
Polyacrylate–Cholesterol Amphiphilic Derivative: Formulation Development and Scale-up for Health Care Applications

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PAAbCH Derivative
2.3. PAAbCH NMR Solid-State Characterization
2.4. Cell Viability Analysis
2.5. Industrial-Scale Evaluation of Solubility Enhancing Properties
2.6. Industrial-Scale Preparation of PAAbCH-Based Emulsions
2.7. Emulsion Characterization
2.8. PAAbCH Film Preparation and Characterization
2.9. PAAbCH Polyelectrolyte Gel with Chitosan
2.10. PAAbCH/Chitosan Nanosystems
3. Results and Discussion
3.1. PAA Functionalization with CH
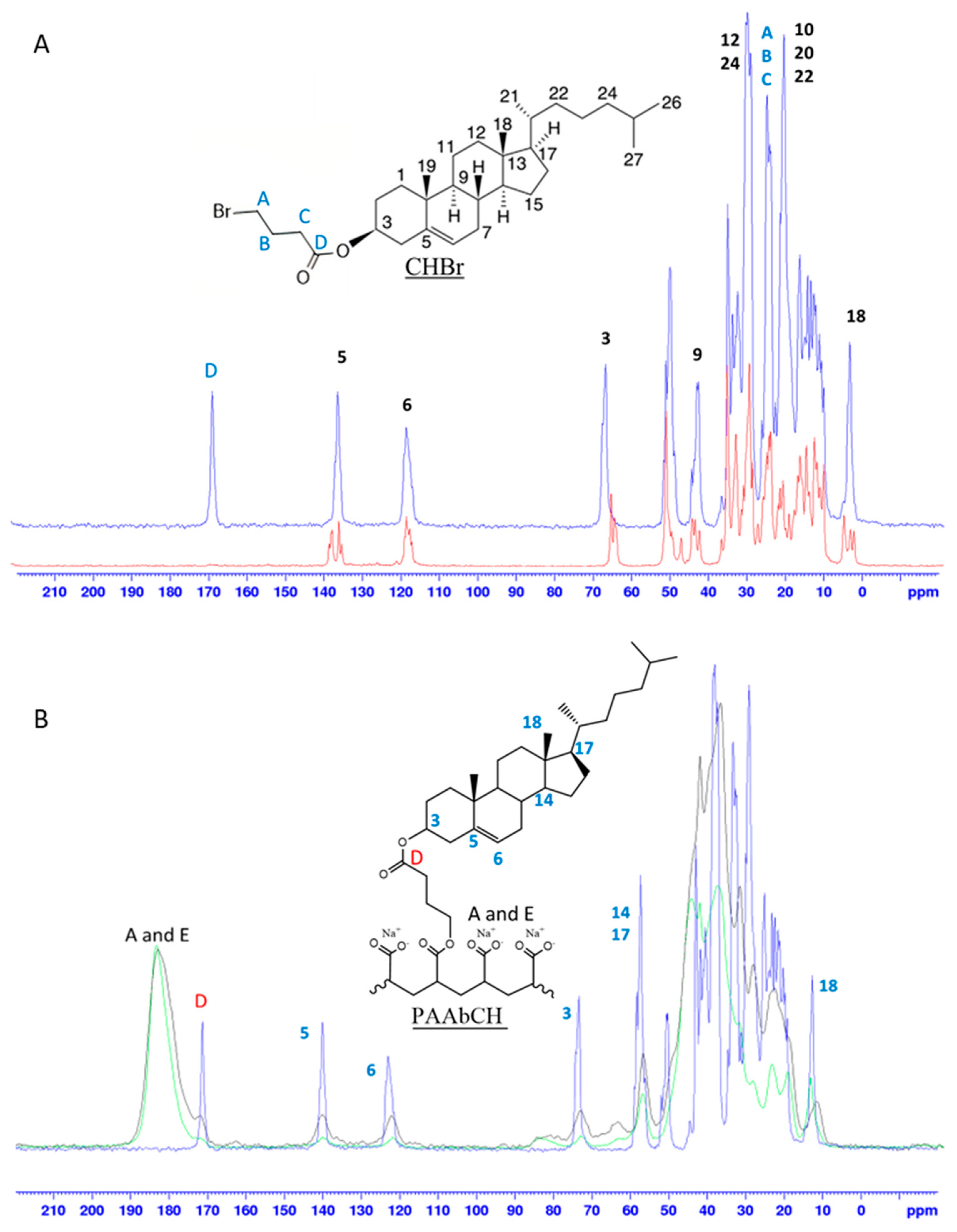
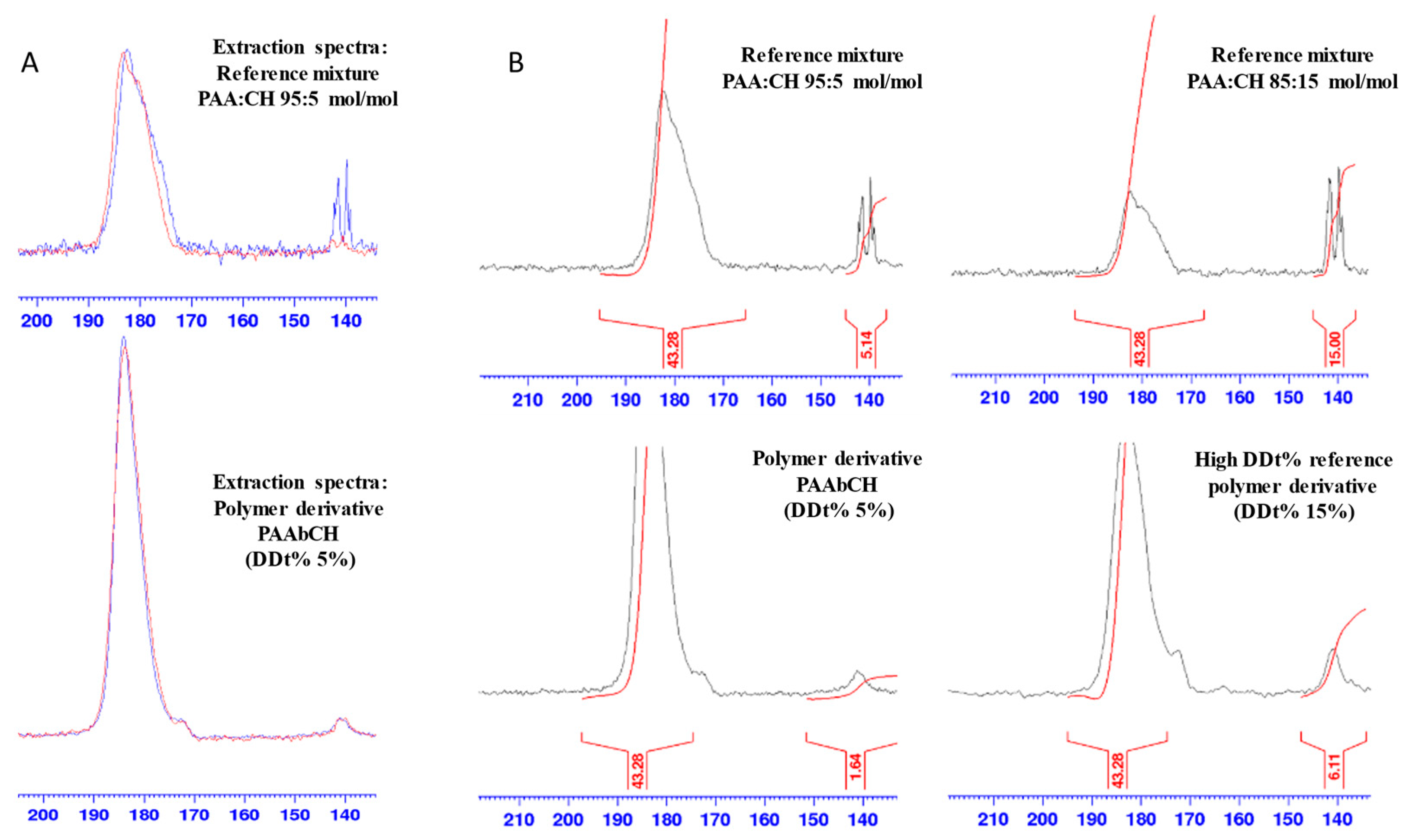
3.2. Solid-State NMR Polymer Characterization
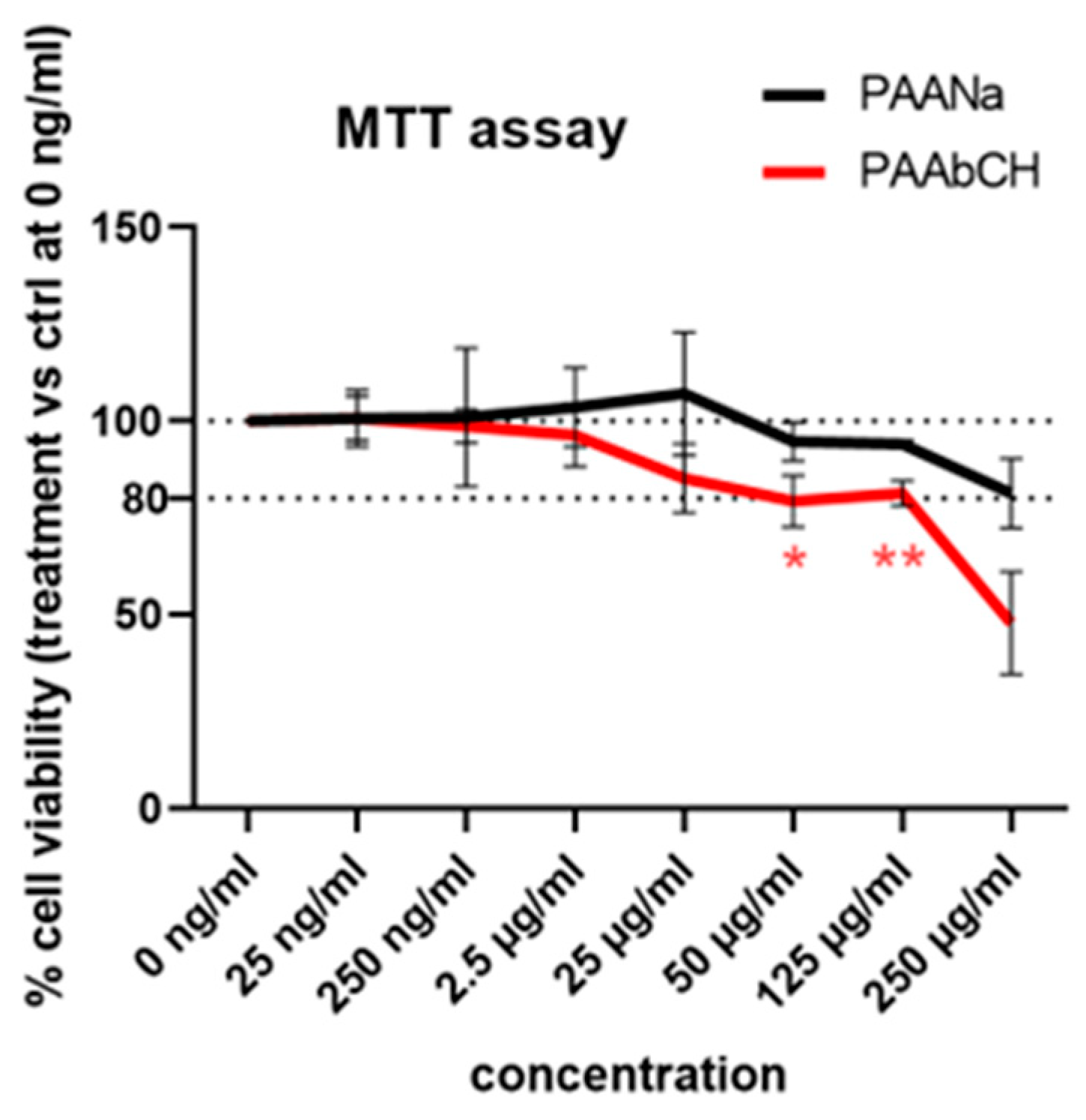
3.3. Cell Viability Analysis
3.4. Industrial Scale Evaluation of Solubility Enhancing Properties
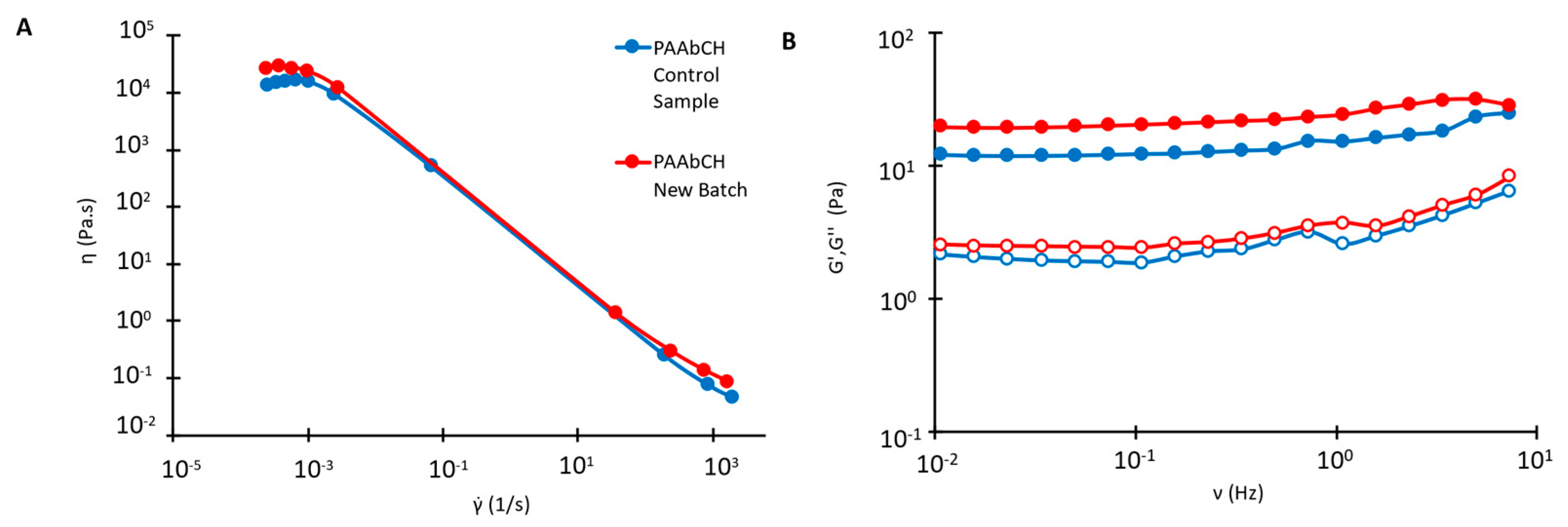
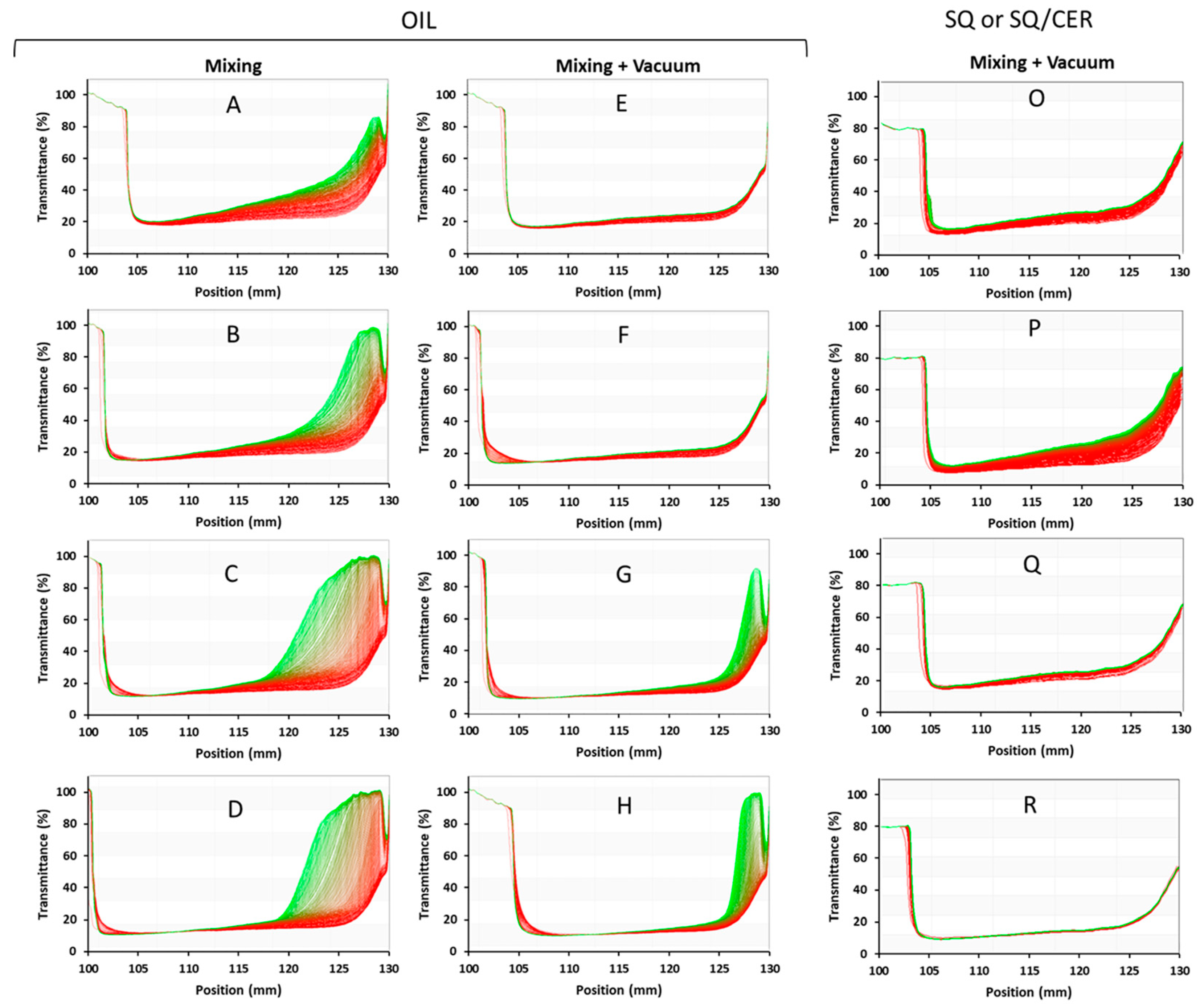
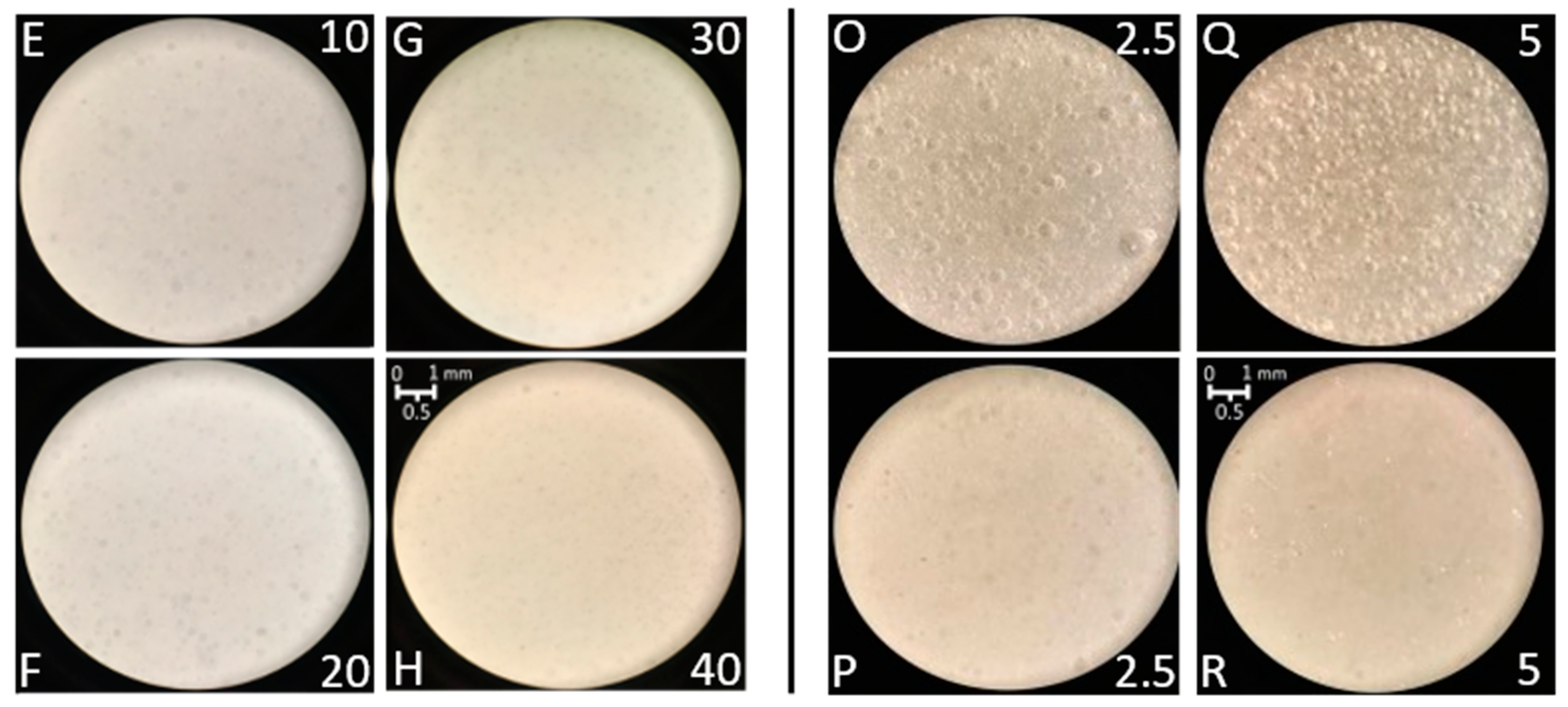
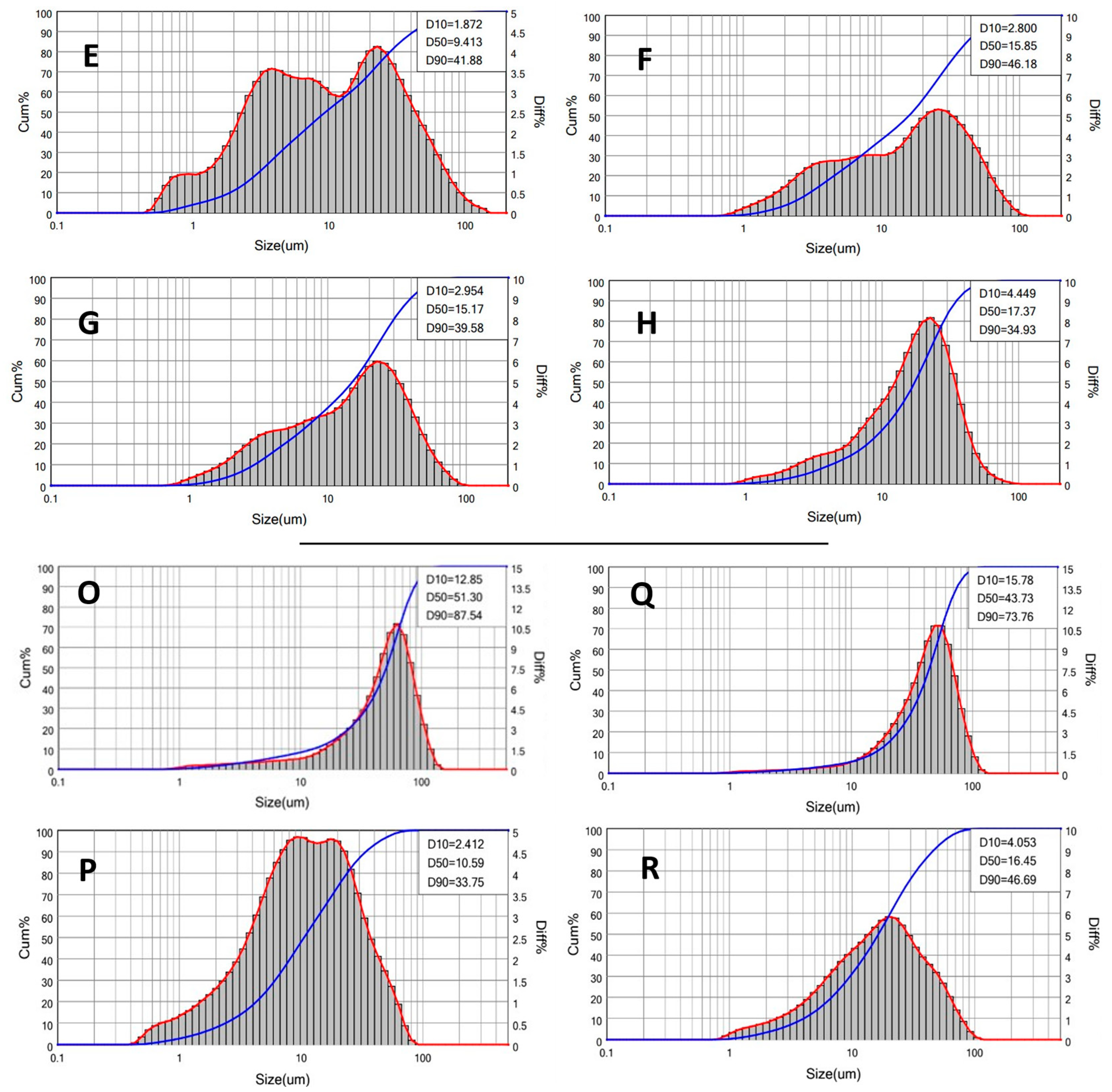
3.5. Preparation and Characterization of PAAbCH-Based Emulsions
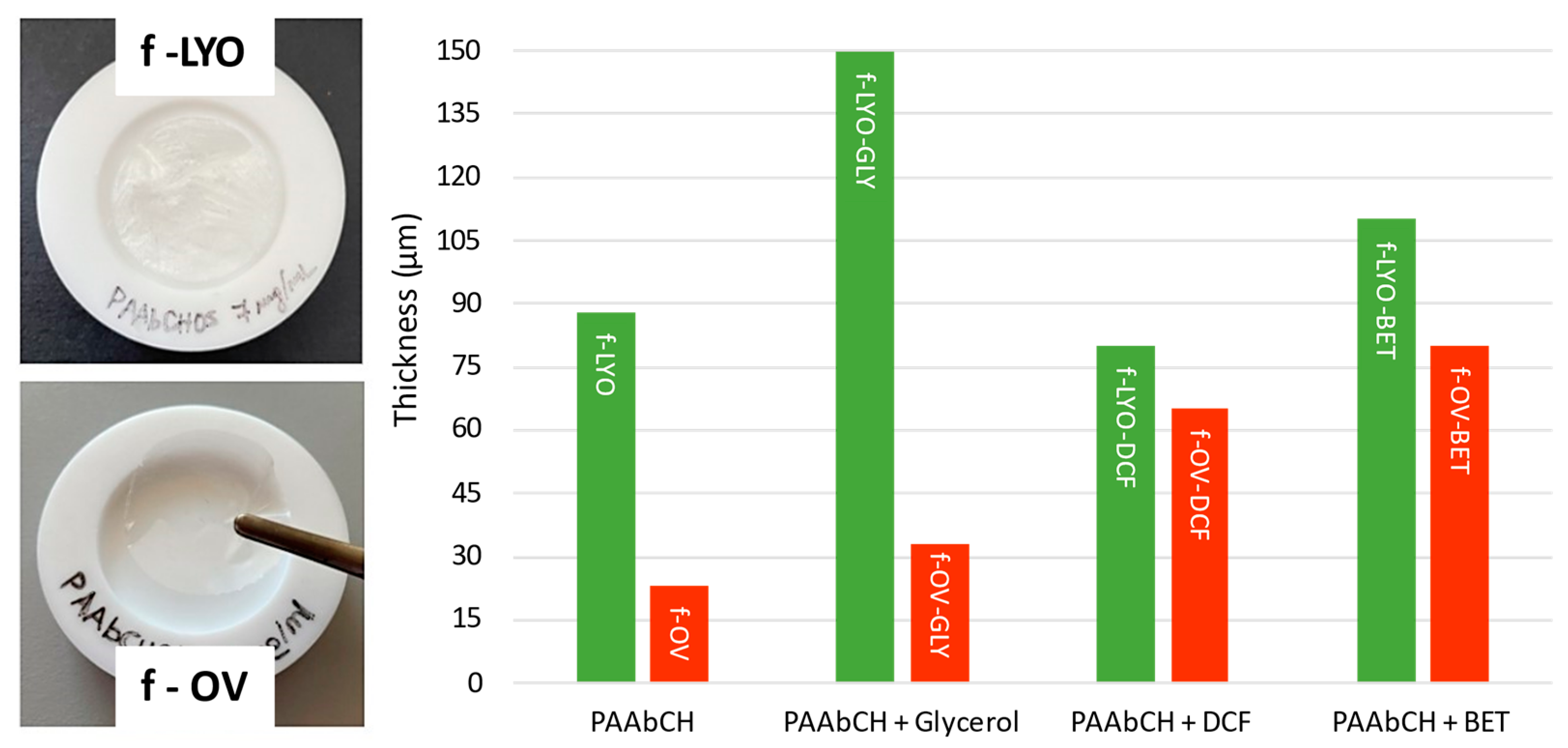
3.6. PAAbCH Film Preparation and Characterization
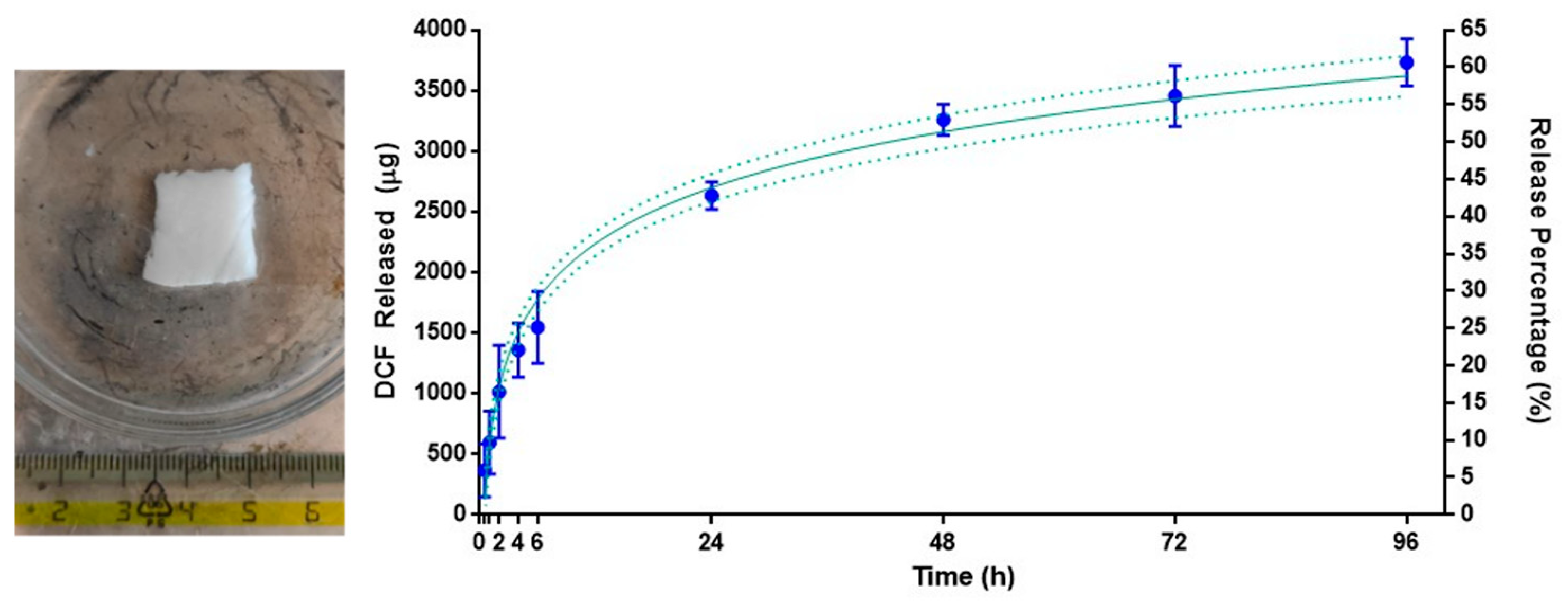
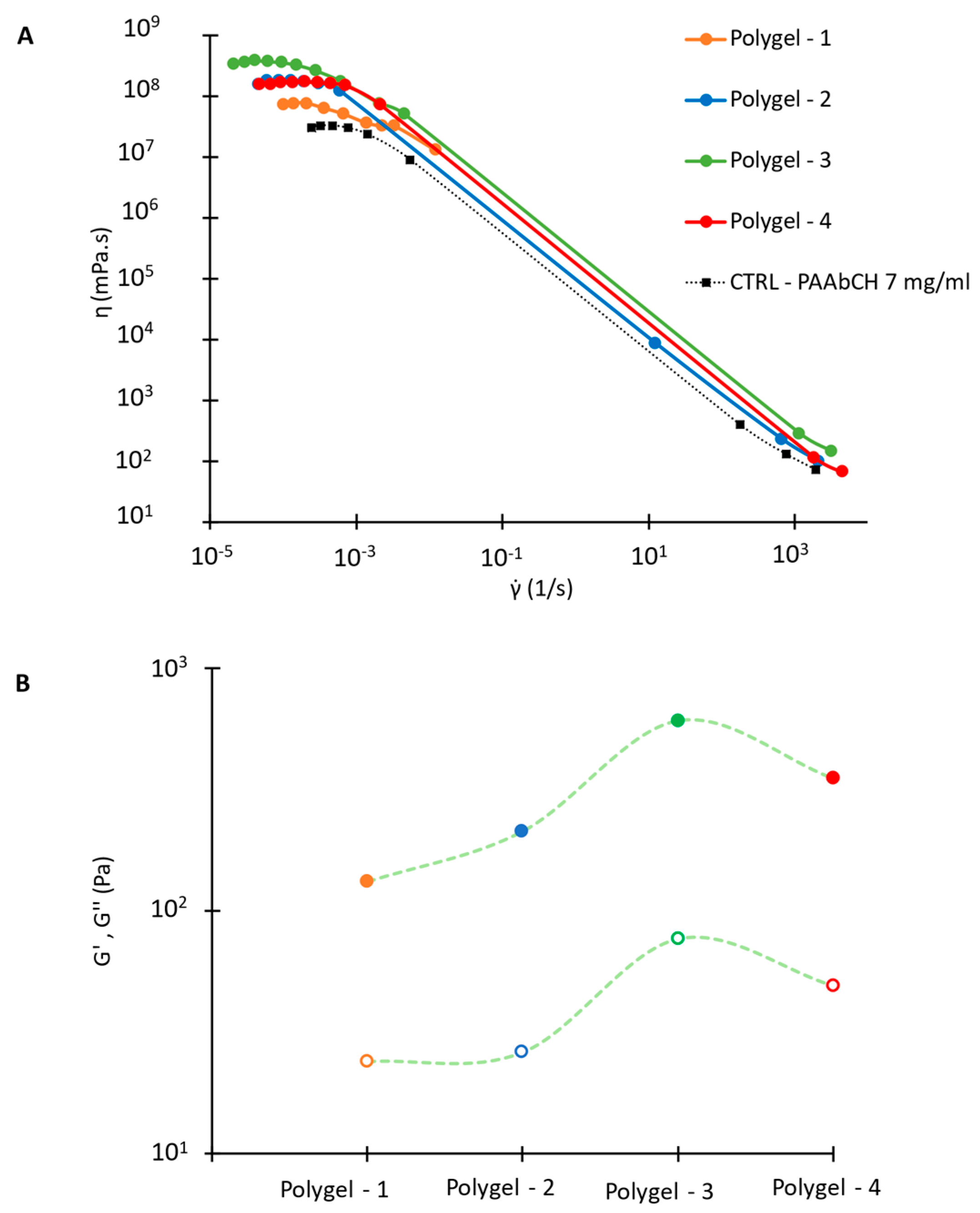
3.7. PAAbCH Polyelectrolyte Gel with Chitosan
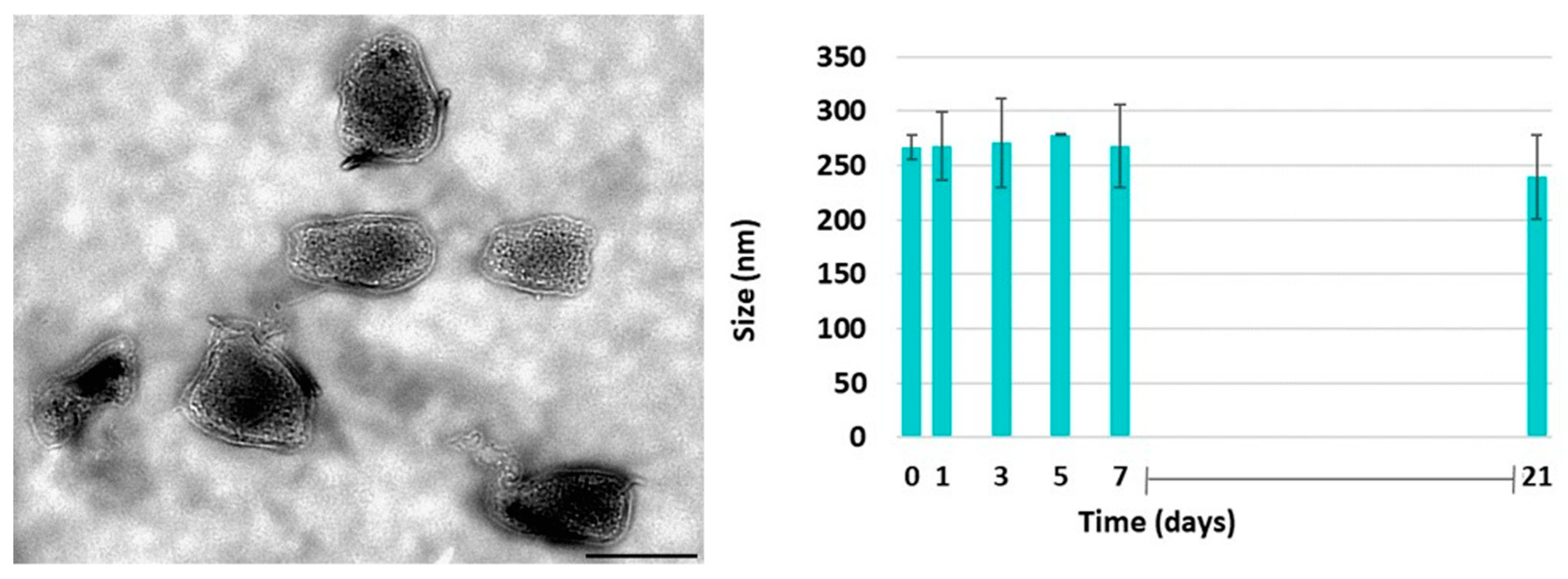
3.8. PAAbCH/Chitosan Nanosystems Preparation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ohara, T.; Sato, T.; Shimizu, N.; Prescher, G.; Schwind, H.; Weiberg, O.; Marten, K.; Greim, H.; Shaffer, T.D.; Nandi, P. Acrylic Acid and Derivatives. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2020; pp. 1–21. [Google Scholar]
- Ahmed, E.M. Hydrogel: Preparation, Characterization, and Applications: A Review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Aroca, Á.; Deb, S. Acrylic-Based Materials for Biomedical and Bioengineering Applications. In Acrylate Polymers for Advanced Applications; IntechOpen: London, UK, 2020; Volume 1, pp. 5–15. [Google Scholar]
- Punia, K.; Punia, A.; Chatterjee, K.; Mukherjee, S.; Fata, J.; Banerjee, P.; Raja, K.; Yang, N.L. Rapid Bactericidal Activity of an Amphiphilic Polyacrylate Terpolymer System Comprised of Same-Centered Comonomers with 2-Carbon and 6-Carbon Spacer Arms and an Uncharged Repeat Unit. RSC Adv. 2017, 7, 10192–10199. [Google Scholar] [CrossRef]
- Gratzl, G.; Paulik, C.; Hild, S.; Guggenbichler, J.P.; Lackner, M. Antimicrobial Activity of Poly(Acrylic Acid) Block Copolymers. Mater. Sci. Eng. C 2014, 38, 94–100. [Google Scholar] [CrossRef]
- De Somer, P.; De Clercq, E.; Billiau, A.; Schonne, E.; Claesen, M. Antiviral Activity of Polyacrylic and Polymethacrylic Acids—Part I. J. Virol. 1968, 2, 878–885. [Google Scholar] [CrossRef] [PubMed]
- De Somer, P.; De Clercq, E.; BIlliau, A.; Schonne, E.; Claesen, M. Antiviral Activity of Polyacrylic and Polymethacrylic Acids—Part II. J. Virol. 1968, 2, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Arkaban, H.; Barani, M.; Akbarizadeh, M.R.; Chauhan, N.P.S.; Jadoun, S.; Soltani, M.D.; Zarrintaj, P. Polyacrylic Acid Nanoplatforms: Antimicrobial, Tissue Engineering, and Cancer Theranostic Applications. Polymers 2022, 14, 1259. [Google Scholar] [CrossRef]
- Zohuriaan-Mehr, M.J.; Kabiri, K. Superabsorbent Polymer Materials: A Review. Iran. Polym. J. 2008, 17, 451–477. [Google Scholar]
- Qureshi, M.A.; Nishat, N.; Jadoun, S.; Ansari, M.Z. Polysaccharide Based Superabsorbent Hydrogels and Their Methods of Synthesis: A Review. Carbohydr. Polym. Technol. Appl. 2020, 1, 100014. [Google Scholar] [CrossRef]
- Dalei, G.; Das, S. Polyacrylic Acid-Based Drug Delivery Systems: A Comprehensive Review on the State-of-Art. J. Drug Deliv. Sci. Technol. 2022, 78, 103988. [Google Scholar] [CrossRef]
- Gaytán, I.; Burelo, M.; Loza-Tavera, H. Current Status on the Biodegradability of Acrylic Polymers: Microorganisms, Enzymes and Metabolic Pathways Involved. Appl. Microbiol. Biotechnol. 2021, 105, 991–1006. [Google Scholar] [CrossRef]
- The Cosmetic Ingredient Review Expert Panel, Annual Review of Cosmetic Ingredient Safety Assessments-2004/2005. Int. J. Toxicol. 2006, 25 (Suppl. 2), 1–89. [CrossRef] [PubMed]
- The Cosmetic Ingredient Review Expert Panel, Final Report on the Safety Assessment of Cholesterol. J. Am. Coll. Toxicol. 1986, 5, 491–516. [CrossRef]
- Loomis, C.R.; Shipley, G.G.; Small, D.M. The Phase Behavior of Hydrated Cholesterol. J. Lipid Res. 1979, 20, 525–535. [Google Scholar] [CrossRef]
- Saad, H.Y.; Higuchi, W.I. Water Solubility of Cholesterol. J. Pharm. Sci. 1965, 54, 1205–1206. [Google Scholar] [CrossRef] [PubMed]
- Renshaw, P.F.; Janoff, A.S.; Miller, K.W. On the Nature of Dilute Aqueous Cholesterol Suspensions. J. Lipid Res. 1983, 24, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Róg, T.; Pasenkiewicz-Gierula, M.; Vattulainen, I.; Karttunen, M. Ordering Effects of Cholesterol and Its Analogues. Biochim. Biophys. Acta Biomembr. 2009, 1788, 97–121. [Google Scholar] [CrossRef]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and Regulation of Cholesterol Homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef]
- Zampelas, A.; Magriplis, E. New Insights into Cholesterol Functions: A Friend or an Enemy? Nutrients 2019, 11, 1645. [Google Scholar] [CrossRef]
- Schmid-Wendtner, M.H.; Korting, H.C. The PH of the Skin Surface and Its Impact on the Barrier Function. Skin. Pharmacol. Physiol. 2006, 19, 296–302. [Google Scholar] [CrossRef]
- Camera, E.; Ludovici, M.; Galante, M.; Sinagra, J.L.; Picardo, M. Comprehensive Analysis of the Major Lipid Classes in Sebum by Rapid Resolution High-Performance Liquid Chromatography and Electrospray Mass Spectrometry. J. Lipid Res. 2010, 51, 3377–3388. [Google Scholar] [CrossRef]
- Smith, K.R.; Thiboutot, D.M. Thematic Review Series: Skin Lipids. Sebaceous Gland Lipids: Friend or Foe? J. Lipid Res. 2008, 49, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Murota, H.; Itoi, S.; Terao, M.; Matsui, S.; Kawai, H.; Satou, Y.; Suda, K.; Katayama, I. Topical Cholesterol Treatment Ameliorates Hapten-Evoked Cutaneous Hypersensitivity by Sustaining Expression of 11β-HSD1 in Epidermis. Exp. Dermatol. 2014, 23, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Vaz, W.L.C. Model Systems, Lipid Rafts, and Cell Membranes. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 269–295. [Google Scholar] [CrossRef]
- Hosta-Rigau, L.; Zhang, Y.; Teo, B.M.; Postma, A.; Städler, B. Cholesterol—A Biological Compound as a Building Block in Bionanotechnology. Nanoscale 2013, 5, 89–109. [Google Scholar] [CrossRef] [PubMed]
- Suksiriworapong, J.; Achayawat, C.; Juangrattanakamjorn, P.; Taresco, V.; Crucitti, V.C.; Sakchaisri, K.; Bunsupa, S. Modification of Poly(Glycerol Adipate) with Tocopherol and Cholesterol Modulating Nanoparticle Self-Assemblies and Cellular Responses of Triple-Negative Breast Cancer Cells to SN-38 Delivery. Pharmaceutics 2023, 15, 2100. [Google Scholar] [CrossRef]
- Abesekara, M.S.; Chau, Y. Recent Advances in Surface Modification of Micro- and Nano-Scale Biomaterials with Biological Membranes and Biomolecules. Front. Bioeng. Biotechnol. 2022, 10, 972790. [Google Scholar] [CrossRef]
- Ruwizhi, N.; Aderibigbe, B.A. The Efficacy of Cholesterol-Based Carriers in Drug Delivery. Molecules 2020, 25, 4330. [Google Scholar] [CrossRef]
- Kathe, K.; Kathpalia, H. Film Forming Systems for Topical and Transdermal Drug Delivery. Asian J. Pharm. Sci. 2017, 12, 487–497. [Google Scholar] [CrossRef]
- Pünnel, L.C.; Lunter, D.J. Film-Forming Systems for Dermal Drug Delivery. Pharmaceutics 2021, 13, 932. [Google Scholar] [CrossRef]
- Viola, M.; Migliorini, C.; Matricardi, P.; Di Meo, C. Synthesis and Characterization of a Novel Amphiphilic Polyacrylate-Cholesterol Derivative as Promising Material for Pharmaceutical and Cosmetic Applications. Eur. Polym. J. 2023, 184, 111774. [Google Scholar] [CrossRef]
- Chang, R.K.; Raw, A.; Lionberger, R.; Yu, L. Generic Development of Topical Dermatologic Products: Formulation Development, Process Development, and Testing of Topical Dermatologic Products. AAPS J. 2013, 15, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Mugglestone, C.J.; Mariz, S.; Lane, M.E. The Development and Registration of Topical Pharmaceuticals. Int. J. Pharm. 2012, 435, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Kesharwani, P.; Taurin, S.; Greish, K. Theory and Applications of Nonparenteral Nanomedicines; Kesharwani, P., Taurin, S., Greish, K., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2020; Volume 1, ISBN 978-012820466-5/978-012820467-2. [Google Scholar]
- Zoratto, N.; Forcina, L.; Matassa, R.; Mosca, L.; Familiari, G.; Musarò, A.; Mattei, M.; Coviello, T.; Di Meo, C.; Matricardi, P. Hyaluronan-Cholesterol Nanogels for the Enhancement of the Ocular Delivery of Therapeutics. Pharmaceutics 2021, 13, 1781. [Google Scholar] [CrossRef] [PubMed]
- D’Arrigo, G.; di Meo, C.; Gaucci, E.; Chichiarelli, S.; Coviello, T.; Capitani, D.; Alhaique, F.; Matricardi, P. Self-Assembled Gellan-Based Nanohydrogels as a Tool for Prednisolone Delivery. Soft Matter 2012, 8, 11557–11564. [Google Scholar] [CrossRef]
- Montanari, E.; Capece, S.; di Meo, C.; Meringolo, M.; Coviello, T.; Agostinelli, E.; Matricardi, P. Hyaluronic Acid Nanohydrogels as a Useful Tool for BSAO Immobilization in the Treatment of Melanoma Cancer Cells. Macromol. Biosci. 2013, 13, 1185–1194. [Google Scholar] [CrossRef]
- Paoletti, L.; Zoratto, N.; Benvenuto, M.; Nardozi, D.; Angiolini, V.; Mancini, P.; Masuelli, L.; Bei, R.; Frajese, G.V.; Matricardi, P.; et al. Hyaluronan-Estradiol Nanogels as Potential Drug Carriers to Target ER+ Breast Cancer Cell Line. Carbohydr. Polym. 2023, 314, 120900. [Google Scholar] [CrossRef] [PubMed]
- El Hariri El Nokab, M.; van der Wel, P.C.A. Use of Solid-State NMR Spectroscopy for Investigating Polysaccharide-Based Hydrogels: A Review. Carbohydr. Polym. 2020, 240, 116276. [Google Scholar] [CrossRef]
- Cheng, H.N.; English, A.D. Advances in the NMR Spectroscopy of Polymers: An Overview. In NMR Spectroscopy of Polymers in Solution and in the Solid State; American Chemical Society: Washington, DC, USA, 2003; Volume 1, pp. 3–20. ISBN 9780841238084. [Google Scholar]
- Zhang, R.; Miyoshi, T.; Sun, P. NMR Methods for the Characterization of Synthetic and Natural Polymers; Royal Society of Chemistry: London, UK, 2019; Volume 1, ISBN 978-1-78801-400-7. [Google Scholar]
- Reif, B.; Ashbrook, S.E.; Emsley, L.; Hong, M. Solid-State NMR Spectroscopy. Nat. Rev. Methods Prim. 2021, 1, 2. [Google Scholar] [CrossRef]
- Della Ripa, L.A.; Petros, Z.A.; Cioffi, A.G.; Piehl, D.W.; Courtney, J.M.; Burke, M.D.; Rienstra, C.M. Solid-State NMR of Highly 13C-Enriched Cholesterol in Lipid Bilayers. Methods 2018, 138–139, 47–53. [Google Scholar] [CrossRef]
- Johnson, R.L. Schmidt-Rohr Klaus Quantitative Solid-State 13C NMR with Signal Enhancement. J. Magn. Reson. 2014, 239, 44–49. [Google Scholar] [CrossRef]
- Rusanov, A.L.; Luzgina, N.G.; Lisitsa, A.V. Sodium Dodecyl Sulfate Cytotoxicity towards HaCaT Keratinocytes: Comparative Analysis of Methods for Evaluation of Cell Viability. Bull. Exp. Biol. Med. 2017, 163, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, R.Y.; Rulison, C.J. An Investigation of the Mechanism by Which Hydrophobically Modified Hydrophilic Polymers Act as Primary Emulsifiers for Oil-in-Water Emulsions. Colloids Surf. A Physicochem. Eng. Asp. 1994, 88, 27–32. [Google Scholar] [CrossRef]
- Medina-Torres, L.; Calderas, F.; Sanchez-Olivares, G.; Nuñez-Ramirez, D.M. Rheology of Sodium Polyacrylate as an Emulsifier Employed in Cosmetic Emulsions. Ind. Eng. Chem. Res. 2014, 53, 18346–18351. [Google Scholar] [CrossRef]
- Narayan Bhilwade, H.; Tatewaki, N.; Nishida, H.; Konishi, T. Squalene as Novel Food Factor. Curr. Pharm. Biotechnol. 2010, 11, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.M.; Lee, B.M. Safety and Risk Assessment of Ceramide 3 in Cosmetic Products. Food Chem. Toxicol. 2015, 84, 8–17. [Google Scholar] [CrossRef]
- Sobisch, T.; Lerche, D. Thickener Performance Traced by Multisample Analytical Centrifugation. Colloids Surf. A Physicochem. Eng. Asp. 2008, 331, 114–118. [Google Scholar] [CrossRef]
- Lerche, D.; Sobisch, T. Direct and Accelerated Characterization of Formulation Stability. J. Dispers. Sci. Technol. 2011, 32, 1799–1811. [Google Scholar] [CrossRef]
- Frederiksen, K.; Guy, R.H.; Petersson, K. Formulation Considerations in the Design of Topical, Polymeric Film-Forming Systems for Sustained Drug Delivery to the Skin. Eur. J. Pharm. Biopharm. 2015, 91, 9–15. [Google Scholar] [CrossRef]
- Hou, Z.; Wen, Q.; Zhou, W.; Yan, P.; Zhang, H.; Ding, J. Topical Delivery of Ketorolac Tromethamine via Cataplasm for Inflammatory Pain Therapy. Pharmaceutics 2023, 15, 1405. [Google Scholar] [CrossRef]
- Nguyen, D.C.; Dang, Q.A.; Nguyen, T.D.; Bui, V.T.; Chi, S.C.; Do, Q.; Tung, N.T. DoE-Based Formulation, Physicochemical Properties, and Anti-Inflammatory Investigation of a Topical Patch Preparing by Partially Neutralized Polyacrylate-Based Adhesive Hydrogel. Mater. Today Commun. 2023, 35, 105606. [Google Scholar] [CrossRef]
- Gong, L.; Xiang, L.; Zhang, J.; Chen, J.; Zeng, H. Fundamentals and Advances in the Adhesion of Polymer Surfaces and Thin Films. Langmuir 2019, 35, 15914–15936. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.K.; Singh, L.; Rastogi, V. Complexation of Oppositely Charged Polymer: Novel Approaches for Drug Delivery. Curr. Drug Deliv. 2021, 18, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Cazorla-Luna, R.; Martín-Illana, A.; Notario-Pérez, F.; Ruiz-Caro, R.; Veiga, M.D. Naturally Occurring Polyelectrolytes and Their Use for the Development of Complex-Based Mucoadhesive Drug Delivery Systems: An Overview. Polymers 2021, 13, 2241. [Google Scholar] [CrossRef] [PubMed]
- Alyamani, A.A.; Al-Musawi, M.H.; Albukhaty, S.; Sulaiman, G.M.; Ibrahim, K.M.; Ahmed, E.M.; Jabir, M.S.; Al-Karagoly, H.; Aljahmany, A.A.; Mohammed, M.K.A. Electrospun Polycaprolactone/Chitosan Nanofibers Containing Cordia Myxa Fruit Extract as Potential Biocompatible Antibacterial Wound Dressings. Molecules 2023, 28, 2501. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.A.; Azharuddin, S.M.; Ali, S.; Patil, C.C. Studies on Different Chitosan Polyelectrolyte Complex Hydrogels for Modified Release of Diltiazem Hyrochloride. Int. J. Pharm. Pharm. Sci. 2010, 2, 6467–6479. [Google Scholar]
- Moustafine, R.I.; Margulis, E.B.; Sibgatullina, L.F.; Kemenova, V.A.; Mooter, G. van den Comparative Evaluation of Interpolyelectrolyte Complexes of Chitosan with Eudragit® L100 and Eudragit® L100-55 as Potential Carriers for Oral Controlled Drug Delivery. Eur. J. Pharm. Biopharm. 2008, 70, 215–225. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, J.; Jiang, S.; Wang, Y.; Zhang, X.; Ding, J.; Yu, T.; Mao, S. Insights into the Mechanisms of Chitosan-Anionic Polymers-Based Matrix Tablets for Extended Drug Release. Int. J. Pharm. 2014, 476, 253–265. [Google Scholar] [CrossRef]
- He, M.; Shi, L.; Wang, G.; Cheng, Z.; Han, L.; Zhang, X.; Wang, C.; Wang, J.; Zhou, P.; Wang, G. Biocompatible and Biodegradable Chitosan/Sodium Polyacrylate Polyelectrolyte Complex Hydrogels with Smart Responsiveness. Int. J. Biol. Macromol. 2020, 155, 1245–1251. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ambient Pressure | Reduced Pressure | Water: Oil (% v/v) | |||
| PolEm-A | PolEm-E | 90:10 | |||
| PolEm-B | PolEm-F | 80:20 | |||
| PolEm-C | PolEm-G | 70:30 | |||
| PolEm-D | PolEm-H | 60:40 | |||
| Ambient Pressure | PAAbCH (mg/mL) | CER | Reduced Pressure | PAAbCH (mg/mL) | CER |
| PolSQ-I | 2.5 | n | PolSQ-O | 2.5 | n |
| PolSQ-L | 2.5 | y | PolSQ-P | 2.5 | y |
| PolSQ-M | 5 | n | PolSQ-Q | 5 | n |
| PolSQ-N | 5 | y | PolSQ-R | 5 | y |
| Samples | FREEZE-DRIED | Oven-Dried |
|---|---|---|
| PAAbCH | f-LYO | f-OV |
| PAAbCH + Glycerol | f-LYO-GLY | f-OV-GLY |
| PAAbCH + DCF | f-LYO-DCF | f-OV-DCF |
| PAAbCH + BET | f-LYO-BET | f-OV-BET |
| Label | Chit: PAAbCH Ratio | Chit (mg) | PAAbCH (mg) |
|---|---|---|---|
| Polygel-1 | 1:1 | 14 | 14 |
| Polygel-2 | 1:2 | 9.3 | 18.7 |
| Polygel-3 | 1:3 | 7.0 | 21.0 |
| Polygel-4 | 1:4 | 5.6 | 22.4 |
| Label | Chit: PAAbCH Ratio | Chit (mg) | PAAbCH (mg) |
|---|---|---|---|
| Nano-1 | 3:2 | 1.2 | 0.8 |
| Nano-2 | 1:1 | 1 | 1 |
| Nano-3 | 2:3 | 0.8 | 1.2 |
| Derivatives | DDt% | DDr% by De-Esterification | DDr% by NMR |
|---|---|---|---|
| PAAbCH | 5 | 1.8 ± 0.2 | 1.6 |
| PAAbCH15 | 15 | 5.9 ± 0.2 | 6.1 |
| Mixing Method | PIR Amount (mg/mL) | PAAbCH Concentration (mg/mL) | PIR SP (%) | PIR Solubilized (µg/mL) | SE |
| Planetary mixer | 1 (powder) | 3 | 29 ± 6 | 285 ± 70 | 12× |
| Magnetic stirring [32] | 1 (film) | 3 | 53 ± 11 | 528 ± 106 | 23× |
| Mixing Method | BET Amount (mg/mL) | PAAbCH Concentration (mg/mL) | BET SP (%) | BET Solubilized (µg/mL) | SE |
| Planetary mixer | 1 (powder) | 3 | 81 ± 4 | 814 ± 32 | 116× |
| Magnetic stirring [32] | 1 (film) | 3 | 57 ± 8 | 568 ± 81 | 81× |
| Samples PolEM | Oil v/v% | D10 | D50 | D90 | Span Index |
| E | 10 | 1.87 | 9.41 | 41.88 | 4.25 |
| F | 20 | 2.8 | 15.85 | 46.18 | 2.73 |
| G | 30 | 2.95 | 15.17 | 39.58 | 2.41 |
| H | 40 | 4.45 | 17.37 | 34.93 | 1.75 |
| Samples PolSQ | SQ or SQ/CER | D10 | D50 | D90 | Span Index |
| O | SQ | 12.85 | 51.3 | 87.54 | 1.46 |
| P | SQ/CER | 2.41 | 10.59 | 33.75 | 2.96 |
| Q | SQ | 15.78 | 43.73 | 73.76 | 1.33 |
| R | SQ/CER | 4.05 | 16.45 | 46.69 | 2.59 |
| Nano-1 | Nano-2 | Nano-3 | ||||
|---|---|---|---|---|---|---|
| Chit: PAAbCH Ratio | 3:2 | 1:1 | 2:3 | |||
| Size (nm) | #1 100% | 267 ± 11 | #1 93% | 817 ± 61 | #1 90% | 915 ± 175 |
| #2 7% | 125 ± 50 | #2 10% | 1.7 × 103 | |||
| Count rate | 365 | 500 | 579 | |||
| PDI | 0.002 | - | - | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viola, M.; Migliorini, C.; Ziarelli, F.; Viel, S.; Cencetti, C.; Di Risola, D.; Mosca, L.; Masuelli, L.; Matricardi, P.; Di Meo, C. Polyacrylate–Cholesterol Amphiphilic Derivative: Formulation Development and Scale-up for Health Care Applications. J. Funct. Biomater. 2023, 14, 482. https://0-doi-org.brum.beds.ac.uk/10.3390/jfb14090482
Viola M, Migliorini C, Ziarelli F, Viel S, Cencetti C, Di Risola D, Mosca L, Masuelli L, Matricardi P, Di Meo C. Polyacrylate–Cholesterol Amphiphilic Derivative: Formulation Development and Scale-up for Health Care Applications. Journal of Functional Biomaterials. 2023; 14(9):482. https://0-doi-org.brum.beds.ac.uk/10.3390/jfb14090482
Chicago/Turabian StyleViola, Marco, Claudia Migliorini, Fabio Ziarelli, Stéphane Viel, Claudia Cencetti, Daniel Di Risola, Luciana Mosca, Laura Masuelli, Pietro Matricardi, and Chiara Di Meo. 2023. "Polyacrylate–Cholesterol Amphiphilic Derivative: Formulation Development and Scale-up for Health Care Applications" Journal of Functional Biomaterials 14, no. 9: 482. https://0-doi-org.brum.beds.ac.uk/10.3390/jfb14090482