.; Nashan, D.; Weller, R.B.; Castellsagué, X. Skin Cancer: Epidemiology, Disease Burden, Pathophysiology, Diagnosis, and Therapeutic Approaches. Dermatol. Ther. 2017, 7, 5–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br. J. Dermatol. 2012, 166, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.; Wernham, A.; Patel, A. When to suspect a non-melanoma skin cancer. BMJ 2020, 368, m692. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, V.; Madan, V. Nonmelanoma skin cancer. J. Cutan. Aesthet. Surg. 2012, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, J.; Saltzstein, S.L.; Sadler, G.R.; Tahir, Z.; Blair, S. A comparison of Merkel cell carcinoma and melanoma: Results from the California Cancer Registry. Clin. Med. Oncol. 2008, 2, 327–333. [Google Scholar] [CrossRef]
- Yanofsky, V.R.; Mercer, S.E.; Phelps, R.G. Histopathological Variants of Cutaneous Squamous Cell Carcinoma: A Review. J. Skin Cancer 2011, 2011, 210813. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.H.; Armstrong, A.W. Nonmelanoma Skin Cancer. Dermatol. Clin. 2012, 30, 125–139. [Google Scholar] [CrossRef]
- Wong, C.S.M. Basal cell carcinoma. BMJ 2003, 327, 794–798. [Google Scholar] [CrossRef]
- Dacosta Byfield, S.; Chen, D.; Yim, Y.M.; Reyes, C. Age distribution of patients with advanced non-melanoma skin cancer in the United States. Arch. Dermatol. Res. 2013, 305, 845–850. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.C.; Eberl, M.; Vagnozzi, A.N.; Belkadi, A.; Veniaminova, N.A.; Verhaegen, M.E.; Bichakjian, C.K.; Ward, N.L.; Dlugosz, A.A.; Wong, S.Y. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 2015, 16, 400–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwasniak, L.A.; Garcia-Zuazaga, J. Basal cell carcinoma: Evidence-based medicine and review of treatment modalities. Int. J. Dermatol. 2011, 50, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vigil, T.; Vázquez-López, F.; Perez-Oliva, N. Recurrence rates of primary basal cell carcinoma in facial risk areas treated with curettage and electrodesiccation. J. Am. Acad. Dermatol. 2007, 56, 91–95. [Google Scholar] [CrossRef]
- Habif, T.P. Clinical Dermatology: A Color Guide to Diagnosis and Therapy; Elsevier: St. Louis, MO, USA, 2016; ISBN 9780323266079. [Google Scholar]
- Yan, W.; Wistuba, I.I.; Emmert-Buck, M.R.; Erickson, H.S. Squamous Cell Carcinoma—Similarities and Differences among Anatomical Sites. Am. J. Cancer Res. 2011, 1, 275–300. [Google Scholar] [PubMed]
- Paulson, K.G.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Lebbé, C.; Zur Hausen, A.; Avril, M.-F.; Hariharan, S.; Bharmal, M.; Rgen, J.; Becker, C.; Bharmal, M.; De, J.B.-H.; et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. Eur. J. Cancer 2017, 71, 53–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunder, E.J.; Stern, R.S. Merkel-cell carcinomas in patients treated with methoxsalen and ultraviolet a radiation. N. Engl. J. Med. 1998, 339, 1247–1248. [Google Scholar] [CrossRef]
- Aw, D.; Silva, A.B.; Palmer, D.B. Immunosenescence: Emerging challenges for an ageing population. Immunology 2007, 120, 435–446. [Google Scholar] [CrossRef]
- Engels, E.A.; Frisch, M.; Goedert, J.J.; Biggar, R.J.; Miller, R.W. Merkel cell carcinoma and HIV infection. Lancet 2002, 359, 497–498. [Google Scholar] [CrossRef] [Green Version]
- Toker, C. Trabecular carcinoma of the skin. Arch. Dermatol. 1972, 105, 107–110. [Google Scholar] [CrossRef]
- Coggshall, K.; Tello, T.L.; North, J.P.; Yu, S.S. Merkel cell carcinoma: An update and review: Pathogenesis, diagnosis, and staging. J. Am. Acad. Dermatol. 2018, 78, 433–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duprat, J.P.; Landman, G.; Salvajoli, J.V.; Brechtbühl, E.R. A review of the epidemiology and treatment of Merkel cell carcinoma. Clinics 2011, 66, 1817–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, C.M.; Furniss, M.; Mackay-Wiggan, J.M. Basal cell carcinoma: An evidence-based treatment update. Am. J. Clin. Dermatol. 2014, 15, 197–216. [Google Scholar] [CrossRef]
- Bromfield, G.; Dale, D.; De, P.; Newman, K.; Rahal, R.; Shaw, A. Canadian Cancer Statistics 2015 Special Topic: Predictions of the Future Burden of Cancer in Canada; Canadian Cancer Society, Government of Canada: Toronto, ON, Canada, 2015.
- Non-Melanoma Skin Cancer Mortality Statistics | Cancer Research UK. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/non-melanoma-skin-cancer/mortality#heading-Two (accessed on 21 March 2020).
- Chren, M.M.; Torres, J.S.; Stuart, S.E.; Bertenthal, D.; Labrador, R.J.; Boscardin, W.J. Recurrence after treatment of nonmelanoma skin cancer: A prospective cohort study. Arch. Dermatol. 2011, 147, 540–546. [Google Scholar] [CrossRef]
- Chren, M.M.; Linos, E.; Torres, J.S.; Stuart, S.E.; Parvataneni, R.; Boscardin, W.J. Tumor recurrence 5 years after treatment of cutaneous basal cell carcinoma and squamous cell carcinoma. J. Investig. Dermatol. 2013, 133, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Kaldor, J.; Shugg, D.; Young, B.; Dwyer, T.; Wang, Y.G. Non-melanoma skin cancer: Ten years of cancer-registry-based surveillance. Int. J. Cancer 1993, 53, 886–891. [Google Scholar] [CrossRef]
- Didona, D.; Paolino, G.; Bottoni, U.; Cantisani, C. Non melanoma skin cancer pathogenesis overview. Biomedicines 2018, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Silverberg, M.J.; Leyden, W.; Warton, E.M.; Quesenberry, C.P.; Engels, E.A.; Asgari, M.M. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J. Natl. Cancer Inst. 2013, 105, 350–360. [Google Scholar] [CrossRef]
- Chew, M.H.; Yeh, Y.T.; Toh, E.L.; Sumarli, S.A.; Chew, G.K.; Lee, L.S.; Tan, M.H.; Hennedige, T.P.; Ng, S.Y.; Lee, S.K.; et al. Critical evaluation of contemporary management in a new Pelvic Exenteration Unit: The first 25 consecutive cases. World J. Gastrointest. Oncol. 2017, 9, 218–227. [Google Scholar] [CrossRef]
- Nikolaou, V.; Stratigos, A.J.; Tsao, H. Hereditary Nonmelanoma Skin Cancer. Semin. Cutan. Med. Surg. 2012, 31, 204–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarasinghe, V.; Madan, V.; Lear, J.T. Focus on Basal cell carcinoma. J. Skin Cancer 2011, 2011, 328615. [Google Scholar] [CrossRef] [PubMed]
- Smeets, N.W.J.; Kuijpers, D.I.M.; Nelemans, P.; Ostertag, J.U.; Verhaegh, M.E.J.M.; Krekels, G.A.M.; Neumann, H.A.M. Mohs’ micrographic surgery for treatment of basal cell carcinoma of the face—Results of a retrospective study and review of the literature. Br. J. Dermatol. 2004, 151, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Welsh, M.M.; Karagas, M.R.; Kuriger, J.K.; Houseman, A.; Spencer, S.K.; Perry, A.E.; Nelson, H.H. Genetic Determinants of UV-Susceptibility in Non-Melanoma Skin Cancer. PLoS ONE 2011, 6, e20019. [Google Scholar] [CrossRef]
- Saijo, S.; Kodari, E.; Kripke, M.L.; Strickland, F.M. UVB irradiation decreases the magnitude of the Th1 response to hapten but does not increase the Th2 response. Photodermatol. Photoimmunol. Photomed. 1996, 12, 145–153. [Google Scholar] [CrossRef]
- Morison, W.L.; Bucana, C.; Kripke, M.L. Systemic suppression of contact hypersensitivity by UVB radiation is unrelated to the UVB-induced alterations in the morphology and number of Langerhans cells. Immunology 1984, 52, 299–306. [Google Scholar]
- McGillis, S.T.; Fein, H. Topical treatment strategies for non-melanoma skin cancer and precursor lesions. Semin. Cutan. Med. Surg. 2004, 23, 174–183. [Google Scholar] [CrossRef]
- Christensen, S.R. Recent advances in field cancerization and management of multiple cutaneous squamous cell carcinomas [version 1; referees: 2 approved]. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Köhler, B.; Schönicke, A.; Scharwächter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef]
- Pellegrini, C.; Maturo, M.G.; Di Nardo, L.; Ciciarelli, V.; Gutiérrez García-Rodrigo, C.; Fargnoli, M.C. Understanding the molecular genetics of basal cell carcinoma. Int. J. Mol. Sci. 2017, 18, 2485. [Google Scholar] [CrossRef] [Green Version]
- Dotto, G.P.; Rustgi, A.K. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell 2016, 29, 622–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crum, C.P.; McKeon, F.D. p63 in Epithelial Survival, Germ Cell Surveillance, and Neoplasia. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 349–371. [Google Scholar] [CrossRef] [PubMed]
- Weina, K.; Utikal, J. SOX2 and cancer: Current research and its implications in the clinic. Clin. Transl. Med. 2014, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, M.; Werner, S. Nrf2—A regulator of keratinocyte redox signaling. Free Radic. Biol. Med. 2015, 88, 243–252. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Sadeqzadeh, E.; De Bock, C.E.; Thorne, R.F. Sleeping Giants: Emerging Roles for the Fat Cadherins in Health and Disease. Med. Res. Rev. 2014, 34, 190–221. [Google Scholar] [CrossRef]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006, 110, 525–541. [Google Scholar] [CrossRef] [Green Version]
- Biliris, K.A.; Koumantakis, E.; Dokianakis, D.N.; Sourvinos, G.; Spandidos, D.A. Human papillomavirus infection of non-melanoma skin cancers in immunocompetent hosts. Cancer Lett. 2000, 161, 83–88. [Google Scholar] [CrossRef]
- Arbiser, J.L. Implications of Epstein-Barr Virus (EBV)-induced carcinogenesis on cutaneous inflammation and carcinogenesis: Evidence of recurring patterns of angiogenesis and signal transduction. J. Investig. Dermatol. 2005, 124, xi. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolstov, Y.L.; Pastrana, D.V.; Feng, H.; Becker, J.C.; Jenkins, F.J.; Moschos, S.; Chang, Y.; Buck, C.B.; Moore, P.S. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int. J. Cancer 2009, 125, 1250–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Hedman, L.; Mattila, P.S.; Jartti, T.; Ruuskanen, O.; Söderlund-Venermo, M.; Hedman, K. Serological evidence of Merkel cell polyomavirus primary infections in childhood. J. Clin. Virol. 2011, 50, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Kiss, M.; Szabó, K.; Kemény, L. Detection of merkel cell polyomavirus DNA in merkel cell carcinomas. Br. J. Dermatol. 2009, 161, 930–932. [Google Scholar] [CrossRef]
- Sastre-Garau, X.; Peter, M.; Avril, M.F.; Laude, H.; Couturier, J.; Rozenberg, F.; Almeida, A.; Boitier, F.; Carlotti, A.; Couturaud, B.; et al. Merkel cell carcinoma of the skin: Pathological and molecular evidence for a causative role of MCV in oncogenesis. J. Pathol. 2009, 218, 48–56. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Zehnbauer, B.A.; Pfeifer, J.D. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod. Pathol. 2009, 22, 516–521. [Google Scholar] [CrossRef]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Böhling, T.; Joensuu, H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin. Cancer Res. 2011, 17, 4806–4813. [Google Scholar] [CrossRef] [Green Version]
- Wendzicki, J.A.; Moore, P.S.; Chang, Y. Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol. 2015, 11, 38–43. [Google Scholar] [CrossRef] [Green Version]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Eberl, M.; Wilbert, D.M.; Meireles, J.; Bichakjian, C.K.; Saunders, T.L.; Wong, S.Y.; Dlugosz, A.A. Merkel cell polyomavirus small T antigen initiates merkel cell carcinoma-like tumor development in mice. Cancer Res. 2017, 77, 3151–3157. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; DeCaprio, J.A.; Fluck, M.M.; Schaffhausen, B.S. Cellular transformation by Simian Virus 40 and Murine Polyoma Virus T antigens. Semin. Cancer Biol. 2009, 19, 218–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spurgeon, M.E.; Lambert, P.F. Merkel cell polyomavirus: A newly discovered human virus with oncogenic potential. Virology 2013, 435, 118–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telomere Shortening—An Overview | ScienceDirect Topics. Available online: https://0-www-sciencedirect-com.brum.beds.ac.uk/topics/neuroscience/telomere-shortening (accessed on 25 March 2020).
- Richter, T.; von Zglinicki, T. A continuous correlation between oxidative stress and telomere shortening in fibroblasts. Exp. Gerontol. 2007, 42, 1039–1042. [Google Scholar] [CrossRef]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef]
- Tsatsakis, A.; Tsoukalas, D.; Fragkiadaki, P.; Vakonaki, E.; Tzatzarakis, M.; Sarandi, E.; Nikitovic, D.; Tsilimidos, G.; Alegakis, A.K. Developing BIOTEL: A Semi-Automated Spreadsheet for Estimating Telomere Length and Biological Age. Front. Genet. 2019, 10, 84. [Google Scholar] [CrossRef]
- Nikolouzakis, T.K.; Vassilopoulou, L.; Fragkiadaki, P.; Sapsakos, T.M.; Papadakis, G.Z.; Spandidos, D.A.; Tsatsakis, A.M.; Tsiaoussis, J. Improving diagnosis, prognosis and prediction by using biomarkers in CRC patients (Review). Oncol. Rep. 2018, 39, 2455–2472. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Zhang, Y.; Zhang, L.; Ma, J.L.; Zhou, T.; Li, Z.X.; Liu, W.D.; Li, W.Q.; Deng, D.J.; You, W.C.; et al. Telomere Length of Circulating Cell-Free DNA and Gastric Cancer in a Chinese Population at High-Risk. Front. Oncol. 2019, 9, 1434. [Google Scholar] [CrossRef]
- Bailey, S.M.; Murnane, J.P. Telomeres, chromosome instability and cancer. Nucleic Acids Res. 2006, 34, 2408–2417. [Google Scholar] [CrossRef] [Green Version]
- Leufke, C.; Leykauf, J.; Krunic, D.; Jauch, A.; Holtgreve-Grez, H.; Böhm-Steuer, B.; Bröcker, E.B.; Mauch, C.; Utikal, J.; Hartschuh, W.; et al. The telomere profile distinguishes two classes of genetically distinct cutaneous squamous cell carcinomas. Oncogene 2014, 33, 3506–3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caini, S.; Raimondi, S.; Johansson, H.; De Giorgi, V.; Zanna, I.; Palli, D.; Gandini, S. Telomere length and the risk of cutaneous melanoma and non-melanoma skin cancer: A review of the literature and meta-analysis. J. Dermatol. Sci. 2015, 80, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Yamada-Hishida, H.; Nobeyama, Y.; Nakagawa, H. Correlation of telomere length to malignancy potential in non-melanoma skin cancers. Oncol. Lett. 2018, 15, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainwright, L.J.; Rees, J.L.; Middleton, P.G. Changes in mean telomere length in basal cell carcinomas of the skin. Genes Chromosom. Cancer 1995, 12, 45–49. [Google Scholar] [CrossRef]
- Han, J.; Qureshi, A.A.; Prescott, J.; Guo, Q.; Ye, L.; Hunter, D.J.; De Vivo, I. A prospective study of telomere length and the risk of skin cancer. J. Investig. Dermatol. 2009, 129, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Anic, G.M.; Sondak, V.K.; Messina, J.L.; Fenske, N.A.; Zager, J.S.; Cherpelis, B.S.; Lee, J.H.; Fulp, W.J.; Epling-Burnette, P.K.; Park, J.Y.; et al. Telomere length and risk of melanoma, squamous cell carcinoma, and basal cell carcinoma. Cancer Epidemiol. 2013, 37, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Qureshi, A.A.; Guo, Q.; De Vivo, I.; Han, J. No association between telomere length in peripheral blood leukocytes and the risk of nonmelanoma skin cancer. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1043–1045. [Google Scholar] [CrossRef] [Green Version]
- Daniel, M.; Peek, G.W.; Tollefsbol, T.O. Regulation of the human catalytic subunit of telomerase (hTERT). Gene 2012, 498, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Akincilar, S.C.; Unal, B.; Tergaonkar, V. Reactivation of telomerase in cancer. Cell. Mol. Life Sci. 2016, 73, 1659–1670. [Google Scholar] [CrossRef] [Green Version]
- Hoffmeyer, K.; Raggioli, A.; Rudloff, S.; Anton, R.; Hierholzer, A.; Del Valle, I.; Hein, K.; Vogt, R.; Kemler, R. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science 2012, 336, 1549–1554. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.W.; Hou, P.S.; Tseng, S.F.; Chien, C.L.; Wu, K.J.; Chen, H.F.; Ho, H.N.; Kyo, S.; Teng, S.C. Krüppel-like transcription factor 4 contributes to maintenance of telomerase activity in stem cells. Stem Cells 2010, 28, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- Burnworth, B.; Arendt, S.; Muffler, S.; Steinkraus, V.; Bröcker, E.B.; Birek, C.; Hartschuh, W.; Jauch, A.; Boukamp, P. The multi-step process of human skin carcinogenesis: A role for p53, cyclin D1, hTERT, p16, and TSP-1. Eur. J. Cell Biol. 2007, 86, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Pellegrini, C.; Cardelli, L.; Rocco, T.; Ciciarelli, V.; Peris, K.; Fargnoli, M.C. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int. J. Mol. Sci. 2019, 20, 1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parris, C.N.; Jezzard, S.; Silver, A.; MacKie, R.; McGregor, J.M.; Newbold, R.F. Telomerase activity in melanoma and non-melanoma skin cancer. Br. J. Cancer 1999, 79, 47–53. [Google Scholar] [CrossRef]
- Boldrini, L.; Loggini, B.; Gisfredi, S.; Zucconi, Y.; Di Quirico, D.; Biondi, R.; Cervadoro, G.; Barachini, P.; Basolo, F.; Pingitore, R.; et al. Evaluation of telomerase in non-melanoma skin cancer. Int. J. Mol. Med. 2003, 11, 607–611. [Google Scholar] [CrossRef]
- Griewank, K.G.; Murali, R.; Schilling, B.; Schimming, T.; Möller, I.; Moll, I.; Schwamborn, M.; Sucker, A.; Zimmer, L.; Schadendorf, D.; et al. TERT promoter mutations are frequent in cutaneous basal cell carcinoma and squamous cell carcinoma. PLoS ONE 2013, 8, e80354. [Google Scholar] [CrossRef]
- Scott, G.A.; Laughlin, T.S.; Rothberg, P.G. Mutations of the TERT promoter are common in basal cell carcinoma and squamous cell carcinoma. Mod. Pathol. 2014, 27, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Penta, D.; Somashekar, B.S.; Meeran, S.M. Epigenetics of skin cancer: Interventions by selected bioactive phytochemicals. Photodermatol. Photoimmunol. Photomed. 2018, 34, 42–49. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Rodríguez-Paredes, M.; Bormann, F.; Raddatz, G.; Gutekunst, J.; Lucena-Porcel, C.; Köhler, F.; Wurzer, E.; Schmidt, K.; Gallinat, S.; Wenck, H.; et al. Methylation profiling identifies two subclasses of squamous cell carcinoma related to distinct cells of origin. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Rodríguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome-biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Salemi, R.; Travali, S.; Scalisi, A.; McCubrey, J.A.; Candido, S.; Libra, M. MMP-9 overexpression is associated with intragenic hypermethylation of MMP9 gene in melanoma. Aging 2016, 8, 933–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candido, S.; Parasiliti Palumbo, G.A.; Pennisi, M.; Russo, G.; Sgroi, G.; Di Salvatore, V.; Libra, M.; Pappalardo, F. EpiMethEx: A tool for large-scale integrated analysis in methylation hotspots linked to genetic regulation. BMC Bioinform. 2019, 19, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Napoli, S.; Scuderi, C.; Gattuso, G.; Di Bella, V.; Candido, S.; Basile, M.S.; Libra, M.; Falzone, L. Functional Roles of Matrix Metalloproteinases and Their Inhibitors in Melanoma. Cells 2020, 9, 1151. [Google Scholar] [CrossRef]
- Hervás-Marín, D.; Higgins, F.; Sanmartín, O.; López-Guerrero, J.A.; Bañó, M.C.; Igual, J.C.; Quilis, I.; Sandoval, J. Genome wide DNA methylation profiling identifies specific epigenetic features in high-risk cutaneous squamous cell carcinoma. PLoS ONE 2019, 14, e0223341. [Google Scholar] [CrossRef]
- Brown, V.L.; Harwood, C.A.; Crook, T.; Cronin, J.G.; Kelsell, D.R.; Proby, C.M. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2004, 122, 1284–1292. [Google Scholar] [CrossRef] [Green Version]
- Chiles, M.C.; Ai, L.; Zuo, C.; Fan, C.Y.; Smoller, B.R. E-Cadherin Promoter Hypermethylation in Preneoplastic and Neoplastic Skin Lesions. Mod. Pathol. 2003, 16, 1014–1018. [Google Scholar] [CrossRef]
- Murao, K.; Kubo, Y.; Ohtani, N.; Hara, E.; Arase, S. Epigenetic abnormalities in cutaneous squamous cell carcinomas: Frequent inactivation of the RB1/p16 and p53 pathways. Br. J. Dermatol. 2006, 155, 999–1005. [Google Scholar] [CrossRef]
- Takeuchi, T.; Liang, S.B.; Matsuyoshi, N.; Zhou, S.; Miyachi, Y.; Sonobe, H.; Ohtsuki, Y. Loss of T-cadherin (CDH13, H-cadherin) expression in cutaneous squamous cell carcinoma. Lab. Investig. 2002, 82, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Venza, I.; Visalli, M.; Tripodo, B.; De Grazia, G.; Loddo, S.; Teti, D.; Venza, M. FOXE1 is a target for aberrant methylation in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2010, 162, 1093–1097. [Google Scholar] [CrossRef]
- Liang, J.; Kang, X.; Halifu, Y.; Zeng, X.; Jin, T.; Zhang, M.; Luo, D.; Ding, Y.; Zhou, Y.; Yakeya, B.; et al. Secreted frizzled-related protein promotors are hypermethylated in cutaneous squamous carcinoma compared with normal epidermis. BMC Cancer 2015, 15, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darr, O.A.; Colacino, J.A.; Tang, A.L.; McHugh, J.B.; Bellile, E.L.; Bradford, C.R.; Prince, M.P.; Chepeha, D.B.; Rozek, L.S.; Moyer, J.S. Epigenetic alterations in metastatic cutaneous carcinoma. Head Neck 2015, 37, 994–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, K.; Drexler, S.K.; Eberle, F.C.; Lefort, K.; Yazdi, A.S. Silencing of ASC in cutaneous squamous cell carcinoma. PLoS ONE 2016, 11, e0164742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobeyama, Y.; Watanabe, Y.; Nakagawa, H. Silencing of G0/G1 switch gene 2 in cutaneous squamous cell carcinoma. PLoS ONE 2017, 12, e0187047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Jiang, M.; Feng, Q.; Kiviat, N.B.; Stern, J.E.; Hawes, S.; Cherne, S.; Lu, H. Aberrant Methylation Changes Detected in Cutaneous Squamous Cell Carcinoma of Immunocompetent Individuals. Cell Biochem. Biophys. 2015, 72, 599–604. [Google Scholar] [CrossRef]
- Toll, A.; Salgado, R.; Espinet, B.; Díaz-Lagares, A.; Hernández-Ruiz, E.; Andrades, E.; Sandoval, J.; Esteller, M.; Pujol, R.M.; Hernández-Muñoz, I. MiR-204 silencing in intraepithelial to invasive cutaneous squamous cell carcinoma progression. Mol. Cancer 2016, 15, 1. [Google Scholar] [CrossRef] [Green Version]
- Venza, M.; Visalli, M.; Catalano, T.; Beninati, C.; Teti, D.; Venza, I. DSS1 promoter hypomethylation and overexpression predict poor prognosis in melanoma and squamous cell carcinoma patients. Hum. Pathol. 2017, 60, 137–146. [Google Scholar] [CrossRef]
- Goldberg, M.; Rummelt, C.; Laerm, A.; Helmbold, P.; Holbach, L.M.; Ballhausen, W.G. Epigenetic silencing contributes to frequent loss of the fragile histidine triad tumour suppressor in basal cell carcinomas. Br. J. Dermatol. 2006, 155, 1154–1158. [Google Scholar] [CrossRef]
- Heitzer, E.; Bambach, I.; Dandachi, N.; Horn, M.; Wolf, P. PTCH promoter methylation at low level in sporadic basal cell carcinoma analysed by three different approaches. Exp. Dermatol. 2010, 19, 926–928. [Google Scholar] [CrossRef]
- Greenberg, E.S.; Chong, K.K.; Huynh, K.T.; Tanaka, R.; Hoon, D.S.B. Epigenetic biomarkers in skin cancer. Cancer Lett. 2014, 342, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salemi, R.; Falzone, L.; Madonna, G.; Polesel, J.; Cinà, D.; Mallardo, D.; Ascierto, P.A.; Libra, M.; Candido, S. MMP-9 as a Candidate Marker of Response to BRAF Inhibitors in Melanoma Patients With BRAFV600E Mutation Detected in Circulating-Free DNA. Front. Pharmacol. 2018, 9, 856. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Lin, J.C.Y.; Wei, V.; Yoo, C.; Cheng, J.C.; Nguyen, C.T.; Weisenberger, D.J.; Egger, G.; Takai, D.; Gonzales, F.A.; et al. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc. Natl. Acad. Sci. USA 2004, 101, 7357–7362. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (−)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p 16 INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Smits, M.; Van Rijn, S.; Hulleman, E.; Biesmans, D.; Van Vuurden, D.G.; Kool, M.; Haberler, C.; Aronica, E.; Vandertop, W.P.; Noske, D.P.; et al. EZH2-regulated DAB2IP is a medulloblastoma tumor suppressor and a positive marker for survival. Clin. Cancer Res. 2012, 18, 4048–4058. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.C.; Chan, M.P.; Andrews, C.A.; Kahana, A. EZH2, proliferation rate, and aggressive tumor subtypes in cutaneous basal cell carcinoma. JAMA Oncol. 2016, 2, 962–963. [Google Scholar] [CrossRef]
- Rao, R.C.; Chan, M.P.; Andrews, C.A.; Kahana, A. Epigenetic markers in basal cell carcinoma: Universal themes in oncogenesis and tumor stratification?—A short report. Cell. Oncol. 2018, 41, 693–698. [Google Scholar] [CrossRef]
- Harms, K.L.; Chubb, H.; Zhao, L.; Fullen, D.R.; Bichakjian, C.K.; Johnson, T.M.; Carskadon, S.; Palanisamy, N.; Harms, P.W. Increased expression of EZH2 in Merkel cell carcinoma is associated with disease progression and poorer prognosis. Hum. Pathol. 2017, 67, 78–84. [Google Scholar] [CrossRef]
- Orouji, E.; Utikal, J. Tackling malignant melanoma epigenetically: Histone lysine methylation. Clin. Epigenetics 2018, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Raman, A.T.; Rai, K. Loss of histone acetylation and H3K4 methylation promotes melanocytic malignant transformation. Mol. Cell. Oncol. 2018, 5, e1359229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, S. Posttranscriptional Upregulation by MicroRNAs. Wiley Interdiscip. Rev. RNA 2012, 3, 311–330. [Google Scholar] [CrossRef]
- Makarova, J.A.; Shkurnikov, M.U.; Wicklein, D.; Lange, T.; Samatov, T.R.; Turchinovich, A.A.; Tonevitsky, A.G. Intracellular and extracellular microRNA: An update on localization and biological role. Prog. Histochem. Cytochem. 2016, 51, 33–49. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Creemers, E.E.; Tijsen, A.J.; Pinto, Y.M. Circulating MicroRNAs: Novel biomarkers and extracellular communicators in cardiovascular disease? Circ. Res. 2012, 110, 483–495. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doukas, S.G.; Vageli, D.P.; Lazopoulos, G.; Spandidos, D.A.; Sasaki, C.T.; Tsatsakis, A. The Effect of NNK, A Tobacco Smoke Carcinogen, on the miRNA and Mismatch DNA Repair Expression Profiles in Lung and Head and Neck Squamous Cancer Cells. Cells 2020, 9, 1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silantyev, A.; Falzone, L.; Libra, M.; Gurina, O.; Kardashova, K.; Nikolouzakis, T.; Nosyrev, A.; Sutton, C.; Mitsias, P.; Tsatsakis, A. Current and Future Trends on Diagnosis and Prognosis of Glioblastoma: From Molecular Biology to Proteomics. Cells 2019, 8, 863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filetti, V.; Falzone, L.; Rapisarda, V.; Caltabiano, R.; Eleonora Graziano, A.C.; Ledda, C.; Loreto, C. Modulation of microRNA expression levels after naturally occurring asbestiform fibers exposure as a diagnostic biomarker of mesothelial neoplastic transformation. Ecotoxicol. Environ. Saf. 2020, 198, 110640. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Lupo, G.; La Rosa, G.R.M.; Crimi, S.; Anfuso, C.D.; Salemi, R.; Rapisarda, E.; Libra, M.; Candido, S. Identification of Novel MicroRNAs and Their Diagnostic and Prognostic Significance in Oral Cancer. Cancers 2019, 11, 610. [Google Scholar] [CrossRef] [Green Version]
- Falzone, L.; Romano, G.L.; Salemi, R.; Bucolo, C.; Tomasello, B.; Lupo, G.; Anfuso, C.D.; Spandidos, D.A.; Libra, M.; Candido, S. Prognostic significance of deregulated microRNAs in uveal melanomas. Mol. Med. Rep. 2019, 19, 2599–2610. [Google Scholar] [CrossRef] [Green Version]
- Falzone, L.; Scola, L.; Zanghì, A.; Biondi, A.; Di Cataldo, A.; Libra, M.; Candido, S. Integrated analysis of colorectal cancer microRNA datasets: Identification of microRNAs associated with tumor development. Aging 2018, 10, 1000–1014. [Google Scholar] [CrossRef]
- Falzone, L.; Candido, S.; Salemi, R.; Basile, M.S.; Scalisi, A.; McCubrey, J.A.; Torino, F.; Signorelli, S.S.; Montella, M.; Libra, M. Computational identification of microRNAs associated to both epithelial to mesenchymal transition and NGAL/MMP-9 pathways in bladder cancer. Oncotarget 2016, 7, 72758–72766. [Google Scholar] [CrossRef] [Green Version]
- Hafsi, S.; Candido, S.; Maestro, R.; Falzone, L.; Soua, Z.; Bonavida, B.; Spandidos, D.A.; Libra, M. Correlation between the overexpression of Yin Yang 1 and the expression levels of miRNAs in Burkitt’s lymphoma: A computational study. Oncol. Lett. 2016, 11, 1021–1025. [Google Scholar] [CrossRef] [Green Version]
- Sand, M.; Bechara, F.G.; Gambichler, T.; Sand, D.; Friedländer, M.R.; Bromba, M.; Schnabel, R.; Hessam, S. Next-generation sequencing of the basal cell carcinoma miRNome and a description of novel microRNA candidates under neoadjuvant vismodegib therapy: An integrative molecular and surgical case study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 332–338. [Google Scholar] [CrossRef]
- Yi, R.; Poy, M.N.; Stoffel, M.; Fuchs, E. A skin microRNA promotes differentiation by repressing “stemness”. Nature 2008, 452, 225–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonkoly, E.; Lovén, J.; Xu, N.; Meisgen, F.; Wei, T.; Brodin, P.; Jaks, V.; Kasper, M.; Shimokawa, T.; Harada, M.; et al. MicroRNA-203 functions as a tumor suppressor in basal cell carcinoma. Oncogenesis 2012, 1, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, P.; Ma, L.; Wu, Z.; Zheng, G.; Li, J. Expression of miR-34a in basal cell carcinoma patients and its relationship with prognosis. J. BUON. 2019, 24, 1283–1288. [Google Scholar] [PubMed]
- Mizrahi, A.; Barzilai, A.; Gur-Wahnon, D.; Ben-Dov, I.Z.; Glassberg, S.; Meningher, T.; Elharar, E.; Masalha, M.; Jacob-Hirsch, J.; Tabibian-Keissar, H.; et al. Alterations of microRNAs throughout the malignant evolution of cutaneous squamous cell carcinoma: The role of miR-497 in epithelial to mesenchymal transition of keratinocytes. Oncogene 2018, 37, 218–230. [Google Scholar] [CrossRef]
- Yu, X.; Li, Z. The role of miRNAs in cutaneous squamous cell carcinoma. J. Cell. Mol. Med. 2016, 20, 3–9. [Google Scholar] [CrossRef] [Green Version]
- García-Sancha, N.; Corchado-Cobos, R.; Pérez-Losada, J.; Cañueto, J. MicroRNA dysregulation in cutaneous squamous cell carcinoma. Int. J. Mol. Sci. 2019, 20, 2181. [Google Scholar]
- Cañueto, J.; Cardeñoso-Álvarez, E.; García-Hernández, J.L.; Galindo-Villardón, P.; Vicente-Galindo, P.; Vicente-Villardón, J.L.; Alonso-López, D.; De Las Rivas, J.; Valero, J.; Moyano-Sanz, E.; et al. MicroRNA (miR)-203 and miR-205 expression patterns identify subgroups of prognosis in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2017, 177, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Stojadinovic, O.; Ramirez, H.; Pastar, I.; Gordon, K.A.; Stone, R.; Choudhary, S.; Badiavas, E.; Nouri, K.; Tomic-Canic, M. MiR-21 and miR-205 are induced in invasive cutaneous squamous cell carcinomas. Arch. Dermatol. Res. 2017, 309, 133–139. [Google Scholar] [CrossRef]
- Gong, Z.H.; Zhou, F.; Shi, C.; Xiang, T.; Zhou, C.K.; Wang, Q.Q.; Jiang, Y.S.; Gao, S.F. miRNA-221 promotes cutaneous squamous cell carcinoma progression by targeting PTEN. Cell. Mol. Biol. Lett. 2019, 24, 9. [Google Scholar] [CrossRef]
- Zhang, L.; Xiang, P.; Han, X.; Wu, L.; Li, X.; Xiong, Z. Decreased expression of microRNA-20a promotes tumor progression and predicts poor prognosis of cutaneous squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 11446–11451. [Google Scholar] [PubMed]
- Ning, M.S.; Kim, A.S.; Prasad, N.; Levy, S.E.; Zhang, H.; Andl, T. Characterization of the Merkel Cell Carcinoma miRNome. J. Skin Cancer 2014, 2014, 289548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veija, T.; Sahi, H.; Koljonen, V.; Bohling, T.; Knuutila, S.; Mosakhani, N. miRNA-34a underexpressed in Merkel cell polyomavirus-negative Merkel cell carcinoma. Virchows Arch. 2015, 466, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Renwick, N.; Cekan, P.; Masry, P.A.; McGeary, S.E.; Miller, J.B.; Hafner, M.; Li, Z.; Mihailovic, A.; Morozov, P.; Brown, M.; et al. Multicolor microRNA FISH effectively differentiates tumor types. J. Clin. Investig. 2013, 123, 2694–2702. [Google Scholar] [CrossRef]
- Konstatinell, A.; Coucheron, D.H.; Sveinbjørnsson, B.; Moens, U. MicroRNAs as potential biomarkers in merkel cell carcinoma. Int. J. Mol. Sci. 2018, 19, 1873. [Google Scholar]
- Tuaeva, N.O.; Falzone, L.; Porozov, Y.B.; Nosyrev, A.E.; Trukhan, V.M.; Kovatsi, L.; Spandidos, D.A.; Drakoulis, N.; Kalogeraki, A.; Mamoulakis, C.; et al. Translational Application of Circulating DNA in Oncology: Review of the Last Decades Achievements. Cells 2019, 8, 1251. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Lee, L.; Caramuta, S.; Höög, A.; Browaldh, N.; Björnhagen, V.; Larsson, C.; Lui, W.O. MicroRNA expression patterns related to merkel cell polyomavirus infection in human Merkel cell carcinoma. J. Investig. Dermatol. 2014, 134, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, S.; Qu, L.; Wang, Y.; Chen, H.D.; He, C. Identification of a five-miRNA signature predicting survival in cutaneous melanoma cancer patients. PeerJ 2019, 2019, e7831. [Google Scholar] [CrossRef] [Green Version]
- Hanniford, D.; Zhong, J.; Koetz, L.; Gaziel-Sovran, A.; Lackaye, D.J.; Shang, S.; Pavlick, A.; Shapiro, R.; Berman, R.; Darvishian, F.; et al. A miRNA-based signature detected in primary melanoma tissue predicts development of brain metastasis. Clin. Cancer Res. 2015, 21, 4903–4912. [Google Scholar] [CrossRef] [Green Version]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Mateuca, R.; Lombaert, N.; Aka, P.V.; Decordier, I.; Kirsch-Volders, M. Chromosomal changes: Induction, detection methods and applicability in human biomonitoring. Biochimie 2006, 88, 1515–1531. [Google Scholar] [CrossRef] [PubMed]
- Pardini, B.; Viberti, C.; Naccarati, A.; Allione, A.; Oderda, M.; Critelli, R.; Preto, M.; Zijno, A.; Cucchiarale, G.; Gontero, P.; et al. Increased micronucleus frequency in peripheral blood lymphocytes predicts the risk of bladder cancer. Br. J. Cancer 2017, 116, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Nikolouzakis, T.K.; Stivaktakis, P.D.; Apalaki, P.; Kalliantasi, K.; Sapsakos, T.M.; Spandidos, D.A.; Tsatsakis, A.; Souglakos, J.; Tsiaoussis, J. Effect of systemic treatment on the micronuclei frequency in the peripheral blood of patients with metastatic colorectal cancer. Oncol. Lett. 2019, 17, 2703–2712. [Google Scholar] [CrossRef] [PubMed]
- Diem, C.; Rünger, T.M. Processing of three different types of DNA damage in cell lines of a cutaneous squamous cell carcinoma progression model. Carcinogenesis 1997, 18, 657–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emri, G.; Wenczl, E.; Van Erp, P.; Jans, J.; Roza, L.; Horkay, I.; Schothorst, A.A. Low doses of UVB or UVA induce chromosomal aberrations in cultured human skin cells. J. Investig. Dermatol. 2000, 115, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford, K.K.; Parshad, R.; Price, F.M.; Tarone, R.E.; Thompson, J.; Guerry, D. Radiation-induced chromatid breaks and DNA repair in blood lymphocytes of patients with dysplastic nevi and/or cutaneous melanoma. J. Investig. Dermatol. 1997, 109, 546–549. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Gene Target | Methylation Status | Type of NMSC | Cellular Effect | Reference |
|---|---|---|---|---|
| CDKN2A | Hypermethylated | SCC | Cell cycle deregulation | Brown et al. [102] |
| CDH1 | Hypermethylated | SCC | Cellular environment deregulation | Chiles et al. [103] Murao et al. [104] |
| CDH13 | Hypermethylated | SCC | Cellular environment deregulation | Takeuchi et al. [105] |
| FOXE1 | Hypermethylated | SCC | Modulator of Wnt signaling | Venza et al. [106] |
| SFRPs | Hypermethylated | SCC | Modulator of Wnt signaling | Liang et al. [107] |
| FRZB | Hypermethylated | SCC | Modulator of Wnt signaling | Darr et al. [108] |
| ASC | Hypermethylated | SCC | Deregulation of apoptosis | Meier et al. [109] |
| G0S2 | Hypermethylated | SCC | Deregulation of apoptosis | Nobeyama et al. [110] |
| DAPK1 | Hypermethylated | SCC | Deregulation of apoptosis | Li et al. [111] |
| miRNA-204 | Hypermethylated | SCC | Deregulation of apoptosis | Toll et al. [112] |
| DSS1 | Hypomethylation | SCC | Deregulated post-translational protein modification | Venza et al. [113] |
| Global DNA | Hypomethylation | SCC (benign) | Restricted genomic silencing | Hervás-Marín et al. [101] |
| Global DNA | Hypermethylation | SCC (aggressive) | Extensive genomic silencing | |
| FHIT promoter | Hypomethylated | BCC | Replication stress and DNA damage | Goldberg et al. [114] |
| PTCH promoter | Hypermethylated | BCC (small number of cases) | Deactivation of tumor suppressor genes | Heitzer et al. [115] |
| MYCL2 | Hypomethylated | BCC (metastatic) | Activation of proto-oncogene | Darr et al. [108] |
| p14-ARK | Hypermethylated | MCC | Deactivation of tumor suppressor genes | Greenberg et al. [116] |
| DUSP2, CDKN2A promoter | Hypermethylated | MCC | Deactivation of tumor suppressor genes | Harms et al. [117] |
| miRNA | Expression Status | Type of NMSC | Possible Significance | Reference |
|---|---|---|---|---|
| hsa-miR-223-3p, | Upregulated | BCC | Diagnosis | Sand et al. [145] |
| hsa-miR-197-3p, | ||||
| hsa-miR-342-3p, | ||||
| hsa-miR-505-3p, | ||||
| hsa-miR-204-5p, | ||||
| hsa-miR-941, | ||||
| hsa-miR-145-5p, | ||||
| hsa-miR-301b-3p, | ||||
| hsa-miR-452-5p, | ||||
| hsa-miR-191-5p, | ||||
| miR203 | Downregulated | BCC | Diagnosis, Therapy | Yi et al. [146] |
| miR-34a | Downregulated | BCC | Prognosis | Hu et al. [149] |
| miR-21, | Upregulated | SCC | Diagnosis | Mizrahi et al. [150], Yu et al. [151] |
| miR-205, | ||||
| miR-365, | ||||
| miR-31, | ||||
| miR-135b, | ||||
| miR-424, | ||||
| miR-320, | ||||
| miR-222 | ||||
| miR-15a, | ||||
| miR-142 | ||||
| miR-186 | ||||
| miR-20a, | Downregulated | SCC | Diagnosis | García-Sancha et al. [152] |
| miR-203, | ||||
| miR-181a, miR-125b, miR-34a, | ||||
| miR-148a, miR-214, | ||||
| miR-124, | ||||
| miR-204, | ||||
| miR-199a | ||||
| miR-205 | Upregulated | SCC | Diagnosis, Prognosis | Cañueto et al. [153], Stojadinovic et al. [154] |
| miR-221 | Upregulated | SCC | Diagnosis, therapy | Gong et al. [155] |
| miR-203 | varied | SCC | Prognosis | Cañueto et al. [153] |
| miR-20a | Varied | SCC | Prognosis | Zhang et al. [156] |
| miR-502-3p, | Upregulated | MCC | Diagnosis | Ning et al. [157] |
| miR-9, | ||||
| miR-7, | ||||
| miR-340 | ||||
| miR-182, | ||||
| miR-190b, | ||||
| miR-873, | ||||
| miR-183 | ||||
| miR-3170, | Downregulated | |||
| miR-125b, | ||||
| miR-374c | ||||
| miR-182, | Downregulated in MCPyV-negative cell line | |||
| miR-183, | ||||
| miR-190b, | ||||
| miR-340 | ||||
| miR-30a | Upregulated | MCPyV-positive MCCs | Diagnosis | Veija et al. [158] |
| miR-190b, | ||||
| miR-142-3p, | ||||
| miR-1539 | ||||
| miR-181d | MCPyV-negative MCCs | |||
| miR-375 | Upregulated | MCC | Diagnosis | Renwick et al. [159] |
| miR-30a, | Upregulated | MCC | Diagnosis | Moens group [160] |
| miR-125b, | ||||
| miR-183, | ||||
| miR-190b | ||||
| miR-375 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolouzakis, T.K.; Falzone, L.; Lasithiotakis, K.; Krüger-Krasagakis, S.; Kalogeraki, A.; Sifaki, M.; Spandidos, D.A.; Chrysos, E.; Tsatsakis, A.; Tsiaoussis, J. Current and Future Trends in Molecular Biomarkers for Diagnostic, Prognostic, and Predictive Purposes in Non-Melanoma Skin Cancer. J. Clin. Med. 2020, 9, 2868. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9092868
Nikolouzakis TK, Falzone L, Lasithiotakis K, Krüger-Krasagakis S, Kalogeraki A, Sifaki M, Spandidos DA, Chrysos E, Tsatsakis A, Tsiaoussis J. Current and Future Trends in Molecular Biomarkers for Diagnostic, Prognostic, and Predictive Purposes in Non-Melanoma Skin Cancer. Journal of Clinical Medicine. 2020; 9(9):2868. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9092868
Chicago/Turabian StyleNikolouzakis, Taxiarchis Konstantinos, Luca Falzone, Konstantinos Lasithiotakis, Sabine Krüger-Krasagakis, Alexandra Kalogeraki, Maria Sifaki, Demetrios A. Spandidos, Emmanuel Chrysos, Aristidis Tsatsakis, and John Tsiaoussis. 2020. "Current and Future Trends in Molecular Biomarkers for Diagnostic, Prognostic, and Predictive Purposes in Non-Melanoma Skin Cancer" Journal of Clinical Medicine 9, no. 9: 2868. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9092868