What Can Machine Learning Approaches in Genomics Tell Us about the Molecular Basis of Amyotrophic Lateral Sclerosis?

 , and
, and
Abstract
:1. Introduction
1.1. Current Knowledge of Molecular Pathways Implicated by the Functions of Known ALS-Linked Genes
- Oxidative stress can also be derived from a stimulation of NADPH oxidase, as observed with ATXN2 mutations [31], or from deficiency in the elimination of Reactive Oxygen Species (ROS) as observed with some SOD1 mutations in familial cases [27,32,33]. It then may contribute to DNA damage. Interestingly, other mutations on the ALS-associated genes NEK1 [34], SETX [35] and C21orf2 [36] are suspected to alter the DNA repair machinery, leading to an accumulation of oxidative damage over time. Consequently, these events could ultimately lead to motor neuron death [34,37].
- Disrupted axonal transport has been directly linked to a mutation in the C-terminal of the ALS-associated gene, KIF5A [12,38], and to mutations in genes encoding for neurofilaments (NEFH), microtubules and motor proteins (PFN1,TUBA4A, DCTN1) [27,39]. Consequently, organelle transport, protein degradation, and RNA transport are affected, disrupting cellular homeostasis. Similarly, axonal transport disruptions have been observed in fALS patients harboring mutations in non-cytoskeletal-related genes such as SOD1 [38].
- Protein degradation is suspected to be a key pathway that is defective in ALS. This can be a direct consequence of mutations in ALS-associated genes involved in proteasome activity and the autophagy pathway, such as UBQLN2, VCP, SQSTM1/P62, OPTN, FIG4, Spg11, or TBK1 [40], and may lead to an accumulation of misfolded and non-functional proteins [27,41]. It can also be an indirect consequence of other mutations leading to the formation of protein aggregates such as SOD1, FUS, TDP43, C9orf72-derived DPR - aggregates that in turn impair the proteasome and autophagic degradation pathways [42], thus exacerbating the accumulation of misfolded proteins. Consequently, the blockade of autophagy pathways may affect vesicle secretion [43,44]. Interestingly, some ALS-associated genes are known to be directly or indirectly involved in exosome biogenesis such as CHMP2B [45] or C9orf72 [46], respectively.
- RNA processing and metabolism is another key pathway affected in ALS. For example, mutations to RNA-binding proteins encoded by FUS, TDP-43, hnRNPA1, hnRNPA2B1, and MATR3, result in altered mRNA splicing, RNA nucleocytoplasmic transport and translation [27,50,51,52,53,54,55], as well as in the generation and accumulation of toxic stress granules [56]. Similarly, accumulation of toxic RNA foci can be observed in motor neurons in the context of C9orf72 mutations, and may lead to the sequestration of splicing proteins, thus affecting RNA maturation and translation [57]. The biogenesis of microRNA is also directly affected by mutated FUS, TDP-43, or C9orf72-mediated DPRs, thus having an impact on the expression of genes involved in motor neuron survival [27,58].
1.2. The Genetic Architecture of ALS
2. ALS-Specific GWAS Challenges and Limitations
3. Facing the Challenges
3.1. A Brief Overview of Machine Learning Concepts
3.2. Recent Feature Preparation and Selection Approaches in ALS Genomic Studies
3.3. Experimental Design and Results of the ALS Gene Prioritization Approaches
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef]
- Chiò, A.; Logroscino, G.; Traynor, B.; Collins, J.; Simeone, J.; Goldstein, L.; White, L. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Al-Chalabi, A.; Van Den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [Green Version]
- Nicaise, C.; Mitrecic, D.; Pochet, R. Brain and spinal cord affected by amyotrophic lateral sclerosis induce differential growth factors expression in rat mesenchymal and neural stem cells. Neuropathol. Appl. Neurobiol. 2011, 37, 179–188. [Google Scholar] [CrossRef]
- McLaughlin, L.R.; Vajda, A.; Hardiman, O. Heritability of amyotrophic lateral sclerosis insights from disparate numbers. Jama Neurol. 2015, 72, 857–858. [Google Scholar] [CrossRef]
- Bush, W.S.; Moore, J.H. Chapter 11: Genome-Wide Association Studies. PLoS Comput. Biol. 2012, 8, e1002822. [Google Scholar] [CrossRef] [Green Version]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef]
- Klein, R.J.; Xu, X.; Mukherjee, S.; Willis, J.; Hayes, J. Successes of Genome-wide association studies. Cell 2010, 142, 350–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, A.; Kenna, K.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Rasheed, A.; Tikkanen, E.; Lee, J.J.; Butterworth, A.S.; Howson, J.M.; Assimes, T.L.; Chowdhury, R.; Orho-Melander, M.; Damrauer, S.; et al. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat. Genet. 2017, 49, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Duncan, L.; Yilmaz, Z.; Gaspar, H.; Walters, R.; Goldstein, J.; Anttila, V.; Bulik-Sullivan, B.; Ripke, S.; Thornton, L.; Hinney, A.; et al. Significant locus and metabolic genetic correlations revealed in genome-wide association study of anorexia nervosa. Am. J. Psychiatry 2017, 174, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Van Rheenen, W.; Pulit, S.L.; Dekker, A.M.; Al Khleifat, A.; Brands, W.J.; Iacoangeli, A.; Kenna, K.P.; Kavak, E.; Kooyman, M.; McLaughlin, R.L.; et al. Project MinE: Study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur. J. Hum. Genet. 2018, 26, 1537–1546. [Google Scholar] [CrossRef] [Green Version]
- Mailman, M.D.; Feolo, M.; Jin, Y.; Kimura, M.; Tryka, K.; Bagoutdinov, R.; Hao, L.; Kiang, A.; Paschall, J.; Phan, L.; et al. The NCBI dbGaP database of genotypes and phenotypes. Nat. Genet. 2007, 39, 1181–1186. [Google Scholar] [CrossRef] [Green Version]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef]
- Vijayakumar, U.G.; Milla, V.; Stafford, M.Y.C.; Bjourson, A.J.; Duddy, W.; Duguez, S.M.R. A systematic review of suggested molecular strata, biomarkers and their tissue sources in ALS. Front. Neurol. 2019, 10, 400. [Google Scholar] [CrossRef] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3. [Google Scholar] [CrossRef]
- Turner, M.R.; Al-Chalabi, A.; Chio, A.; Hardiman, O.; Kiernan, M.C.; Rohrer, J.D.; Rowe, J.; Seeley, W.; Talbot, K. Genetic screening in sporadic ALS and FTD. J. Neurol. Neurosurg. Psychiatry 2017, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 2018, 30, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.Y.; Zhou, Z.R.; Che, C.H.; Liu, C.Y.; He, R.L.; Huang, H.P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- Connolly, O.; Le Gall, L.; McCluskey, G.; Donaghy, C.G.; Duddy, W.J.; Duguez, S. A Systematic Review of Genotype–Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS. J. Pers. Med. 2020, 10, 58. [Google Scholar] [CrossRef]
- Morgan, S.; Duguez, S.; Duddy, W. Personalized Medicine and Molecular Interaction Networks in Amyotrophic Lateral Sclerosis (ALS): Current Knowledge. J. Pers. Med. 2018, 8, 44. [Google Scholar] [CrossRef] [Green Version]
- Gall, L.L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duguez, S. Molecular and cellular mechanisms affected in ALS. J. Pers. Med 2020, 10, 101. [Google Scholar] [CrossRef]
- Volonté, C.; Morello, G.; Spampinato, A.G.; Amadio, S.; Apolloni, S.; D’Agata, V.; Cavallaro, S. Omics-based exploration and functional validation of neurotrophic factors and histamine as therapeutic targets in ALS. Ageing Res. Rev. 2020, 62, 101121. [Google Scholar] [CrossRef]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11, 1005357. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Lan, M.; Mojsilovic-Petrovic, J.; Choi, W.H.; Safren, N.; Barmada, S.; Lee, M.J.; Kalb, R. The proline/arginine dipeptide from hexanucleotide repeat expanded C9ORF72 inhibits the proteasome. eNeuro 2017, 4, 249–265. [Google Scholar] [CrossRef] [Green Version]
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Kong, Q.; Shan, X.; Tian, G.; Ilieva, H.; Cleveland, D.W.; Rothstein, J.D.; Borchelt, D.R.; Wong, P.C.; Lin, C.L.G. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE 2008, 3, e2849. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Lin, H.; Wang, X.; Zuo, Q.; Qin, J.; Zhang, P. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim. Biophys. Sin. 2015, 47, 834–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; Van Der Spek, R.A.; Võsa, U.; De Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higelin, J.; Catanese, A.; Semelink-Sedlacek, L.L.; Oeztuerk, S.; Lutz, A.K.; Bausinger, J.; Barbi, G.; Speit, G.; Andersen, P.M.; Ludolph, A.C.; et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 2018, 30, 150–162. [Google Scholar] [CrossRef]
- De vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; Mcloughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef] [Green Version]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate invitro and invivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Greg Stacey, R.; Grad, L.I.; et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal exocytosis, exosome release and secretory autophagy: The autophagic- and endo-lysosomal systems go extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson, N.; Ince, P.G.; Smith, M.O.; Highley, R.; Skibinski, G.; Andersen, P.M.; Morrison, K.E.; Pall, H.S.; Hardiman, O.; Collinge, J.; et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006, 67, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Blanc, L.; Vidal, M. New insights into the function of Rab GTPases in the context of exosomal secretion. Small GTPases 2018, 9, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laslo, P.; Lipski, J.; Nicholson, L.F.; Miles, G.B.; Funk, G.D. GluR2 AMPA Receptor Subunit Expression in Motoneurons at Low and High Risk for Degeneration in Amyotrophic Lateral Sclerosis. Exp. Neurol. 2001, 169, 461–471. [Google Scholar] [CrossRef]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: A reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Milanese, M.; Zappettini, S.; Onofri, F.; Musazzi, L.; Tardito, D.; Bonifacino, T.; Messa, M.; Racagni, G.; Usai, C.; Benfenati, F.; et al. Abnormal exocytotic release of glutamate in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2011, 116, 1028–1042. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Ebmeier, C.C.; Podell, E.R.; Heimiller, J.; Taatjes, D.J.; Cech, T.R. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 2012, 26, 2690–2695. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E.; Baralle, F.E. Characterization and Functional Implications of the RNA Binding Properties of Nuclear Factor TDP-43, a Novel Splicing Regulator of CFTR Exon 9. J. Biol. Chem. 2001, 276, 36337–36343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leblond, C.S.; Gan-Or, Z.; Spiegelman, D.; Laurent, S.B.; Szuto, A.; Hodgkinson, A.; Dionne-Laporte, A.; Provencher, P.; de Carvalho, M.; Orrù, S.; et al. Replication study of MATR3 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2016, 37, 17–209. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, D.; Akinyi, M.V.; Mechtersheimer, J.; Frilander, M.J.; Ruepp, M.D. The emerging role of minor intron splicing in neurological disorders. Cell Stress 2018, 2, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobágyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef]
- Kumar, V.; Hasan, G.M.; Hassan, M.I. Unraveling the role of RNA mediated toxicity of C9orf72 repeats in C9-FTD/ALS. Front. Neurosci. 2017, 11, 711. [Google Scholar] [CrossRef]
- Kawahara, Y.; Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352. [Google Scholar] [CrossRef] [Green Version]
- Al-Chalabi, A.; Fang, F.; Hanby, M.F.; Leigh, P.N.; Shaw, C.E.; Ye, W.; Rijsdijk, F. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1324–1326. [Google Scholar] [CrossRef]
- Dion, P.A.; Daoud, H.; Rouleau, G.A. Genetics of motor neuron disorders: New insights into pathogenic mechanisms. Nat. Rev. Genet. 2009, 10, 769–782. [Google Scholar] [CrossRef]
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef]
- Myers, R.H. Huntington’s Disease Genetics. NeuroRx 2004, 1, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Loh, P.R.; Bhatia, G.; Gusev, A.; Finucane, H.K.; Bulik-Sullivan, B.K.; Pollack, S.J.; Lee, H.; Wray, N.R.; Kendler, K.S.; O’donovan, M.C.; et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet. 2015, 47, 1385. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.; Heverin, M.; Elamin, M.; Bede, P.; Lynch, C.; Kenna, K.; MacLaughlin, R.; Walsh, C.; Al Chalabi, A.; Hardiman, O. Aggregation of neurologic and neuropsychiatric disease in amyotrophic lateral sclerosis kindreds: A population-based case-control cohort study of familial and sporadic amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Majounie, E.; Abramzon, Y.; Renton, A.E.; Perry, R.; Bassett, S.S.; Pletnikova, O.; Troncoso, J.C.; Hardy, J.; Singleton, A.B.; Traynor, B.J. Repeat expansion in C9ORF72 in Alzheimer’s disease. N. Engl. J. Med. 2012, 366, 283. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Le Ber, I.; Condroyer, C.; Broussolle, E.; Gabelle, A.; Thobois, S.; Pasquier, F.; Mondon, K.; Dion, P.A.; Rochefort, D.; et al. C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain 2013, 136, 385–391. [Google Scholar] [CrossRef]
- Majoor-Krakauer, D.; Ottman, R.; Johnson, W.G.; Rowland, L.P. Familial aggregation of amyotrophic lateral sclerosis, dementia, and Parkinson’s disease: Evidence of shared genetic susceptibility. Neurology 1994, 44, 1872–1877. [Google Scholar] [CrossRef]
- Karch, C.M.; Wen, N.; Fan, C.C.; Yokoyama, J.S.; Kouri, N.; Ross, O.A.; Höglinger, G.; Müller, U.; Ferrari, R.; Hardy, J.; et al. Selective genetic overlap between amyotrophic lateral sclerosis and diseases of the frontotemporal dementia spectrum. JAMA Neurol. 2018, 75, 860–875. [Google Scholar] [CrossRef]
- O’Brien, M.; Burke, T.; Heverin, M.; Vajda, A.; McLaughlin, R.; Gibbons, J.; Byrne, S.; Pinto-Grau, M.; Elamin, M.; Pender, N.; et al. Clustering of neuropsychiatric disease in first-degree and second-degree relatives of patients with amyotrophic lateral sclerosis. JAMA Neurol. 2017, 74, 1425–1430. [Google Scholar] [CrossRef]
- Stearns, F.W. One hundred years of pleiotropy: A retrospective. Genetics 2010, 186, 767–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Liu, H.; Xiong, H.; Cao, J.; Chen, J. K-means-based consensus clustering: A unified view. IEEE Trans. Knowl. Data Eng. 2015, 27, 155–169. [Google Scholar] [CrossRef]
- Wagstaff, K.; Cardie, C.; Rogers, S.; Schroedl, S. Constrained K-means Clustering with Background Knowledge. Proc. Eighteenth Int. Conf. Mach. Learn. 2001, 1, 577–584. [Google Scholar]
- MacQueen, J.B. Some methods for classification and analysis of multivariate observations. In Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability; University of California Press: Berkeley, CA, USA, 1967; pp. 281–297. [Google Scholar]
- Schymick, J.C.; Scholz, S.W.; Fung, H.C.; Britton, A.; Arepalli, S.; Gibbs, J.R.; Lombardo, F.; Matarin, M.; Kasperaviciute, D.; Hernandez, D.G.; et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: First stage analysis and public release of data. Lancet Neurol. 2007, 6, 322–328. [Google Scholar] [CrossRef]
- Zuk, O.; Hechter, E.; Sunyaev, S.R.; Lander, E.S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA 2012, 109, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.A.; Pettersson, F.H.; Clarke, G.M.; Cardon, L.R.; Morris, P.; Zondervan, K.T. Data quality control in genetic case-control association studies. Nat. Protoc. 2011, 5, 1564–1573. [Google Scholar] [CrossRef] [Green Version]
- Marees, A.T.; de Kluiver, H.; Stringer, S.; Vorspan, F.; Curis, E.; Marie-Claire, C.; Derks, E.M. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 2018, 27, 1–10. [Google Scholar] [CrossRef]
- Verma, S.S.; de Andrade, M.; Tromp, G.; Kuivaniemi, H.; Pugh, E.; Namjou-Khales, B.; Mukherjee, S.; Jarvik, G.P.; Kottyan, L.C.; Burt, A.; et al. Imputation and quality control steps for combining multiple genome-wide datasets. Front. Genet. 2014, 5, 1–15. [Google Scholar] [CrossRef]
- Laurie, C.C.; Doheny, K.F.; Mirel, D.B.; Pugh, E.W.; Bierut, L.J.; Bhangale, T.; Boehm, F.; Caporaso, N.E.; Cornelis, M.C.; Edenberg, H.J.; et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet. Epidemiol. 2011, 34, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Dudbridge, F.; Gusnanto, A. Estimation of significance thresholds for genomewide association scans. Genet. Epidemiol. 2008, 32, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Culverhouse, R.; Suarez, B.K.; Lin, J.; Reich, T. A perspective on epistasis: Limits of models displaying no main effect. Am. J. Hum. Genet. 2002, 70, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemani, G.; Knott, S.; Haley, C. An Evolutionary Perspective on Epistasis and the Missing Heritability. PLoS Genet. 2013, 9, 1003295. [Google Scholar] [CrossRef] [Green Version]
- Bateson, W.; Saunders, E.; Punnett, R.; Sons, C.H.U.H. Reports to the Evolution Committee of the Royal Society, Report II. London. R. Soc. 1905, 2, 5–131. [Google Scholar]
- de Visser, J.A.G.M.; Cooper, T.F.; Elena, S.F. The causes of epistasis. Proc. R. Soc. B Biol. Sci. 2011, 278, 3617–3624. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Hu, T.; Moore, J.H. Epistasis, Complexity, and Multifactor Dimensionality Reduction; Humana Press: Totowa, NJ, USA, 2013; pp. 465–477. [Google Scholar] [CrossRef]
- Churchill, G.A. Epistasis. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 505–507. [Google Scholar] [CrossRef]
- Goudey, B.; Rawlinson, D.; Wang, Q.; Shi, F.; Ferra, H.; Campbell, R.M.; Stern, L.; Inouye, M.T.; Ong, C.S.; Kowalczyk, A. GWIS–model-free, fast and exhaustive search for epistatic interactions in case-control GWAS. BMC Genom. 2013, 14 (Suppl. S3), 1–18. [Google Scholar] [CrossRef] [Green Version]
- Yin, B.; Balvert, M.; Van Der Spek, R.A.; Dutilh, B.E.; Bohté, S.; Veldink, J.; Schönhuth, A. Using the structure of genome data in the design of deep neural networks for predicting amyotrophic lateral sclerosis from genotype. Bioinformatics 2019, 35, i538–i547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.C.; Andrews, P.C.; Asselbergs, F.W.; Frost, H.R.; Williams, S.M.; Harris, B.T.; Read, C.; Askland, K.D.; Moore, J.H. Gene ontology analysis of pairwise genetic associations in two genome-wide studies of sporadic ALS. BioData Min. 2012, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Reich, D.E.; Lander, E.S. On the allelic spectrum of human disease. Trends Genet. 2001, 17, 502–510. [Google Scholar] [CrossRef]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [CrossRef] [Green Version]
- Naj, A.C. Genotype Imputation in Genome-Wide Association Studies. Curr. Protoc. Hum. Genet. 2019, 102, e84. [Google Scholar] [CrossRef] [PubMed]
- Pistis, G.; Porcu, E.; Vrieze, S.I.; Sidore, C.; Steri, M.; Danjou, F.; Busonero, F.; Mulas, A.; Zoledziewska, M.; Maschio, A.; et al. Rare variant genotype imputation with thousands of study-specific whole-genome sequences: Implications for cost-effective study designs. Eur. J. Hum. Genet. 2015, 23, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [Green Version]
- Myszczynska, M.A.; Ojamies, P.N.; Lacoste, A.M.; Neil, D.; Saffari, A.; Mead, R.; Hautbergue, G.M.; Holbrook, J.D.; Ferraiuolo, L. Applications of machine learning to diagnosis and treatment of neurodegenerative diseases. Nat. Rev. Neurol. 2020, 16, 440–456. [Google Scholar] [CrossRef] [PubMed]
- Deo, R.C. Machine learning in medicine. Circulation 2015, 132, 1920–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, G.; Sahin, F. A survey on feature selection methods. Comput. Electr. Eng. 2014, 40, 16–28. [Google Scholar] [CrossRef]
- Adadi, A.; Berrada, M. Peeking Inside the Black-Box: A Survey on Explainable Artificial Intelligence (XAI). IEEE Access 2018, 6, 52138–52160. [Google Scholar] [CrossRef]
- Zhang, Q.S.; Zhu, S.C. Visual interpretability for deep learning: A survey. Front. Inf. Technol. Electron. Eng. 2018, 19, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Mirza, B.; Wang, W.; Wang, J.; Choi, H.; Chung, N.C.; Ping, P. Machine learning and integrative analysis of biomedical big data. Genes 2019, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Vitsios, D.; Petrovski, S. Mantis-ml: Disease-Agnostic Gene Prioritization from High-Throughput Genomic Screens by Stochastic Semi-supervised Learning. Am. J. Hum. Genet. 2020, 106, 659–678. [Google Scholar] [CrossRef]
- Yousefian-Jazi, A.; Sung, M.K.; Lee, T.; Hong, Y.H.; Choi, J.K.; Choi, J. Functional fine-mapping of noncoding risk variants in amyotrophic lateral sclerosis utilizing convolutional neural network. Sci. Rep. 2020, 10, 12872. [Google Scholar] [CrossRef] [PubMed]
- Bean, D.M.; Al-Chalabi, A.; Dobson, R.J.B.; Iacoangeli, A. A Knowledge-Based Machine Learning Approach to Gene Prioritisation in Amyotrophic Lateral Sclerosis. Genes 2020, 11, 668. [Google Scholar] [CrossRef] [PubMed]
- Cronin, S.; Berger, S.; Ding, J.; Schymick, J.C.; Washecka, N.; Hernandez, D.G.; Greenway, M.J.; Bradley, D.G.; Traynor, B.J.; Hardiman, O. A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum. Mol. Genet. 2008, 17, 768–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, C.S.; Sinnott-Armstrong, N.A.; Himmelstein, D.S.; Park, P.J.; Moore, J.H.; Harris, B.T. Multifactor dimensionality reduction for graphics processing units enables genome-wide testing of epistasis in sporadic ALS. Bioinformatics 2010, 26, 694–695. [Google Scholar] [CrossRef]
- Sha, Q.; Zhang, Z.; Schymick, J.C.; Traynor, B.J.; Zhang, S. Genome-wide association reveals three SNPs associated with sporadic amyotrophic lateral sclerosis through a two-locus analysis. BMC Med Genet. 2009, 10, 86. [Google Scholar] [CrossRef] [Green Version]
- Köhler, S.; Vasilevsky, N.A.; Engelstad, M.; Foster, E.; McMurry, J.; Aymé, S.; Baynam, G.; Bello, S.M.; Boerkoel, C.F.; Boycott, K.M.; et al. The human phenotype ontology in 2017. Nucleic Acids Res. 2017, 45, D865–D876. [Google Scholar] [CrossRef]
- Van Der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 3, 861. [Google Scholar] [CrossRef]
- Ritchie, M.D.; Hahn, L.W.; Roodi, N.; Bailey, L.R.; Dupont, W.D.; Parl, F.F.; Moore, J.H. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am. J. Hum. Genet. 2001, 69, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Matys, V.; Fricke, E.; Geffers, R.; Gößling, E.; Haubrock, M.; Hehl, R.; Hornischer, K.; Karas, D.; Kel, A.E.; Kel-Margoulis, O.V.; et al. TRANSFAC®: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003, 31, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Bryne, J.C.; Valen, E.; Tang, M.H.E.; Marstrand, T.; Winther, O.; Da piedade, I.; Krogh, A.; Lenhard, B.; Sandelin, A. JASPAR, the open access database of transcription factor-binding profiles: New content and tools in the 2008 update. Nucleic Acids Res. 2008, 36, D102–D106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bean, D.M.; Wu, H.; Dzahini, O.; Broadbent, M.; Stewart, R.; Dobson, R.J. Knowledge graph prediction of unknown adverse drug reactions and validation in electronic health records. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Dudbridge, F. Power and Predictive Accuracy of Polygenic Risk Scores. PLoS Genet. 2013, 9, 1003348. [Google Scholar] [CrossRef]
- Boser, B.E.; Guyon, I.M.; Vapnik, V.N. Training algorithm for optimal margin classifiers. In Proceedings of the Fifth Annual ACM Workshop on Computational Learning Theory; ACM: New York, NY, USA, 1992; pp. 144–152. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Freund, Y.; Schapire, R.E. A Short Introduction to Boosting. J. Jpn. Soc. Artif. Intell. 1999, 14, 771–780. [Google Scholar]
- Friedman, J.; Hastie, T.; Tibshirani, R. Additive logistic regression: A statistical view of boosting (With discussion and a rejoinder by the authors). Ann. Stat. 2000, 28, 337–407. [Google Scholar] [CrossRef]
- Lee, T.; Sung, M.K.; Lee, S.; Yang, W.; Oh, J.; Kim, J.Y.; Hwang, S.; Ban, H.J.; Choi, J.K. Convolutional neural network model to predict causal risk factors that share complex regulatory features. Nucleic Acids Res. 2019, 47, 146. [Google Scholar] [CrossRef] [Green Version]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.R.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Cardona, A.E. The myeloid cells of the central nervous system parenchyma. Nature 2010, 468, 253–262. [Google Scholar] [CrossRef]
- Liu, N.; Yu, Z.; Xun, Y.; Li, M.; Peng, X.; Xiao, Y.; Hu, X.; Sun, Y.; Yang, M.; Gan, S.; et al. TNFAIP1 contributes to the neurotoxicity induced by Aβ25-35 in Neuro2a cells. BMC Neurosci. 2016, 17, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, B.E.; Barve, R.A.; Maloney, S.E.; Strickland, A.; Rensing, N.; Wang, P.L.; Wong, M.; Head, R.; Wozniak, D.F.; Milbrandt, J. Abnormal microglia and enhanced inflammation-related gene transcription in mice with conditional deletion of Ctcf in Camk2a-Cre-expressing neurons. J. Neurosci. 2018, 38, 200–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagamoto-Combs, K.; Combs, C.K. Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells). J. Neurosci. 2010, 30, 9641–9646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limviphuvadh, V.; Tanaka, S.; Goto, S.; Ueda, K.; Kanehisa, M. The commonality of protein interaction networks determined in neurodegenerative disorders (NDDs). Bioinformatics 2007, 23, 2129–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mademan, I.; Harmuth, F.; Giordano, I.; Timmann, D.; Magri, S.; Deconinck, T.; Claaßen, J.; Jokisch, D.; Genc, G.; Di Bella, D.; et al. Multisystemic SYNE1 ataxia: Confirming the high frequency and extending the mutational and phenotypic spectrum. Brain 2016, 139, e46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergemalm, D.; Forsberg, K.; Jonsson, P.A.; Graffmo, K.S.; Brä Nnströ M §, T.; Andersen, P.M.; Antti, H.; Marklund, S.L. Changes in the Spinal Cord Proteome of an Amyotrophic Lateral Sclerosis Murine Model Determined by Differential In-gel Electrophoresis. Mol. Cell. Proteom. 2009, 8, 1306–1317. [Google Scholar] [CrossRef] [Green Version]
- Saris, C.G.; Horvath, S.; van Vught, P.W.; van Es, M.A.; Blauw, H.M.; Fuller, T.F.; Langfelder, P.; DeYoung, J.; Wokke, J.H.; Veldink, J.H.; et al. Weighted gene co-expression network analysis of the peripheral blood from Amyotrophic Lateral Sclerosis patients. BMC Genom. 2009, 10, 405. [Google Scholar] [CrossRef] [Green Version]
- Andrés-Benito, P.; Moreno, J.; Aso, E.; Povedano, M.; Ferrer, I. Amyotrophic lateral sclerosis, gene deregulation in the anterior horn of the spinal cord and frontal cortex area 8: Implications in frontotemporal lobar degeneration. Aging 2017, 9, 823–851. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J.; Wong, M. Aberrant Regulation of DNA Methylation in Amyotrophic Lateral Sclerosis: A New Target of Disease Mechanisms. Neurotherapeutics 2013, 10, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.; Gertz, B.; Chestnut, B.A.; Martin, L.J. Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Front. Cell. Neurosci. 2013, 7, 279. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, T.K.; Zhong, J.; Mathivanan, S.; Karthick, L.; Chandrika, K.N.; Mohan, S.S.; Sharma, S.; Pinkert, S.; Nagaraju, S.; Periaswamy, B.; et al. Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat. Genet. 2006, 38, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Orchard, S. The MIntAct Project and Molecular Interaction Databases. Methods Mol. Biol. 2016, 1415, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N.; et al. The MIntAct project - IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [Green Version]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol. 2015, 11, 1–19. [Google Scholar] [CrossRef]
- Moore, J.H.; Williams, S.M. Epistasis: Methods and Protocols. Epistasis Methods Protoc. 2014, 1253, 1–346. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; Lu, T.P. Gene-gene interaction: The curse of dimensionality. Ann. Transl. Med. 2019, 7, 813. [Google Scholar] [CrossRef]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef]
- Bellot, P.; de los Campos, G.; Pérez-Enciso, M. Can deep learning improve genomic prediction of complex human traits? Genetics 2018, 210, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Felice, B.; Guida, M.; Guida, M.; Coppola, C.; De Mieri, G.; Cotrufo, R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene 2012, 508, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Maragakis, N.J.; Dykes-Hoberg, M.; Rothstein, J.D. Altered Expression of the Glutamate Transporter EAAT2b in Neurological Disease. Ann. Neurol. 2004, 55, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Tzu, J.; Marinkovich, M.P. Bridging structure with function: Structural, regulatory, and developmental role of laminins. Int. J. Biochem. Cell Biol. 2008, 40, 199–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.R.; An, C.H.; Yoo, N.J.; Lee, S.H. Laminin gene LAMB4 is somatically mutated and expressionally altered in gastric and colorectal cancers. APMIS 2015, 123, 65–71. [Google Scholar] [CrossRef]
- Zhang, H.; Pan, Y.Z.; Cheung, M.; Cao, M.; Yu, C.; Chen, L.; Zhan, L.; He, Z.W.; Sun, C.Y. LAMB3 mediates apoptotic, proliferative, invasive, and metastatic behaviors in pancreatic cancer by regulating the PI3K/Akt signaling pathway. Cell Death Dis. 2019, 10, 230. [Google Scholar] [CrossRef] [Green Version]
- Jena, K.K.; Kolapalli, S.P.; Mehto, S.; Nath, P.; Das, B.; Sahoo, P.K.; Ahad, A.; Syed, G.H.; Raghav, S.K.; Senapati, S.; et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018, 37, e98358. [Google Scholar] [CrossRef]
- Grassi, D.A.; Jönsson, M.E.; Brattås, P.L.; Jakobsson, J. TRIM28 and the control of transposable elements in the brain. Brain Res. 2019, 1705, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jin, Y.; Prazak, L.; Hammell, M.; Dubnau, J. Transposable Elements in TDP-43-Mediated Neurodegenerative Disorders. PLoS ONE 2012, 7, e44099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, H.L.; John, C.R.; Watson, D.S.; Munroe, P.B.; Barnes, M.R.; Cabrera, C.P. Reaching the End-Game for GWAS: Machine Learning Approaches for the Prioritization of Complex Disease Loci. Front. Genet. 2020, 11, 350. [Google Scholar] [CrossRef]
- Ritchie, M.D.; Van Steen, K. The search for gene-gene interactions in genome-wide association studies: Challenges in abundance of methods, practical considerations, and biological interpretation. Ann. Transl. Med. 2018, 6, 157. [Google Scholar] [CrossRef]
- Ritchie, M.D. Using Biological Knowledge to Uncover the Mystery in the Search for Epistasis in Genome-Wide Association Studies. Ann. Hum. Genet. 2011, 75, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]



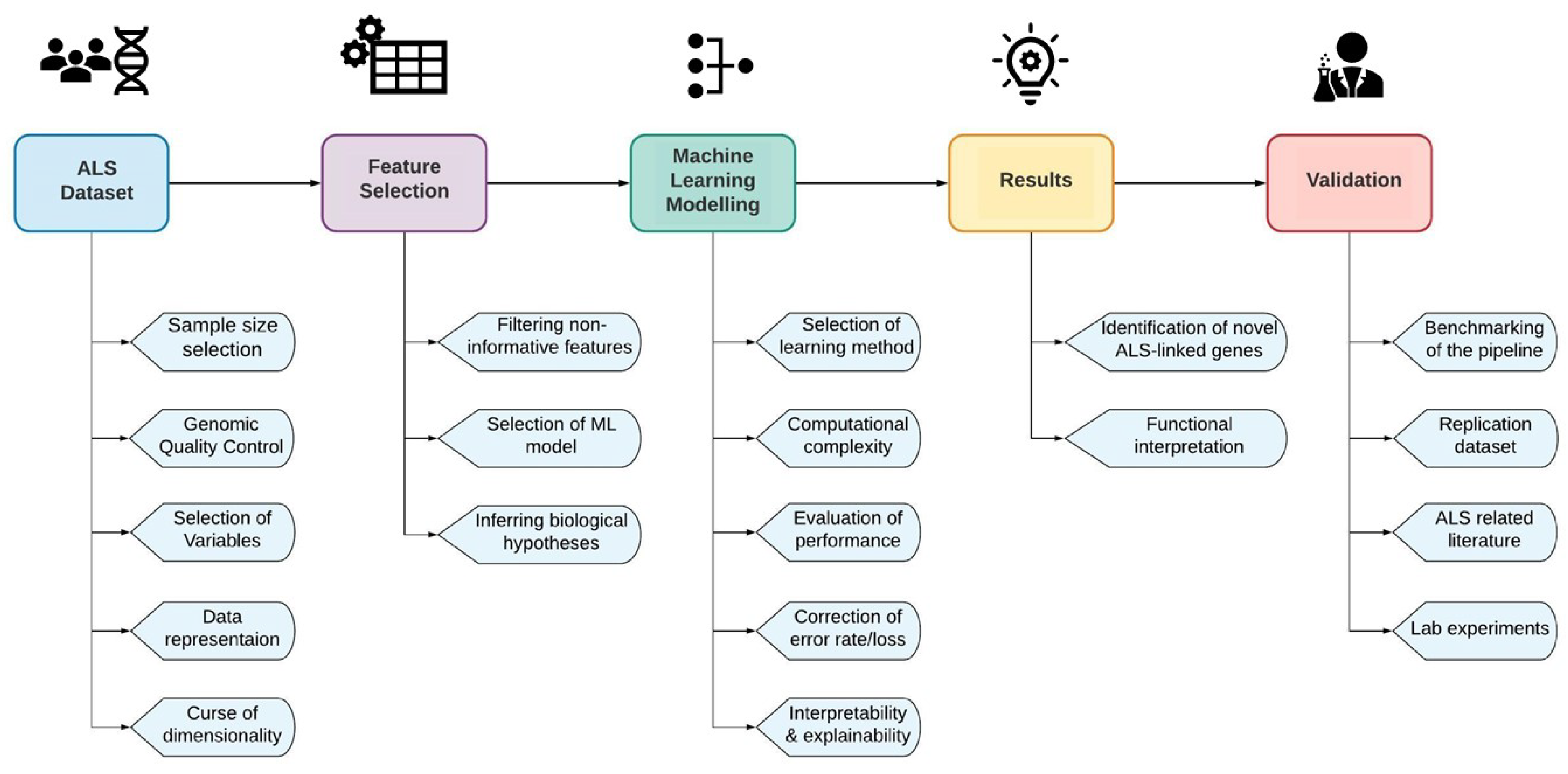{kind=link}
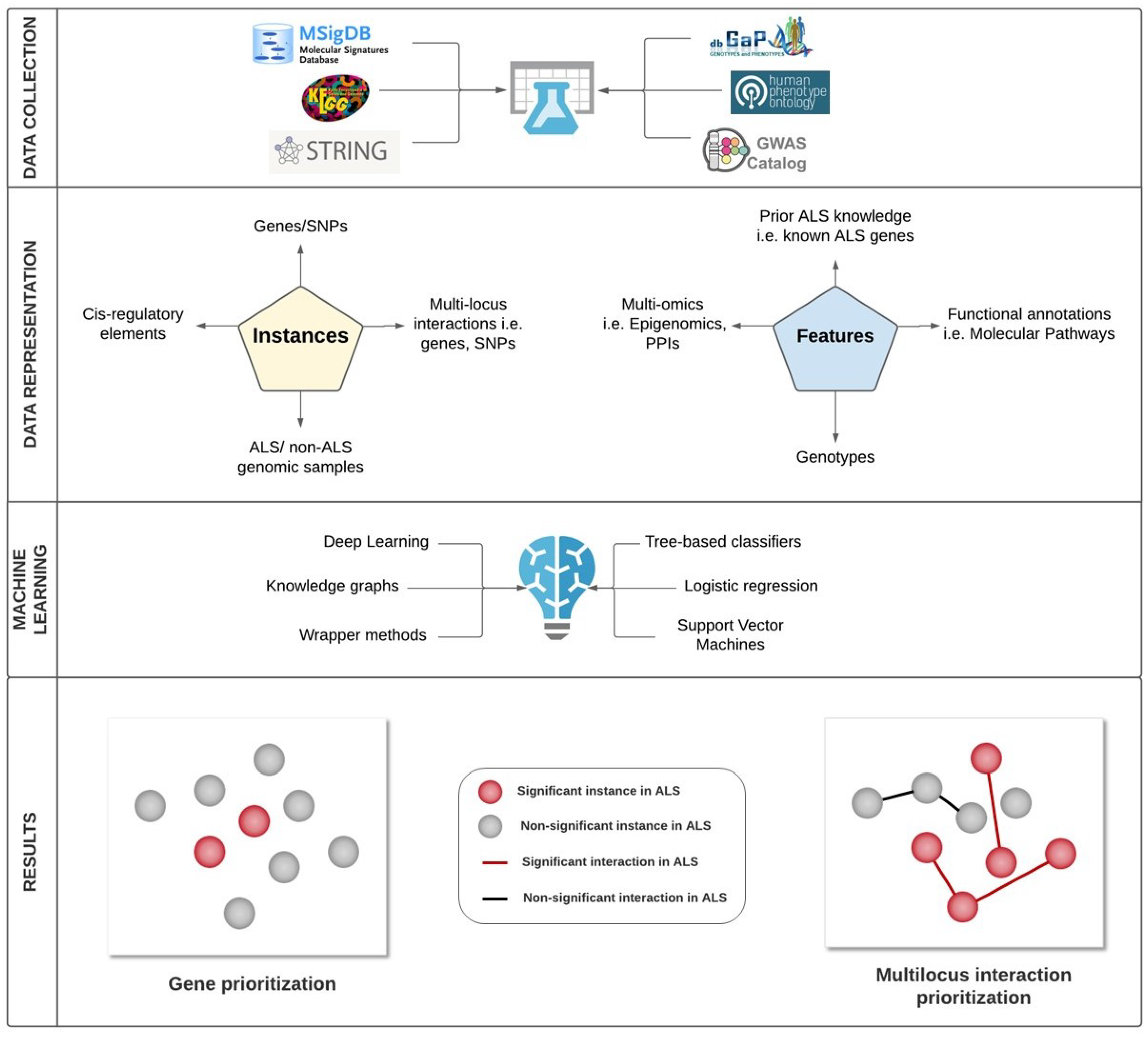{kind=link}
| Study | Data/Instances | Features | Genomic Structure | Epistasis | Cis-Regulatory Elements | ALS-Linked Knowledge | ML Methods |
|---|---|---|---|---|---|---|---|
| Vitsios et al. [103] | 18,626 coding genes (label:positive/unlabelled) | 1,249 gene-annotations: generic, disease- and tissue -specific features | No | No | No | Yes | PCA, t-SNE, UMAP |
| Yousefian et al. [104] | 8,697,640 SNP p-values of 14,791 ALS cases and 26,898 controls [36] | 2,252 functional features: DHS mapping data, histone modifications, target gene functions, and TFBS | Yes | No | Yes | Yes | None |
| Bean et al. [105] | ALS-linked gene lists: DisGeNet: 101 genes ALSoD: 126 genes, ClinVar: 44 genes, Manual list: 40 genes Union: 199 genes | PPIs, disease-gene associations and functional annotations | No | No | No | Yes | None |
| Yin et al. [90] | 4511 cases and 7397 controls [16] | 823,504 SNPs from 7, 9, 17 and 22 chromosomes | Yes | No | Yes | Yes | CNN |
| Kim et al. [91] | SNP pairwise interactions | 550,000 SNPs of 276 cases/271 controls and 211 cases/211 controls [76,106] | No | Yes | No | Yes | MDR |
| Greene et al. [107] | SNP pairwise interactions | 210,382 SNPs of 276 cases/271 controls and 211 cases/211 controls [76,106] | No | Yes | No | No | MDR |
| Sha et al. [108] | SNP pairwise interactions | 555,352 SNPs of 276 cases/271 controls [76] | No | No | No | Yes | None |
| Genomic Structure | Epistasis | Cis-Regulatory Elements | ALS-Linked Knowledge | Functional Annotations | |
|---|---|---|---|---|---|
| Vitsios et al. [103] | No | No | No | Yes | Yes |
| Yousefian et al. [104] | Yes | No | Yes | Yes | Yes |
| Bean et al. [105] | No | No | No | Yes | Yes |
| Yin et al. [90] | Yes | Yes | Yes | No | No |
| Kim et al. [91] | No | Yes | Yes | Yes | Yes |
| Greene et al. [107] | No | Yes | No | No | No |
| Sha et al. [108] | No | Yes | No | Yes | No |
| Study | Machine Learning Models | Model Assessment | Performance |
|---|---|---|---|
| Vitsios et al. [103] | Stochastic Semi-supervised Learning: Stacking, DNN, Gradient Boosting, RF, SVC, XGBoost, ExtraTrees Classifiers | 10-fold cross validation | Stacking: Avg AUC: 0.767, DNN: Avg AUC: 0.774, Gradient Boosting: Avg AUC: 0.79, RF: Avg AUC: 0.798, SVC: Avg AUC: 0.801, XGBoost: Avg AUC: 0.805, ExtraTrees: Avg AUC: 0.814 |
| Yousefian et al. [104] | Convolutional Neural Network | Autoencoder pre-training, Chr 1-10: training set, Chr 11–14: testing set and Chr 15–22: validation set | CNN: AUC: 0.96 F1-score: 0.83 |
| Bean et al. [105] | Knowledge graph edge prediction model [116] | 5-fold cross validation | Fold-Change Enrichment and random guess baseline: ALSoD: 23.33 (23.53), ClinVar: 30.05 (15.64), DisGeNet: 55.90 (81.66), Manual: 84.54 (13.27), Union: 8.92 (4.28) |
| Yin et al. [90] | Deep Neural Network (ALS-Net), Logistic Regression, SVM, Random Forest and Adaboost | 9-fold cross validation | ALS-Net: Acc: 0.769 F1-score: 0.797 LR: Acc: 0.739 F1-score:0.728 SVM: Acc: 0.725 F1-score:0.694 RM: Acc: 0.596 F1-score:0.381 Adaboost: Acc: 0.661 F1-score:0.625 (+PromoterCNN and all 4 chromosomes combined) |
| Kim et al. [91] | Multifactor dimensionality reduction; using a naïve Bayes classifier | 1000 permutation tests | Critical Acc: 0.629 and 0.640 (replication dataset) |
| Greene et al. [107] | Multifactor dimensionality reduction | 1000 permutation tests | Best SNP pairwise model: Acc: 0.6551 and 0.5821 (replication dataset); with p < 0.048 and p < 0.021 (replication dataset) |
| Sha et al. [108] | Two-locus probabilistic models, Multifactor dimensionality reduction, Combinatorial Searching Method | 1000 permutation tests | Two-locus models: rs4363506-rs3733242: p = 0.032 rs4363506-rs16984239: p = 0.042 MDR model: rs4363506-rs12680546: p = 0.156 CSM model: rs4363506-rs12680546: p = 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilopoulou, C.; Morris, A.P.; Giannakopoulos, G.; Duguez, S.; Duddy, W. What Can Machine Learning Approaches in Genomics Tell Us about the Molecular Basis of Amyotrophic Lateral Sclerosis? J. Pers. Med. 2020, 10, 247. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm10040247
Vasilopoulou C, Morris AP, Giannakopoulos G, Duguez S, Duddy W. What Can Machine Learning Approaches in Genomics Tell Us about the Molecular Basis of Amyotrophic Lateral Sclerosis? Journal of Personalized Medicine. 2020; 10(4):247. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm10040247
Chicago/Turabian StyleVasilopoulou, Christina, Andrew P. Morris, George Giannakopoulos, Stephanie Duguez, and William Duddy. 2020. "What Can Machine Learning Approaches in Genomics Tell Us about the Molecular Basis of Amyotrophic Lateral Sclerosis?" Journal of Personalized Medicine 10, no. 4: 247. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm10040247