
Whole Genome Sequencing and Annotation of Naematelia aurantialba (Basidiomycota, Edible-Medicinal Fungi)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strains and Strain Culture
2.2. Extraction of Genome DNA
2.3. De Novo Sequencing and Genome Assembly
2.3.1. De Novo Sequencing
2.3.2. Genome Assembly and Assessment
2.4. Genome Component Prezdiction
2.5. Genome Annotation
2.6. Comparative Genomics Analysis
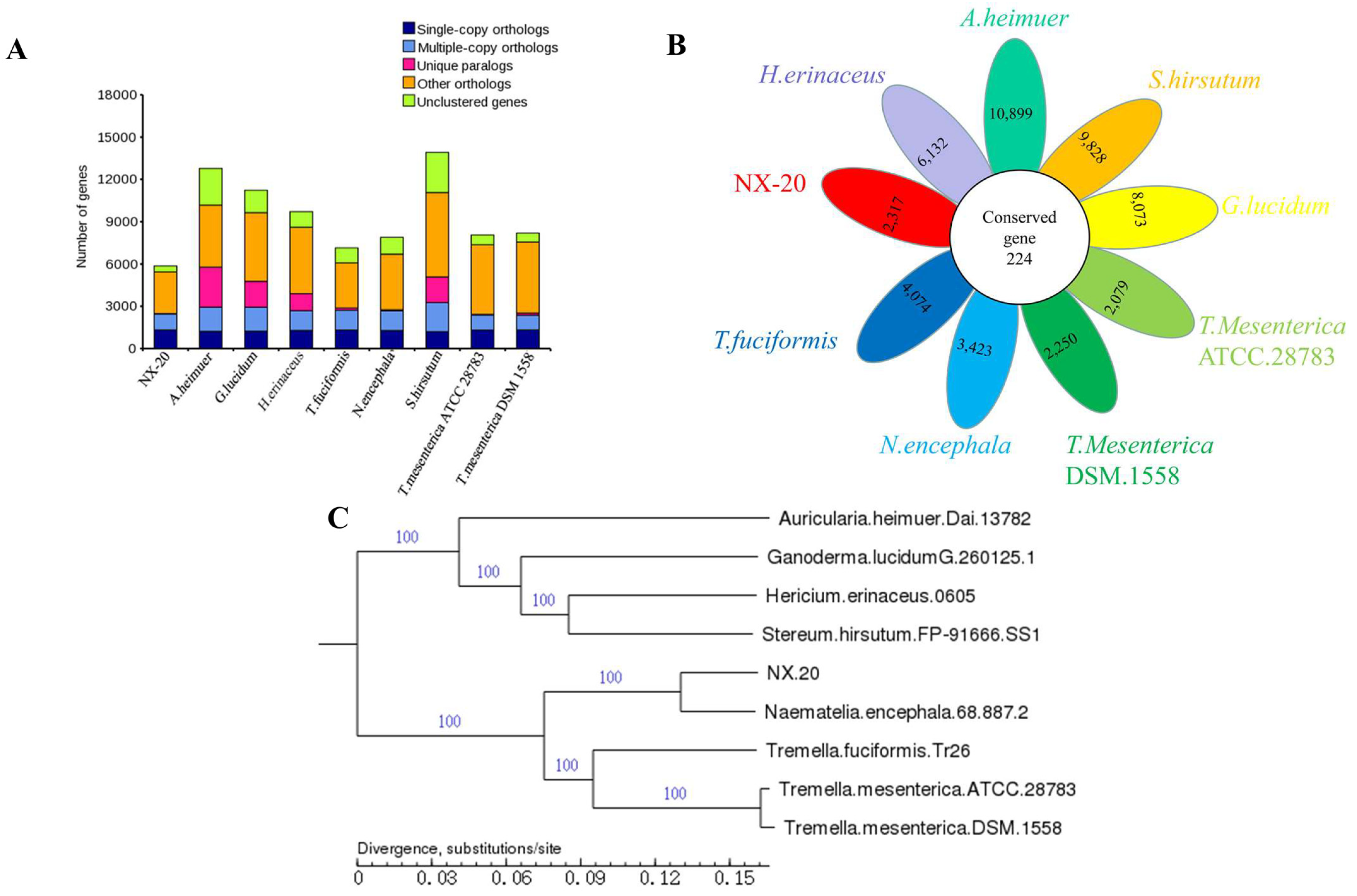
2.6.1. Core-Pan Genome, Phylogenetic, and Gene Family Analysis
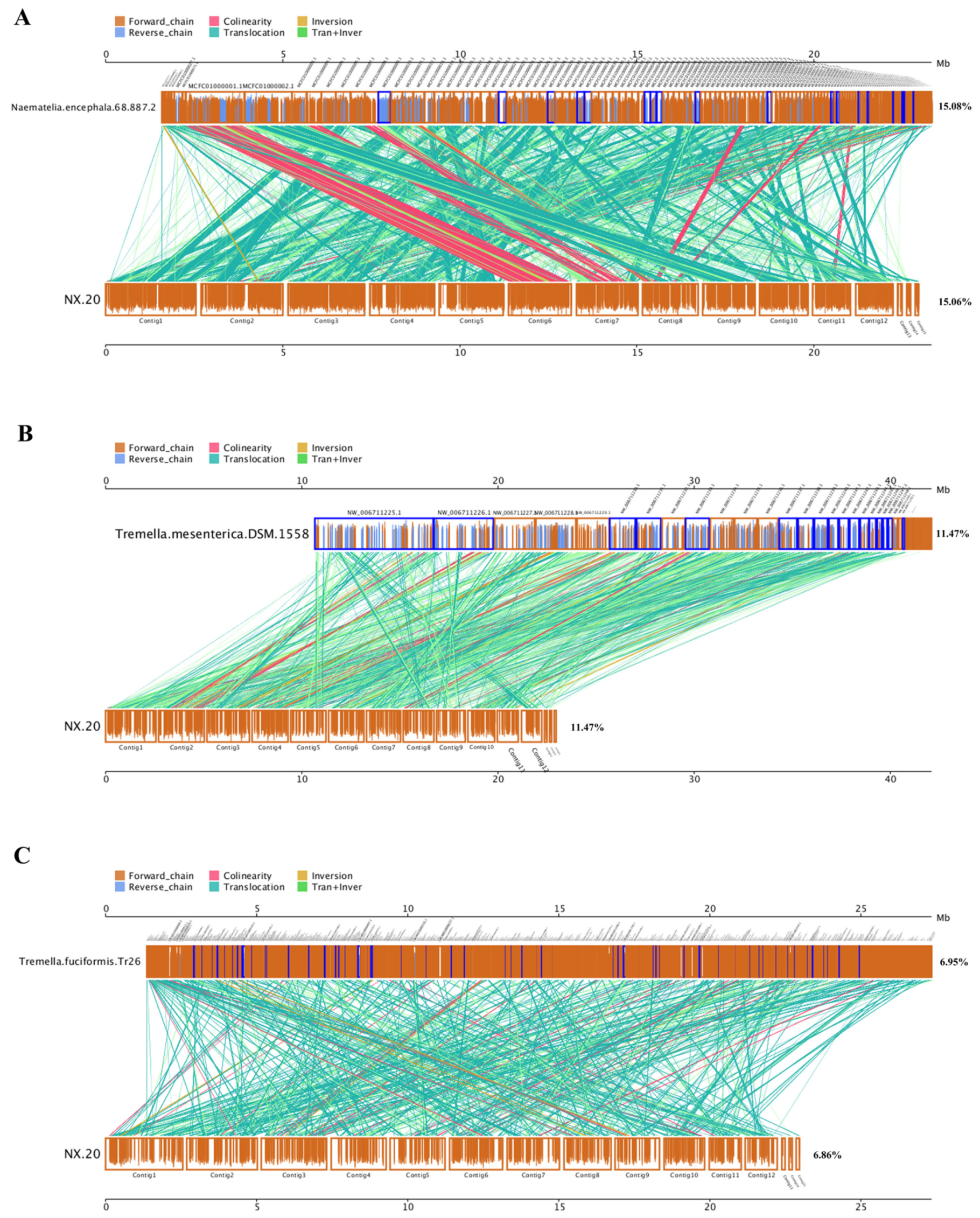
2.6.2. Genomic Synteny
2.7. Other Basidiomycete Genome Sources
3. Results and Discussion
3.1. Sequencing and Assembly Data
3.2. Genomic Features
3.3. Repeat Sequence
3.4. Noncoding RNA
3.5. Gene Function Annotation
3.5.1. KOG Annotations
3.5.2. GO Annotations
3.5.3. KEGG Annotations
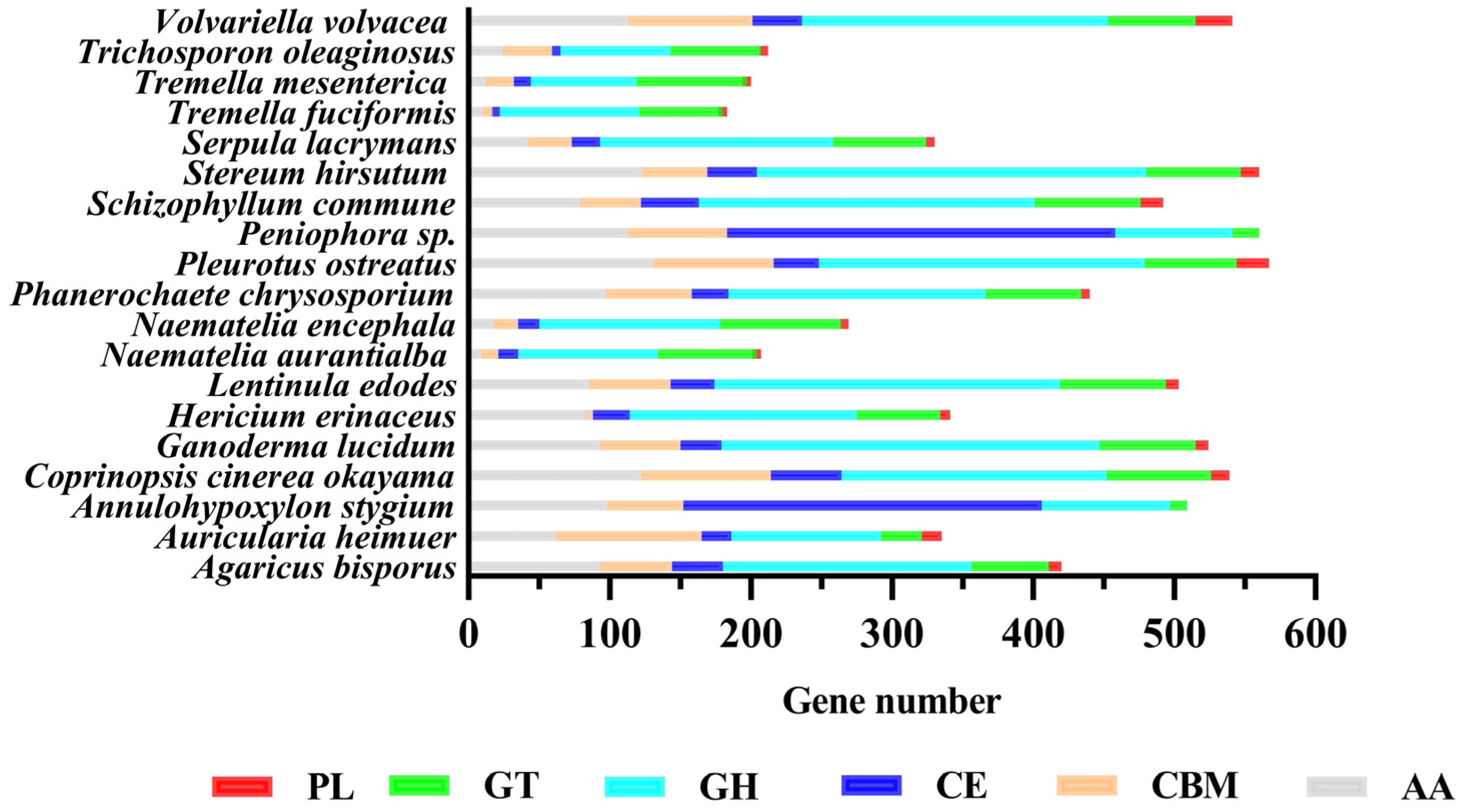
3.5.4. CAZymes
3.5.5. The Cytochromes P450 (CYPs) Family
3.6. Secondary Metabolites
3.7. Synthesis of Polysaccharides
3.7.1. EPS
Synthesis of Nucleotide-Activated Sugars
Linking and Modification of Sugar Chains
Extracellular Export of Polysaccharides
3.7.2. CWPS
3.7.3. OPS
3.8. Biosynthesis of Bioactive Proteins, Vitamins B, Amino Acids, and Unsaturated Fatty Acids
3.9. Comparison with Other Basidiomycete Genomes
3.9.1. Gene Family, Core-Pan, and Phylogenetic Analysis
3.9.2. Genomic Synteny
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, S.; Gao, Q.; Rong, C.; Wang, S.; Zhao, Z.; Liu, Y.; Xu, J. Immunomodulatory effects of edible and medicinal mushrooms and their bioactive immunoregulatory products. J. Fungi 2020, 6, 269. [Google Scholar] [CrossRef]
- Lu, H.; Lou, H.; Hu, J.; Liu, Z.; Chen, Q. Macrofungi: A review of cultivation strategies, bioactivity, and application of mushrooms. Compr. Rev. Food Sci. F 2020, 19, 2333–2356. [Google Scholar] [CrossRef]
- Niazi, A.R.; Ghafoor, A. Different ways to exploit mushrooms: A review. All Life 2021, 14, 450–460. [Google Scholar] [CrossRef]
- Du, X.; Zhang, Y.; Mu, H.; Lv, Z.; Yang, Y.; Zhang, J. Structural elucidation and antioxidant activity of a novel polysaccharide (TAPB1) from Tremella aurantialba. Food Hydrocoll. 2015, 43, 459–464. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhang, X.; Ma, M.; Long, T.; Xiao, C.; Zhang, J.; Liu, J.; Zhao, L. Immunoenhancing glucuronoxylomannan from Tremella aurantialba Bandoni et Zang and its low-molecular-weight fractions by radical depolymerization: Properties, structures and effects on macrophages. Carbohydr. Polym. 2020, 238, 116184. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Sun, Y.; Fu, H.; Zhang, S.; Chen, J.; Xu, X. Antioxidant and immunostimulatory activities of polysaccharides extracted from Tremella aurantialba mycelia. Mol. Med. Rep. 2016, 14, 4857–4864. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Tang, Q.; Zhang, Z.; Li, C.-h.; Cao, H.; Yang, Y.; Zhang, J. Nutritional composition of three domesticated culinary-medicinal mushrooms: Oudemansiella sudmusida, Lentinus squarrosulus, and Tremella aurantialba. Int. J. Med. Mushrooms 2015, 17, 43–49. [Google Scholar] [CrossRef]
- Fan, J.; Chu, Z.; Li, L.; Zhao, T.; Yin, M.; Qin, Y. Physicochemical properties and microbial quality of tremella aurantialba packed in antimicrobial composite films. Molecules 2017, 22, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Wang, X.; Chen, Y.; Tian, S.; Lu, S. Antioxidant activity and oxidative injury rehabilitation of chemically modified polysaccharide (TAPA1) from Tremella aurantialba. Macromol. Res. 2018, 26, 479–483. [Google Scholar] [CrossRef]
- Islam, T.; Yu, X.; Xu, B. Phenolic profiles, antioxidant capacities and metal chelating ability of edible mushrooms commonly consumed in China. LWT 2016, 72, 423–431. [Google Scholar] [CrossRef]
- Bandoni, R.; Zang, M. On an undescribed Tremella from China. Mycologia 1990, 82, 270–273. [Google Scholar] [CrossRef]
- Liu, D.; Pujiana, D.; Wang, Y.; Zhang, Z.; Zheng, L.; Chen, L.; Ma, A. Comparative transcriptomic analysis identified differentially expressed genes and pathways involved in the interaction between Tremella fuciformis and Annulohypoxylon stygium. Antonie Van Leeuwenhoek 2019, 112, 1675–1689. [Google Scholar] [CrossRef]
- Li, H.-J.; Zhang, D.-H.; Yue, T.-H.; Jiang, L.-X.; Yu, X.; Zhao, P.; Li, T.; Xu, J.-W. Improved polysaccharide production in a submerged culture of Ganoderma lucidum by the heterologous expression of Vitreoscilla hemoglobin gene. J. Biotechnol. 2016, 217, 132–137. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Y.; Chen, C.; Ren, A.; Yu, H.; Zhao, M. Effect of the heme oxygenase gene on mycelial growth and polysaccharide synthesis in Ganoderma lucidum. J. Basic Microbiol. 2021, 61, 253–264. [Google Scholar] [CrossRef]
- Zhou, J.; Bai, Y.; Dai, R.; Guo, X.; Liu, Z.-H.; Yuan, S. Improved polysaccharide production by homologous co-overexpression of phosphoglucomutase and UDP glucose pyrophosphorylase genes in the mushroom Coprinopsis cinerea. J. Agric. Food Chem. 2018, 66, 4702–4709. [Google Scholar] [CrossRef]
- Zan, X.-Y.; Zhu, H.-A.; Jiang, L.-H.; Liang, Y.-Y.; Sun, W.-J.; Tao, T.-L.; Cui, F.-J. The role of Rho1 gene in the cell wall integrity and polysaccharides biosynthesis of the edible mushroom Grifola frondosa. Int. J. Biol. Macromol. 2020, 165, 1593–1603. [Google Scholar] [CrossRef]
- Yuan, Y.; Wu, F.; Si, J.; Zhao, Y.-F.; Dai, Y.-C. Whole genome sequence of Auricularia heimuer (Basidiomycota, Fungi), the third most important cultivated mushroom worldwide. Genomics 2019, 111, 50–58. [Google Scholar] [CrossRef]
- Yap, H.-Y.Y.; Muria-Gonzalez, M.J.; Kong, B.-H.; Stubbs, K.A.; Tan, C.-S.; Ng, S.-T.; Tan, N.-H.; Solomon, P.S.; Fung, S.-Y.; Chooi, Y.-H. Heterologous expression of cytotoxic sesquiterpenoids from the medicinal mushroom Lignosus rhinocerotis in yeast. Microb. Cell Factories 2017, 16, 103. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, E.; McGowan, J.; McCarthy, C.G.; Amini, A.; Grogan, H.; Fitzpatrick, D.A. Whole genome sequence of the commercially relevant mushroom strain Agaricus bisporus var. bisporus ARP23. G3 Genes Genomes Genet. 2019, 9, 3057–3066. [Google Scholar]
- Gea, F.J.; Navarro, M.J.; Santos, M.; Diánez, F.; Carrasco, J. Control of Fungal Diseases in Mushroom Crops while Dealing with Fungicide Resistance: A Review. Microorganisms 2021, 9, 585. [Google Scholar] [CrossRef]
- Gong, W.; Wang, Y.; Xie, C.; Zhou, Y.; Zhu, Z.; Peng, Y. Whole genome sequence of an edible and medicinal mushroom, Hericium erinaceus (Basidiomycota, Fungi). Genomics 2020, 112, 2393–2399. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Guo, H.; Zhang, J.; Liu, H.; Wang, K.; Zuo, S.; Xu, P.; Xia, Z.; Zhou, Q.; Zhang, H. The genome of the medicinal macrofungus Sanghuang provides insights into the synthesis of diverse secondary metabolites. Front. Microbiol. 2020, 10, 3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.-H.; Wu, S.-H.; Zhou, L.-W. The First Whole Genome Sequencing of Sanghuangporus sanghuang Provides Insights into Its Medicinal Application and Evolution. J. Fungi 2021, 7, 787. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.P.; Barh, A.; Bairwa, R.K.; Annepu, S.K.; Kumari, B.; Kamal, S. Enoki Mushroom (Flammulina velutipes (Curtis) Singer) Breeding. In Advances in Plant Breeding Strategies: Vegetable Crops; Springer: Cham, Switzerland, 2021; pp. 423–441. [Google Scholar]
- Chen, J.; Li, J.-M.; Tang, Y.-J.; Ma, K.; Li, B.; Zeng, X.; Liu, X.-B.; Li, Y.; Yang, Z.-L.; Xu, W.-N. Genome-wide analysis and prediction of genes involved in the biosynthesis of polysaccharides and bioactive secondary metabolites in high-temperature-tolerant wild Flammulina filiformis. BMC Genom. 2020, 21, 719. [Google Scholar] [CrossRef]
- Luo Xingye, L.; Baxian, C.; Zhisen, L. Yunnan golden fungus (Tremella mesenterica Retz ex Fr.), a rarity in recipe. Yunnan Agric. Sci. Technol. 1987, 2, 44–46. [Google Scholar]
- Liu, X.-Z.; Wang, Q.-M.; Göker, M.; Groenewald, M.; Kachalkin, A.; Lumbsch, H.T.; Millanes, A.; Wedin, M.; Yurkov, A.; Boekhout, T. Towards an integrated phylogenetic classification of the Tremellomycetes. Stud. Mycol. 2015, 81, 85–147. [Google Scholar] [CrossRef] [Green Version]
- Morin, E.; Kohler, A.; Baker, A.R.; Foulongne-Oriol, M.; Lombard, V.; Nagye, L.G.; Ohm, R.A.; Patyshakuliyeva, A.; Brun, A.; Aerts, A.L. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. USA 2012, 109, 17501–17506. [Google Scholar] [CrossRef] [Green Version]
- Stajich, J.E.; Wilke, S.K.; Ahrén, D.; Au, C.H.; Birren, B.W.; Borodovsky, M.; Burns, C.; Canbäck, B.; Casselton, L.A.; Cheng, C. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc. Natl. Acad. Sci. USA 2010, 107, 11889–11894. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, D.; Park, S.-G.; Kim, K.; Bae, W.; Lee, G.W.; Ha, B.-S.; Ro, H.-S.; Kim, M.; Ryoo, R.; Rhee, S.-K. Whole genome de novo sequencing and genome annotation of the world popular cultivated edible mushroom, Lentinula edodes. J. Biotechnol. 2016, 223, 24–25. [Google Scholar] [CrossRef]
- Mondo, S.J.; Dannebaum, R.O.; Kuo, R.C.; Louie, K.B.; Bewick, A.J.; LaButti, K.; Haridas, S.; Kuo, A.; Salamov, A.; Ahrendt, S.R. Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 2017, 49, 964–968. [Google Scholar] [CrossRef]
- Deng, Y.; Huang, X.; Ruan, B.; Xie, B.; van Peer, A.F.; Jiang, Y. Optimal codons in Tremella fuciformis end in C/G, a strong difference with known Tremella species. World J. Microbiol. Biotechnol. 2015, 31, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martínez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Wang, R.; Sun, D.; Li, S.; Xu, H.; Qiu, Y.; Lei, P.; Sun, L.; Xu, X.; Zhu, Y. High-efficiency production of Tremella aurantialba polysaccharide through basidiospore fermentation. Bioresour. Technol. 2020, 318, 124268. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-H.; Lee, W.-H.; Lee, C.-M.; Sim, J.-S.; Won, S.Y.; Han, S.-R.; Kwon, S.-J.; Kim, J.S.; Kim, C.-K.; Oh, T.-J. De novo transcriptome sequence of Senna tora provides insights into anthraquinone biosynthesis. PLoS ONE 2020, 15, e0225564. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Wang, J.; Shi, W.; Du, L.; Sun, Y.; Zhan, W.; Jiang, J.-F.; Wang, Q.; Zhang, B.; Ji, P. Large-scale comparative analyses of tick genomes elucidate their genetic diversity and vector capacities. Cell 2020, 182, 1328–1340.e1313. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, T.; Kang, W.; Tang, D.; Feng, Q. Pathogenic and antimicrobial resistance genes in Streptococcus oralis strains revealed by comparative genome analysis. Genomics 2020, 112, 3783–3793. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 2047–2217X-1-18. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Jackman, S.D.; Vandervalk, B.P.; Mohamadi, H.; Chu, J.; Yeo, S.; Hammond, S.A.; Jahesh, G.; Khan, H.; Coombe, L.; Warren, R.L. ABySS 2.0: Resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 2017, 27, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-H.; Liao, Y.-C. CISA: Contig integrator for sequence assembly of bacterial genomes. PLoS ONE 2013, 8, e60843. [Google Scholar] [CrossRef]
- Berlin, K.; Koren, S.; Chin, C.-S.; Drake, J.P.; Landolin, J.M.; Phillippy, A.M. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 2015, 33, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoff, K.J.; Stanke, M. WebAUGUSTUS—A web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 2013, 41, W123–W128. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Smit, A.F. Repeat-Masker Open-3.0. 2010. Available online: http://www.repeatmasker.org (accessed on 5 August 2021).
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H. Predicting secretory proteins with SignalP. In Protein Function Prediction; Springer: Berlin, Germany, 2017; pp. 59–73. [Google Scholar]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 1, W29–W35. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Vilella, A.J.; Severin, J.; Ureta-Vidal, A.; Heng, L.; Durbin, R.; Birney, E. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009, 19, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA; The Pennsylvania State University: State College, PA, USA, 2007. [Google Scholar]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Zane, L.; Bargelloni, L.; Patarnello, T. Strategies for microsatellite isolation: A review. Mol. Ecol. 2002, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bilgen, M.; Karaca, M.; Onus, A.N.; Ince, A.G. A software program combining sequence motif searches with keywords for finding repeats containing DNA sequences. Bioinformatics 2004, 20, 3379–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.; Zhao, M.; Hsiang, T.; Feng, X.; Zhang, J.; Huang, C. Identification and characterization of small noncoding RNAs in genome sequences of the edible fungus Pleurotus ostreatus. BioMed Res. Int. 2016, 2016, 2503023. [Google Scholar] [CrossRef]
- Yin, J.; Jiang, L.; Wang, L.; Han, X.; Guo, W.; Li, C.; Zhou, Y.; Denton, M.; Zhang, P. A high-quality genome of taro (Colocasia esculenta (L.) Schott), one of the world’s oldest crops. Mol. Ecol. Resour. 2021, 21, 68–77. [Google Scholar] [CrossRef]
- Smit, E.; Kock, J.; Van Der Wsthuizen, J.; Britz, T. Taxonomic relationships of Cryptococcus and Tremella based on fatty acid composition and other phenotypic characters. Microbiology 1988, 134, 2849–2855. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Lu, H.; Chen, H.; Tang, X.; Zhao, J.; Zhang, H.; Chen, Y.Q.; Chen, W. Lipid metabolism research in oleaginous fungus Mortierella alpina: Current progress and future prospects. Biotechnol. Adv. 2021, 107794. [Google Scholar] [CrossRef]
- Akapo, O.O.; Padayachee, T.; Chen, W.; Kappo, A.P.; Yu, J.-H.; Nelson, D.R.; Syed, K. Distribution and diversity of cytochrome P450 monooxygenases in the fungal class Tremellomycetes. Int. J. Mol. Sci. 2019, 20, 2889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rytioja, J.; Hildén, K.; Yuzon, J.; Hatakka, A.; De Vries, R.P.; Mäkelä, M.R. Plant-polysaccharide-degrading enzymes from basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Sinha, K.; Sil, C.P. Cytochrome P450s: Mechanisms and biological implications in drug metabolism and its interaction with oxidative stress. Curr. Drug Metab. 2014, 15, 719–742. [Google Scholar] [CrossRef] [PubMed]
- Aliyu, H.; Gorte, O.; Zhou, X.; Neumann, A.; Ochsenreither, K. In silico proteomic analysis provides insights into phylogenomics and plant biomass deconstruction potentials of the tremelalles. Front. Bioeng. Biotechnol. 2020, 8, 226. [Google Scholar] [CrossRef] [PubMed]
- Kgosiemang, I.K.R.; Syed, K.; Mashele, S.S. Comparative genomics and evolutionary analysis of cytochrome P450 monooxygenases in fungal subphylum Saccharomycotina. J. Pure Appl. Microbiol. 2014, 8, 291–302. [Google Scholar]
- Ogidi, C.O.; Oyetayo, V.O.; Akinyele, B.J. Wild Medicinal Mushrooms: Potential Applications in Phytomedicine and Functional Foods. In An Introduction to Mushroom; IntechOpen: Vienna, Austria, 2020; pp. 118–126. [Google Scholar]
- Zhang, Z.; Lian, B.; Huang, D.; Cui, F. Compare activities on regulating lipid-metabolism and reducing oxidative stress of diabetic rats of Tremella aurantialba broth’s extract (TBE) with its mycelia polysaccharides (TMP). J. Food Sci. 2009, 74, H15–H21. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-J.; Deng, G.-F.; Xu, X.-R.; Wu, S.; Li, S.; Xia, E.-Q.; Li, F.; Chen, F.; Ling, W.-H.; Li, H.-B. Antioxidant capacities, phenolic compounds and polysaccharide contents of 49 edible macro-fungi. Food Funct. 2012, 3, 1195–1205. [Google Scholar] [CrossRef]
- Liu, D.; Gong, J.; Dai, W.; Kang, X.; Huang, Z.; Zhang, H.-M.; Liu, W.; Liu, L.; Ma, J.; Xia, Z. The genome of Ganderma lucidum provide insights into triterpense biosynthesis and wood degradation. PLoS ONE 2012, 7, e36146. [Google Scholar]
- Liang, Y.; Lu, D.; Wang, S.; Zhao, Y.; Gao, S.; Han, R.; Yu, J.; Zheng, W.; Geng, J.; Hu, S. Genome assembly and pathway analysis of edible mushroom Agrocybe cylindracea. Genom. Proteom. Bioinform. 2020, 18, 341–351. [Google Scholar] [CrossRef]
- Wang, W.F.; Xiao, H.; Zhong, J.J. Biosynthesis of a ganoderic acid in Saccharomyces cerevisiae by expressing a cytochrome P450 gene from Ganoderma lucidum. Biotechnol. Bioeng. 2018, 115, 1842–1854. [Google Scholar] [CrossRef]
- Yu, F.; Song, J.; Liang, J.; Wang, S.; Lu, J. Whole genome sequencing and genome annotation of the wild edible mushroom, Russula griseocarnosa. Genomics 2020, 112, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Yap, H.-Y.Y.; Chooi, Y.-H.; Firdaus-Raih, M.; Fung, S.-Y.; Ng, S.-T.; Tan, C.-S.; Tan, N.-H. The genome of the Tiger Milk mushroom, Lignosus rhinocerotis, provides insights into the genetic basis of its medicinal properties. BMC Genom. 2014, 15, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüffler, A. Secondary metabolites of basidiomycetes. In Physiology and Genetics; Springer: Berlin/Heidelberg, Germany, 2018; pp. 231–275. [Google Scholar]
- Fenteany, G.; Standaert, R.F.; Reichard, G.A.; Corey, E.; Schreiber, S.L. A beta-lactone related to lactacystin induces neurite outgrowth in a neuroblastoma cell line and inhibits cell cycle progression in an osteosarcoma cell line. Proc. Natl. Acad. Sci. USA 1994, 91, 3358–3362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhnert, E.; Navarro-Muñoz, J.; Becker, K.; Stadler, M.; Collemare, J.; Cox, R. Secondary metabolite biosynthetic diversity in the fungal family Hypoxylaceae and Xylaria hypoxylon. Stud. Mycol. 2021, 99, 100118. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yang, X.; Zhang, H. Extracellular polysaccharide biosynthesis in Cordyceps. Crit. Rev. Microbiol. 2020, 46, 359–380. [Google Scholar] [CrossRef]
- Rana, S.; Upadhyay, L.S.B. Microbial exopolysaccharides: Synthesis pathways, types and their commercial applications. Int. J. Biol. Macromol. 2020, 157, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Rathore, H.; Prasad, S.; Sharma, S. Mushroom nutraceuticals for improved nutrition and better human health: A review. Pharma. Nutr. 2017, 5, 35–46. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, E.; Geng, Y.; Wang, K.; Chen, D.; Huang, X.; Ouyang, P.; Zuo, Z.; Huang, C.; Fang, J. Complete genome analysis of Vibrio mimicus strain SCCF01, a highly virulent isolate from the freshwater catfish. Virulence 2020, 11, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Nagy, L.G.; Ohm, R.A.; Kovács, G.M.; Floudas, D.; Riley, R.; Gácser, A.; Sipiczki, M.; Davis, J.M.; Doty, S.L.; De Hoog, G.S. Latent homology and convergent regulatory evolution underlies the repeated emergence of yeasts. Nat. Commun. 2014, 5, 4471. [Google Scholar] [CrossRef] [Green Version]
- Nagy, L.G. Evolution: Complex multicellular life with 5500 genes. Curr. Biol. 2017, 27, R609–R612. [Google Scholar] [CrossRef]
- Xu, P.; Wang, H.; Qin, C.; Li, Z.; Lin, C.; Liu, W.; Miao, W. Analysis of the Taxonomy and Pathogenic Factors of Pectobacterium aroidearum L6 Using Whole-Genome Sequencing and Comparative Genomics. Front. Microbiol. 2021, 12, 1770. [Google Scholar] [CrossRef] [PubMed]
- Alcaraz, L.D.; Moreno-Hagelsieb, G.; Eguiarte, L.E.; Souza, V.; Herrera-Estrella, L.; Olmedo, G. Understanding the evolutionary relationships and major traits of Bacillus through comparative genomics. BMC Genom. 2010, 11, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Dominici, L.I.; Saati-Santamaría, Z.; García-Fraile, P. Genome Analysis and Genomic Comparison of the Novel Species Arthrobacter ipsi Reveal Its Potential Protective Role in Its Bark Beetle Host. Microb. Ecol. 2021, 81, 471–482. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Value |
|---|---|
| Genome assembly | |
| Contigs number | 15 |
| Max length (bp) | 2,546,384 |
| N50 length (bp) | 1,814,705 |
| Total length (bp) | 20,998,359 |
| GC (%) | 56.42 |
| Gene prediction | |
| Gene number | 5860 |
| Gene total length (bp) | 8,989,977 |
| Gene average length (bp) | 1534 |
| Gene length/Genome (%) | 42.81 |
| Species | NCBI BioProject | Total Length (Mb) | GC% | Contigs | N50 Length (bp) | Completeb a | Fragmented | Missing |
|---|---|---|---|---|---|---|---|---|
| T. fuciformis Tr26 | PRJNA281519 | 23.6356 | 57.0 | 3502 | 18,448 | 92.4% | 1.4% | 6.2% |
| T. mesenterica DSM 1558 | PRJNA225529 | 28.6399 | 46.8 | 484 | 123,767 | 92.0% | 1.4% | 6.6% |
| T. mesenterica ATCC 28783 | PRJNA207298 | 27.1109 | 41.3 | 1019 | 73,463 | 90.6% | 2.4% | 7.0% |
| N. encephala 68-887.2 | PRJNA330699 | 19.7863 | 49.3 | 151 | 209,500 | 85.5% | 3.4% | 11.1% |
| N. aurantialba NX-20 | PRJNA772294 | 20.9984 | 56.4 | 15 | 1,825,336 | 93.1% | 2.4% | 4.5% |
| Repeat Type | Type | Number of Elements | Length Occupied (bp) | Repeat Size (bp) | Percentage of Genome (%) |
|---|---|---|---|---|---|
| Interspersed repeat | SINE | 9 | 1030 | - | 0.0049 |
| LINEs | 395 | 39,539 | - | 0.1883 | |
| LTR elements | 643 | 115,566 | - | 0.5504 | |
| DNA elements | 418 | 39,329 | - | 0.1873 | |
| RC | 68 | 8542 | - | 0.0407 | |
| Unknown | 16 | 1593 | - | 0.0076 | |
| Tandem repeat | TR | 12,449 | 583,229 | 1~982 | 2.7775 |
| Microsatellite DNA | 1448 | 91,405 | 2~6 | 0.4353 | |
| Minisatellite DNA | 9096 | 453,057 | 10~60 | 2.1576 |
| Type | Number of Elements | Total Length (bp) | Average Length (bp) | Percentage in Genome (%) |
|---|---|---|---|---|
| tRNA | 44 | 3925 | 89 | 0.01869 |
| 5s_rRNA | 9 | 1034 | 115 | 0.00599 |
| 5.8s_rRNA | 0 | 0 | 0 | 0 |
| 18s_rRNA | 1 | 1802 | 1802 | 0.02294 |
| 28s_rRNA | 1 | 3492 | 3492 | 0.05030 |
| sRNA | 0 | 0 | 0 | 0 |
| snRNA | 7 | 677 | 96 | 0.00322 |
| miRNA | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, T.; Zhang, Y.; Jiang, H.; Yang, K.; Wang, S.; Wang, R.; Li, S.; Lei, P.; Xu, H.; Qiu, Y.; et al. Whole Genome Sequencing and Annotation of Naematelia aurantialba (Basidiomycota, Edible-Medicinal Fungi). J. Fungi 2022, 8, 6. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8010006
Sun T, Zhang Y, Jiang H, Yang K, Wang S, Wang R, Li S, Lei P, Xu H, Qiu Y, et al. Whole Genome Sequencing and Annotation of Naematelia aurantialba (Basidiomycota, Edible-Medicinal Fungi). Journal of Fungi. 2022; 8(1):6. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8010006
Chicago/Turabian StyleSun, Tao, Yixuan Zhang, Hao Jiang, Kai Yang, Shiyu Wang, Rui Wang, Sha Li, Peng Lei, Hong Xu, Yibin Qiu, and et al. 2022. "Whole Genome Sequencing and Annotation of Naematelia aurantialba (Basidiomycota, Edible-Medicinal Fungi)" Journal of Fungi 8, no. 1: 6. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8010006