Clonal Hematopoiesis of Indeterminate Potential: A Multidisciplinary Challenge in Personalized Hematology


{kind=link}
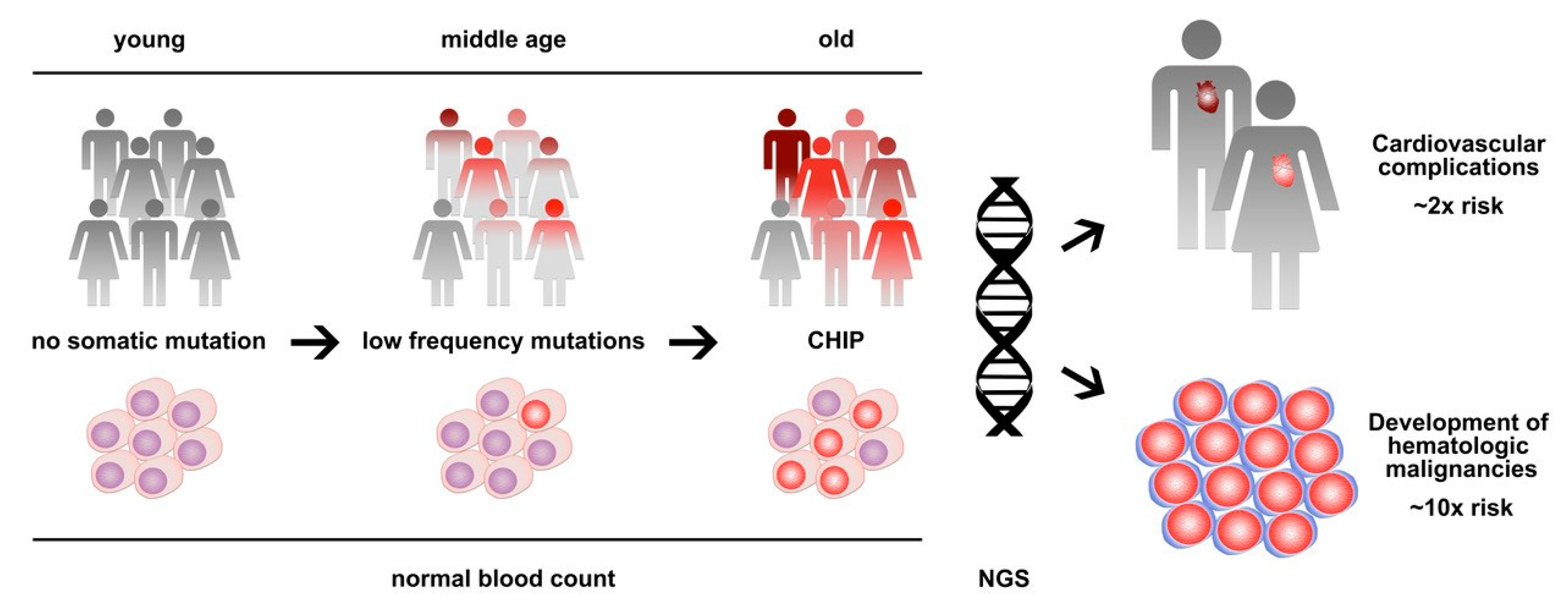
Abstract
:1. Introduction
2. Clonal Hematopoiesis and Hematologic Neoplasms
3. Clonal Hematopoiesis and Cardiovascular Disease
4. Detection of Clonal Hematopoiesis
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Link, D.C.; Walter, M.J. ‘CHIP’ ping away at clonal hematopoiesis. Leukemia 2016, 30, 1633–1635. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Steensma, D.P. Clinical Implications of Clonal Hematopoiesis. Mayo Clin. Proc. 2018, 93, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382.e4. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Valent, P.; Orazi, A.; Steensma, D.P.; Ebert, B.L.; Haase, D.; Malcovati, L.; Van De Loosdrecht, A.A.; Haferlach, T.; Westers, T.M.; Wells, D.A.; et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget 2017, 8, 73483–73500. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Kern, W.; Hoermann, G.; Feenstra, J.D.M.; Sotlar, K.; Pfeilstöcker, M.; Germing, U.; Sperr, W.R.; Reiter, A.; Wolf, D.; et al. Clonal Hematopoiesis with Oncogenic Potential (CHOP): Separation from CHIP and Roads to AML. Int. J. Mol. Sci. 2019, 20, 789. [Google Scholar] [CrossRef] [Green Version]
- Valent, P. ICUS, IDUS, CHIP and CCUS: Diagnostic Criteria, Separation from MDS and Clinical Implications. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2018, 86, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.; Hellström-Lindberg, E.; Gambacorti-Passerini, C.; et al. SomaticSF3B1Mutation in Myelodysplasia with Ring Sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Valent, P.; Horny, H.-P.; Bennett, J.M.; Fonatsch, C.; Germing, U.; Greenberg, P.; Haferlach, T.; Haase, D.; Kolb, H.-J.; Krieger, O.; et al. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: Consensus statements and report from a working conference. Leuk. Res. 2007, 31, 727–736. [Google Scholar] [CrossRef]
- Malcovati, L.; Gallì, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef]
- Kwok, B.; Hall, J.M.; Witte, J.S.; Xu, Y.; Reddy, P.; Lin, K.; Flamholz, R.; Dabbas, B.; Yung, A.; Al-Hafidh, J.; et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood 2015, 126, 2355–2361. [Google Scholar] [CrossRef] [Green Version]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Cohen, N.M.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.E.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef]
- Desai, P.; Trinchant, N.M.; Savenkov, O.; Simon, M.S.; Cheang, G.; Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Ballman, K.V.; et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018, 24, 1015–1023. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.V.; Douville, C.B.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimwade, D.; Ivey, A.; Huntly, B.J.P. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 2016, 127, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höllein, A.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T. NPM1 mutated AML can relapse with wild-type NPM1: Persistent clonal hematopoiesis can drive relapse. Blood Adv. 2018, 2, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Jongen-Lavrencic, M.; Grob, T.; Kavelaars, F.G.; Al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.; Gradowska, P.L.; Meijer, R.; Biemond, B.J.; Kooy, M.V.M.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef]
- Hadzijusufovic, E.; Albrecht-Schgoer, K.; Huber, K.V.M.; Hoermann, G.; Grebien, F.; Eisenwort, G.; Schgoer, W.; Herndlhofer, S.; Kaun, C.; Theurl, M.; et al. Nilotinib-induced vasculopathy: Identification of vascular endothelial cells as a primary target site. Leukemia 2017, 31, 2388–2397. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Hadzijusufovic, E.; Schernthaner, G.-H.; Wolf, D.; Rea, D.; Le Coutre, P. Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood 2015, 125, 901–906. [Google Scholar] [CrossRef]
- Mas-Peiro, S.; Hoffmann, J.; Fichtlscherer, S.; Dorsheimer, L.; Rieger, M.A.; Dimmeler, S.; Vasa-Nicotera, M.; Zeiher, A.M. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur. Hear. J. 2019, 41, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Dorsheimer, L.; Assmus, B.; Rasper, T.; Ortmann, C.A.; Ecke, A.; Abou-El-Ardat, K.; Schmid, T.; Brüne, B.; Wagner, S.; Serve, H.; et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019, 4, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.-L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Abplanalp, W.T.; Mas-Peiro, S.; Cremer, S.; John, D.; Dimmeler, S.; Zeiher, A.M. Association of Clonal Hematopoiesis of Indeterminate Potential With Inflammatory Gene Expression in Patients With Severe Degenerative Aortic Valve Stenosis or Chronic Postischemic Heart Failure. JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.K.; Izukawa, T.; Young, S.; Rosen, G.; Jamali, M.; Zhang, L.; Johnson, D.; Bain, E.; Hilland, J.; Ferrone, C.K.; et al. Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Adv. 2019, 3, 2482–2486. [Google Scholar] [CrossRef] [Green Version]
- Busque, L.; Sun, M.; Buscarlet, M.; Ayachi, S.; Zada, Y.F.; Provost, S.; Bourgoin, V.; Mollica, L.; Meisel, M.; Hinterleitner, R.; et al. High-sensitivity C-reactive protein is associated with clonal hematopoiesis of indeterminate potential. Blood Adv. 2020, 4, 2430–2438. [Google Scholar] [CrossRef]
- Bick, A.G.; Pirruccello, J.P.; Griffin, G.K.; Gupta, N.; Gabriel, S.; Saleheen, D.; Libby, P.; Kathiresan, S.; Natarajan, P. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 2019, 141, 124–131. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J.; Kastelein, J.; Koenig, W.; Genest, J.; Lorenzatti, A.; et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.; Teo, S.-S.; Tiedt, R.; Passweg, J.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation ofJAK2in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Liu, W.; Fidler, T.; Wang, Y.; Tang, Y.; Woods, B.; Welch, C.; Cai, B.; Silvestre-Roig, C.; Ai, D.; et al. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2 (V617F) Mice. Circ. Res. 2018, 123, e35–e47. [Google Scholar] [CrossRef]
- Libby, P.; Ebert, B.L. CHIP (Clonal Hematopoiesis of Indeterminate Potential). Circulation 2018, 138, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Kaasinen, E.; Kuismin, O.; Rajamäki, K.; Ristolainen, H.; Aavikko, M.; Kondelin, J.; Saarinen, S.; Berta, D.G.; Katainen, R.; Hirvonen, E.A.M.; et al. Impact of constitutional TET2 haploinsufficiency on molecular and clinical phenotype in humans. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.-P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragoljevic, D.; Westerterp, M.; Veiga, C.B.; Nagareddy, P.R.; Murphy, A.J. Disordered haematopoiesis and cardiovascular disease: A focus on myelopoiesis. Clin. Sci. 2018, 132, 1889–1899. [Google Scholar] [CrossRef]
- Brunner, A.M.; Blonquist, T.M.; Hobbs, G.S.; Amrein, P.C.; Neuberg, N.S.; Steensma, D.P.; Abel, G.A.; Fathi, A.T. Risk and timing of cardiovascular death among patients with myelodysplastic syndromes. Blood Adv. 2017, 1, 2032–2040. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef] [Green Version]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Crawley, C.; Craig, J.; Scott, M.A.; Understanding Society Scientific Group; et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef]
- Snetsinger, B.; Ferrone, C.K.; Rauh, M.J. Targeted, Amplicon-Based, Next-Generation Sequencing to Detect Age-Related Clonal Hematopoiesis. Methods in Mol. Biol. 2019, 2045, 167–180. [Google Scholar]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Bratman, S.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484. [Google Scholar] [CrossRef] [PubMed]
- Pierre, R.V.; Hoagland, H.C. Age-associated aneuploidy: Loss of Y chromosome from human bone marrow cells with aging. Cancer 1972, 30, 889–894. [Google Scholar] [CrossRef]
- Forsberg, L.A.; Rasi, C.; Malmqvist, N.; Davies, H.; Pasupulati, S.; Pakalapati, G.; Sandgren, J.; De Ståhl, T.D.; Zaghlool, A.; Giedraitis, V.; et al. Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat. Genet. 2014, 46, 624–628. [Google Scholar] [CrossRef] [Green Version]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; VasanthaKumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Wiedmeier, J.E.; Kato, C.; Zhang, Z.; Lee, H.; Dunlap, J.; Nutt, E.; Rattray, R.; McKay, S.; Eide, C.; Press, R.D.; et al. Clonal hematopoiesis as determined by the HUMARA assay is a marker for acquired mutations in epigenetic regulators in older women. Exp. Hematol. 2016, 44, 857–865.e5. [Google Scholar] [CrossRef] [Green Version]
- Laurie, C.C.; Laurie, C.A.; Rice, K.; Doheny, K.F.; Zelnick, L.R.; McHugh, C.P.; Ling, H.; Hetrick, K.N.; Pugh, E.W.; Amos, C.; et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012, 44, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Loh, P.-R.; Genovese, G.; Handsaker, R.E.; Finucane, H.K.; Reshef, Y.A.; Palamara, P.F.; Birmann, B.M.; Talkowski, M.E.; Bakhoum, S.F.; McCarroll, S.A.; et al. Insights into clonal haematopoiesis from 8342 mosaic chromosomal alterations. Nature 2018, 559, 350–355. [Google Scholar] [CrossRef]
- Takahashi, K.; Wang, F.; Kantarjian, H.; Song, X.; Patel, K.; Neelapu, S.; Gumbs, C.; Little, L.; Tippen, S.; Thornton, R.; et al. Copy number alterations detected as clonal hematopoiesis of indeterminate potential. Blood Adv. 2017, 1, 1031–1036. [Google Scholar] [CrossRef]
- Bolton, K.L.; Gillis, N.K.; Coombs, C.C.; Takahashi, K.; Zehir, A.; Bejar, R.; Garcia-Manero, G.; Futreal, A.; Jensen, B.C.; Diaz, L.A.; et al. Managing Clonal Hematopoiesis in Patients With Solid Tumors. J. Clin. Oncol. 2019, 37, 7–11. [Google Scholar] [CrossRef]
- Bolton, K.L.; Zehir, A.; Ptashkin, R.N.; Patel, M.; Gupta, D.; Sidlow, R.; Papaemmanuil, E.; Berger, M.F.; Levine, R.L. The Clinical Management of Clonal Hematopoiesis: Creation of a Clonal Hematopoiesis Clinic. Hematol. Clin. North Am. 2020, 34, 357–367. [Google Scholar] [CrossRef]
- Sidlow, R.; Lin, A.E.; Gupta, D.; Bolton, K.L.; Steensma, D.P.; Levine, R.L.; Ebert, B.L.; Libby, P. The Clinical Challenge of Clonal Hematopoiesis, a Newly Recognized Cardiovascular Risk Factor. JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoermann, G.; Greiner, G.; Griesmacher, A.; Valent, P. Clonal Hematopoiesis of Indeterminate Potential: A Multidisciplinary Challenge in Personalized Hematology. J. Pers. Med. 2020, 10, 94. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm10030094
Hoermann G, Greiner G, Griesmacher A, Valent P. Clonal Hematopoiesis of Indeterminate Potential: A Multidisciplinary Challenge in Personalized Hematology. Journal of Personalized Medicine. 2020; 10(3):94. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm10030094
Chicago/Turabian StyleHoermann, Gregor, Georg Greiner, Andrea Griesmacher, and Peter Valent. 2020. "Clonal Hematopoiesis of Indeterminate Potential: A Multidisciplinary Challenge in Personalized Hematology" Journal of Personalized Medicine 10, no. 3: 94. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm10030094