
Genomic Characteristics and Comparative Genomics Analysis of Parafenestella ontariensis sp. nov.

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Isolation
2.2. Morphological Analysis
2.3. DNA Extraction, PCR Amplification and Sequencing
2.4. Sequence Alignment and Phylogenetic Analysis
2.5. DNA Extraction, Library Preparation, Genome Sequencing and Assembly
2.6. Gene Prediction and Functional Annotation
2.7. Phylogenomic Analysis and Genome Sequence Alignment
3. Results
3.1. Taxonomy
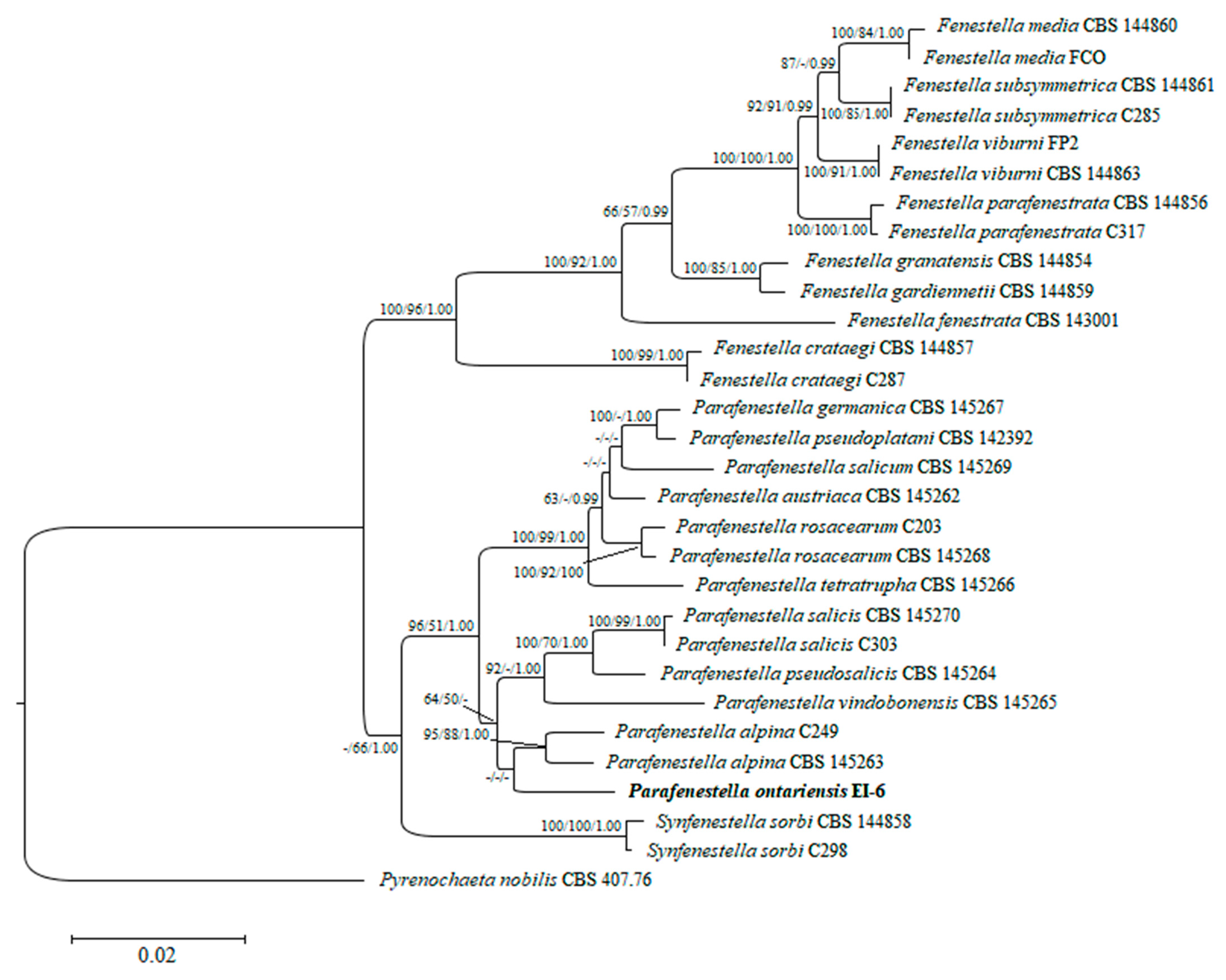
3.2. Phylogenetic and Phylogenomic Analyses
3.3. Genome Sequencing and Assembly Characteristics of Parafenestella ontariensis and Other Closely Related Cucurbitariaceae Species
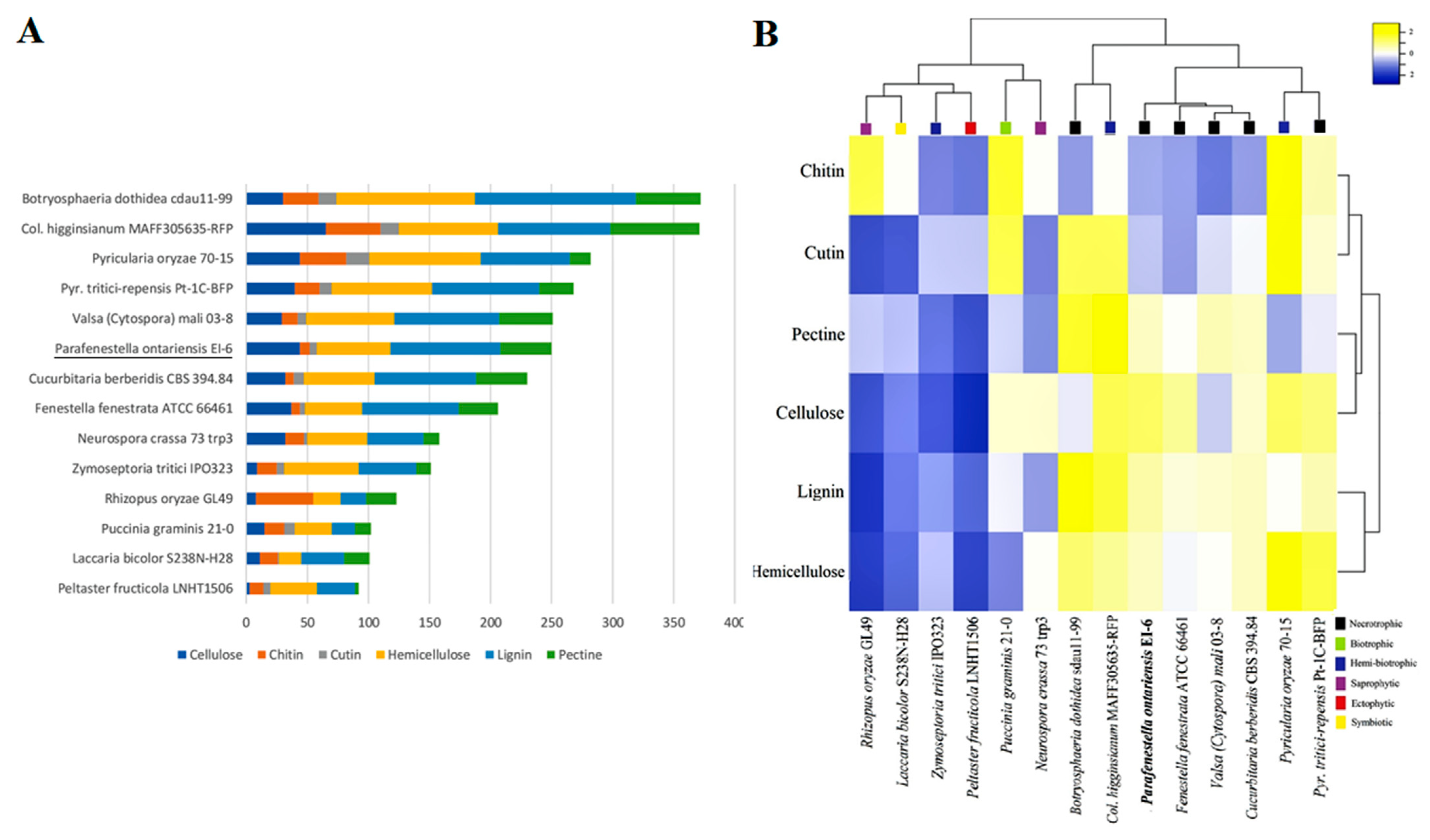
3.4. Secondary Metabolites
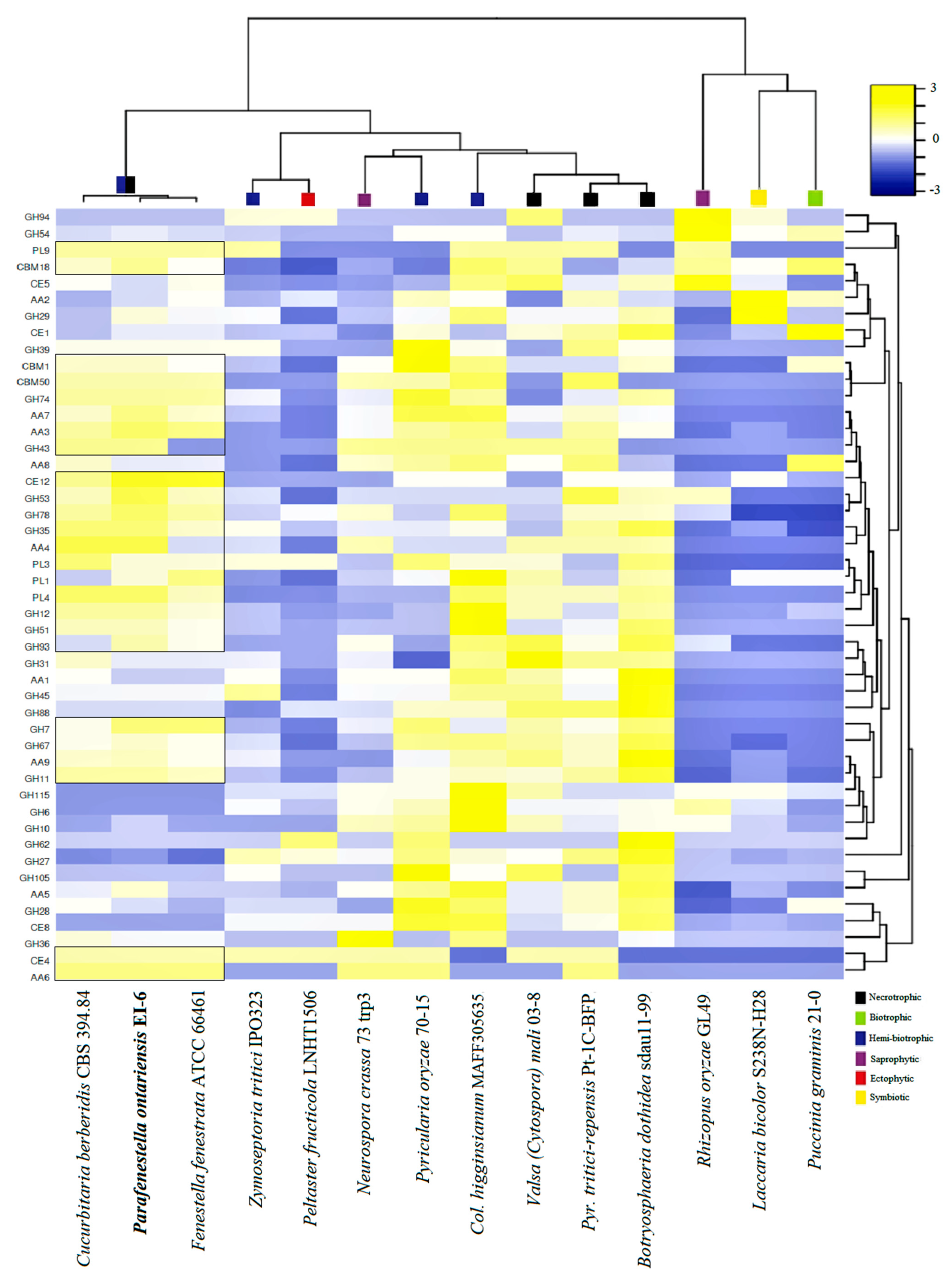
3.5. Carbohydrate Active Enzymes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jaklitsch, W.; Checa, J.; Blanco, M.; Olariaga, I.; Tello, S.; Voglmayr, H. A Preliminary Account of the Cucurbitariaceae. Stud. Mycol. 2018, 90, 71–118. [Google Scholar] [CrossRef] [PubMed]
- Hyde, K.D.; Jones, E.B.G.; Liu, J.-K.; Ariyawansa, H.; Boehm, E.W.A.; Boonmee, S.; Braun, U.; Chomnunti, P.; Crous, P.W.; Dai, D.-Q.; et al. Families of Dothideomycetes. Fungal Divers. 2013, 63, 1–313. [Google Scholar] [CrossRef]
- Crous, P.W.; Wingfield, M.J.; Lombard, L.; Roets, F.; Swart, W.J.; Alvarado, P.; Carnegie, A.J.; Moreno, G.; Luangsa-Ard, J.; Thangavel, R.; et al. Fungal Planet Description Sheets: 951–1041. Persoonia 2019, 43, 223–425. [Google Scholar] [CrossRef] [PubMed]
- Jaklitsch, W.M.; Voglmayr, H. Fenestelloid Clades of the Cucurbitariaceae. Persoonia 2020, 44, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Haridas, S.; Albert, R.; Binder, M.; Bloem, J.; LaButti, K.; Salamov, A.; Andreopoulos, B.; Baker, S.; Barry, K.; Bills, G.; et al. 101 Dothideomycetes Genomes: A Test Case for Predicting Lifestyles and Emergence of Pathogens. Stud. Mycol. 2020, 96, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Dal’Sasso, T.C.D.S.; Rody, H.V.S.; Grijalba, P.E.; Oliveira, L.O. Genome Sequences and In Silico Effector Mining of Corynespora cassiicola CC_29 and Corynespora olivacea CBS 114450. Arch. Microbiol. 2021, 203, 5257–5265. [Google Scholar] [CrossRef]
- Rodrigues, C.M.; Takita, M.A.; Silva, N.V.; Ribeiro-Alves, M.; Machado, M.A. Comparative Genome Analysis of Phyllosticta citricarpa and Phyllosticta capitalensis, Two Fungi Species that Share the Same Host. BMC Genom. 2019, 20, 554. [Google Scholar] [CrossRef]
- Stauber, L.; Prospero, S.; Croll, D. Comparative Genomics Analyses of Lifestyle Transitions at the Origin of an Invasive Fungal Pathogen in the Genus Cryphonectria. Ecol. Evol. Sci. 2020, 5, 00737–20. [Google Scholar] [CrossRef]
- Lind, A.L.; Wisecaver, J.H.; Lameiras, C.; Wiemann, P.; Palmer, J.M.; Keller, N.P.; Rodrigues, F.; Goldman, G.H.; Rokas, A. Drivers of Genetic Diversity in Secondary Metabolic Gene Clusters within a Fungal Species. PLoS Biol. 2017, 15, 2003583. [Google Scholar] [CrossRef] [Green Version]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic Analysis of the Necrotrophic Fungal Pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, 1002230. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Liang, X.; Gleason, M.L.; Zhang, R.; Sun, G. Comparative Genomics of Botryosphaeria dothidea and B. kuwatsukai, Causal Agents of Apple Ring Rot, Reveals both Species Expansion of Pathogenicity–Related Genes and Variations in Virulence Gene Content during Speciation. IMA Fungus 2018, 9, 243–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, V.; Golaconda, R.H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The Carbohydrate–Active Enzymes Database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, R. Comparative Analysis of Fungal Genomes Reveals Different Plant Cell Wall Degrading Capacity in Fungi. BMC Genom. 2013, 14, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aime, M.C.; Miller, A.N.; Aoki, T.; Bensch, K.; Cai, L.; Crous, P.W.; Hawksworth, D.L.; Hyde, K.D.; Kirk, P.M.; Lücking, R.; et al. How to Publish a New Fungal Species, or Name, Version 3.0. IMA Fungus 2021, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Phookamsak, R.; Norphanphoun, C.; Tanaka, K.; Dai, D.-Q.; Luo, Z.-L.; Liu, J.-K.; Su, H.-Y.; Bhat, D.J.; Bahkali, A.H.; Mortimer, P.E.; et al. Towards a Natural Classification of Astrosphaeriella-like Species; Introducing Astrosphaeriellaceae and Pseudoastrosphaeriellaceae fam. nov. and Astrosphaeriellopsis, gen. nov. Fungal Divers. 2015, 74, 143–197. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.D.; Lee, S.; Taylor, J.W. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 312–322. [Google Scholar]
- Vilgalys, R.; Hester, M. Rapid Genetic Identification and Mapping of Enzymatically Amplified Ribosomal DNA from Several Cryptococcus Species. J. Bacteriol. 1990, 172, 4238–4246. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position–Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, S. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian Inference of Phylogenetic Trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Posada, D.; Buckley, T. Model Selection and Model Averaging in Phylogenetics: Advantages of Akaike Information Criterion and Bayesian Approaches Over Likelihood Ratio Tests. Syst. Biol. 2004, 53, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest–NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillis, D.M.; Bull, J.J. An Empirical Test of Bootstrapping as a Method for Assessing Confidence in Phylogenetic Analysis. Syst. Biol. 1993, 42, 182–192. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree, version 1.4.4; Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2018.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and its Applications to Single–Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile Genome Assembly Evaluation with QUAST–LG. Bioinformatics 2018, 34, 142–150. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. 2013–2015 RepeatMasker Open–4.0. Available online: http://www.repeatmasker.org (accessed on 4 February 2022).
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single–Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A Web Server for Gene Prediction in Eukaryotes that Allows User–Defined Constraints. Nucleic Acids Res. 2005, 33, 465–467. [Google Scholar] [CrossRef] [Green Version]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An Easy-to-Use Annotation Pipeline Designed for Emerging Model Organism Genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A Web Resource for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2012, 40, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Shelest, V.; Nath, N.; Shelest, E. CASSIS and SMIPS: Promoter–Based Prediction of Secondary Metabolite Gene Clusters in Eukaryotic Genomes. Bioinformatics 2016, 32, 1138–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.A.; Otillar, R.; Riley, R.; Salamov, A.A.; Zhao, X.; Korzeniewski, F.; et al. MycoCosm Portal: Gearing up for 1000 Fungal Genomes. Nucleic Acids Res. 2014, 42, 699–704. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving Fundamental Biases in Whole Genome Comparisons Dramatically Improves Orthogroup Inference Accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-Approximately Maximum–Likelihood Trees for Large Alignments. PLoS ONE. 2010, 5, e9490. [Google Scholar] [CrossRef]
- Jain, C.; Koren, S.; Dilthey, A.T.; Phillippy, A.M.; Aluru, S. A Fast Adaptive Algorithm for Computing Whole–Genome Homology Maps. Bioinformatics 2018, 34, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Cabanettes, F.; Klopp, C.D. GENIES: Dot Plot Large Genomes in an Interactive, Efficient and Simple Way. PeerJ 2018, 6, 4958. [Google Scholar] [CrossRef]
- Wanasinghe, D.N.; Phookamsak, R.; Jeewon, R.; Li, W.J.; Hyde, K.D.; Gareth Jones, E.B.; Erio, C.; Promputtha, I. A Family Level rDNA Based Phylogeny of Cucurbitariaceae and Fenestellaceae with Descriptions of New Fenestella Species and Neocucurbitaria gen. nov. Mycosphere 2017, 8, 397–414. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Bae, H. The Diversity of Fungal Genome. Biol. Proced. Online 2015, 17, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.; Nandineni, M.R. Genome Sequencing and Comparative Genomics Reveal a Repertoire of Putative Pathogenicity Genes in Chilli Anthracnose Fungus Colletotrichum truncatum. PLoS ONE 2017, 12, 0183567. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Song, Q.; Ji, P.; Cregan, P. Draft Genome Sequence of Phomopsis longicolla Type Strain TWH P74, a Fungus Causing Phomopsis Seed Decay in Soybean. Genome Announc. 2015, 3, e00010-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hane, J.K.; Williams, A.H.; Oliver, R.P. 9 Genomic and Comparative Analysis of the Class Dothideomycetes. In Evolution of Fungi and Fungal–Like Organisms. The Mycota, 14; Pöggeler, S., Wöstemeyer, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 205–229. [Google Scholar]
- Khaldi, N.; Collemare, J.; Lebrun, M.H.; Wolfe, K.H. Evidence for Horizontal Transfer of a Secondary Metabolite Gene Cluster Between Fungi. Genome Biol. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadeawi, S.; Nasution, A.; Hairmansis, A.; Telebanco-Yanoria, M.J.; Obara, M.; Hayashi, N.; Fukuta, Y. Pathogenicity of Isolates of the Rice Blast Pathogen (Pyricularia oryzae) from Indonesia. Plant Dis. 2021, 105, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Kamel, S.; Cherif, M.; Hafez, M.; Despins, T.; Aboukhaddour, R. Pyrenophora tritici–repentis in Tunisia: Race Structure and Effector Genes. Front. Plant Sci. 2019, 10, 1562. [Google Scholar] [CrossRef] [Green Version]
- Oide, S.; Turgeon, B.G. Natural Roles of Nonribosomal Peptide Metabolites in Fungi. Mycoscience 2020, 61, 101–110. [Google Scholar] [CrossRef]
- Wang, X.; Wei, J.; Huang, L.; Kang, Z. Re-evaluation of Pathogens Causing Valsa Canker on Apple in China. Mycologia 2011, 103, 317–324. [Google Scholar] [CrossRef]
- Ohm, R.A.; Feau, N.; Henrissat, B.; Schoch, C.L.; Horwitz, B.A.; Barry, K.W.; Condon, B.J.; Copeland, A.C.; Dhillon, B.; Glaser, F.; et al. Diverse Lifestyles and Strategies of Plant Pathogenesis Encoded in the Genomes of Eighteen Dothideomycetes fungi. PLoS Pathog. 2012, 8, 1003037. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, X.; Huang, P.W.; Dai, C.C. Endophytism or Saprophytism: Decoding the Lifestyle Transition of the Generalist Fungus Phomopsis liquidambari. Microbiol. Res. 2017, 206, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, X.; Cai, J.; Chen, Y. Genomic Characteristics and Comparative Genomics Analysis of the Endophytic Fungus Sarocladium brachiariae. BMC Genom. 2019, 20, 782. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Host | GenBank Accession Numbers | |||
|---|---|---|---|---|---|---|
| ITS | LSU | tef1 | tub2 | |||
| Fenestella crataegi | CBS 144857 * | Crataegus monogyna | MK356282 | MK356282 | MK357555 | MK357599 |
| C287 | C. monogyna | MK356281 | MK356281 | MK357554 | MK357598 | |
| F. fenestrata | CBS 143001 * | Alnus glutinosa | MF795765 | MF795765 | MF795853 | MF795893 |
| F. gardiennetii | CBS 144859 | Acer saccharum | MK356283 | MK356283 | MK357556 | MK357600 |
| F. granatensis | CBS 144854 * | Acer granatense | MK356284 | MK356284 | MK357557 | MK357601 |
| F. media | CBS 144860 * | Corylus avellana | MK356285 | MK356285 | MK357558 | MK357602 |
| FCO | Carpinus orientalis | MK356286 | MK356286 | MK357559 | - | |
| F. parafenestrata | CBS 144856 * | Quercus robur | MK356291 | MK356291 | MK357564 | MK357607 |
| C317 | Salix sp. | MK356292 | MK356292 | MK357565 | MK357608 | |
| F. subsymmetrica | CBS 144861 * | Acer campestre | MK356297 | MK356297 | MK357569 | MK357610 |
| C285 | Juglans regia | MK356293 | MK356293 | MK357566 | - | |
| F. viburni | CBS 144863 * | Viburnum lantana | MK356300 | MK356300 | MK357572 | MK357613 |
| FP2 | V. lantana | MK356299 | MK356299 | MK357571 | MK357612 | |
| Parafenestella alpina | CBS 145263 * | Cotoneaster integerrimus | MK356302 | MK356302 | MK357574 | MK357615 |
| C249 | Salix appendiculata | MK356303 | MK356303 | MK357575 | MK357616 | |
| P. austriaca | CBS 145262 * | Rosa canina | MK356304 | MK356304 | MK357576 | MK357617 |
| P. germanica | CBS 145267 * | Corylus avellana | MK356305 | MK356305 | MK357577 | MK357618 |
| P. ontariensis | EI-6 * | Acer negundo | OM286882 | OM286884 | genome ** | genome ** |
| P. pseudoplatani | CBS 142392 * | Acer pseudoplatanus | MF795788 | MF795788 | MF795876 | MF795914 |
| P. pseudosalicis | CBS 145264 * | Salix cf. alba | MK356307 | MK356307 | MK357579 | MK357620 |
| P. rosacearum | CBS 145268 * | Pyracantha coccinea | MK356311 | MK356311 | MK357583 | MK357624 |
| C203 | Pyrus communis | MK356308 | MK356308 | MK357580 | MK357621 | |
| P. salicis | CBS 145270 * | Salix alba | MK356317 | MK356317 | MK357589 | MK357629 |
| C303 | S. alba | MK356316 | MK356316 | MK357588 | MK357628 | |
| P. salicum | CBS 145269 * | S. alba | MK356318 | MK356318 | MK357590 | MK357630 |
| continued | ||||||
| P. tetratrupha | CBS 145266 * | Alnus glutinosa | MK356319 | MK356319 | MK357591 | MK357631 |
| P. vindobonensis | CBS 145265 * | Salix babylonica | MK356320 | MK356320 | MK357592 | MK357632 |
| Pyrenochaeta nobilis | CBS 407.76 * | Laurus nobilis | MF795792 | MF795792 | MF795880 | MF795916 |
| Synfenestella sorbi | C298 | Sorbus aucuparia | MK356325 | MK356325 | MK357597 | MK357636 |
| CBS 144858 | S. aucuparia | MK356324 | MK356324 | MK357596 | MK357635 | |
| Species Name | Strain | GenBank Accession Number | Host | Country |
|---|---|---|---|---|
| Bimuria novae-zelandiae | CBS 107.79 | GCA_010015655 | NA | NA |
| Bipolaris sorokiniana | ND90Pr | GCA_000338995 | NA | NA |
| Bipolaris maydis | C5 | GCA_000338975 | NA | NA |
| Botryosphaeria dothidea | sdau11-99 | GCA_011503125 | Malus sp. | China |
| Byssothecium circinans | CBS 675.92 | GCA_010015675 | NA | NA |
| Clathrospora elynae | CBS 161.51 | GCA_010015635 | Carex curvula | NA |
| Corynespora cassiicola | Phil v1.0 | GCA_003016335 | NA | Philippines |
| Cucurbitaria berberidis | CBS 394.84 | GCA_010015615 | NA | NA |
| Didymella exigua | CBS 183.55 | GCA_010094145 | Rumex arifolius | France |
| Didymella heteroderae | 28M-1 | GCA_011058895 | Homo sapiens | Brazil |
| Fenestella fenestrata | ATCC 66461 | NA * | Acer sp. | USA |
| Karstenula rhodostoma | CBS 690.94 | GCA_010093485 | NA | NA |
| Lentithecium fluviatile | CBS 122367 | GCA_010405425 | Populus sp. | France |
| Leptosphaeria maculans | JN3 | GCA_000230375 | NA | NA |
| Massarina eburnea | CBS 473.64 | GCA_010093635 | NA | NA |
| Parafenestella ontariensis | EI-6 | GCA_021596325 ** | Acer negundo | Canada |
| Plenodomus tracheiphilus | IPT5 | GCA_010093695 | NA | NA |
| Setospaheria turcica | Et28A | GCA_000359705 | NA | NA |
| Stagonospora nodorum | SN15 | GCA_000146915 | NA | NA |
| Features | C. berberidis CBS 394.84 | F. fenestrata ATCC 66461 | P. ontariensis EI-6 |
|---|---|---|---|
| Sequence coverage (×) | 145.7 | 81.9 | 267.4 |
| Assembly size (MB) | 32.9 | 32.7 | 34.4 |
| Scaffolds (>1000 bp) | 42 | 22 | 162 |
| Scaffold N50 (Kb) | 6 234 | 2 290 | 906 |
| GC content (%) | 49.3 | 51.53 | 50.79 |
| N of gene models | 12 429 | 11 804 | 11 748 |
| Repeat rate (%) | 2.24 | 3.91 | 1.59 |
| BUSCO estimates (%) | 97.2 | 98.1 | 97.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilyukhin, E.; Markovskaja, S.; Elgorban, A.M.; Al-Rejaie, S.S.; Maharachchikumbura, S.S.N. Genomic Characteristics and Comparative Genomics Analysis of Parafenestella ontariensis sp. nov. J. Fungi 2022, 8, 732. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8070732
Ilyukhin E, Markovskaja S, Elgorban AM, Al-Rejaie SS, Maharachchikumbura SSN. Genomic Characteristics and Comparative Genomics Analysis of Parafenestella ontariensis sp. nov. Journal of Fungi. 2022; 8(7):732. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8070732
Chicago/Turabian StyleIlyukhin, Evgeny, Svetlana Markovskaja, Abdallah M. Elgorban, Salim S. Al-Rejaie, and Sajeewa S.N. Maharachchikumbura. 2022. "Genomic Characteristics and Comparative Genomics Analysis of Parafenestella ontariensis sp. nov." Journal of Fungi 8, no. 7: 732. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8070732