
αβ-T Cells Engineered to Express γδ-T Cell Receptors Can Kill Neuroblastoma Organoids Independent of MHC-I Expression

 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Organoids and Culture Conditions
2.2. Effector Cells and Culture Conditions
2.3. Co-Culture Conditions
2.4. Read-Out of Organoid Killing
2.5. Flow Cytometry
2.6. RNA Isolation
2.7. Whole Transcriptome Sequencing
2.8. Data Analysis and Statistics
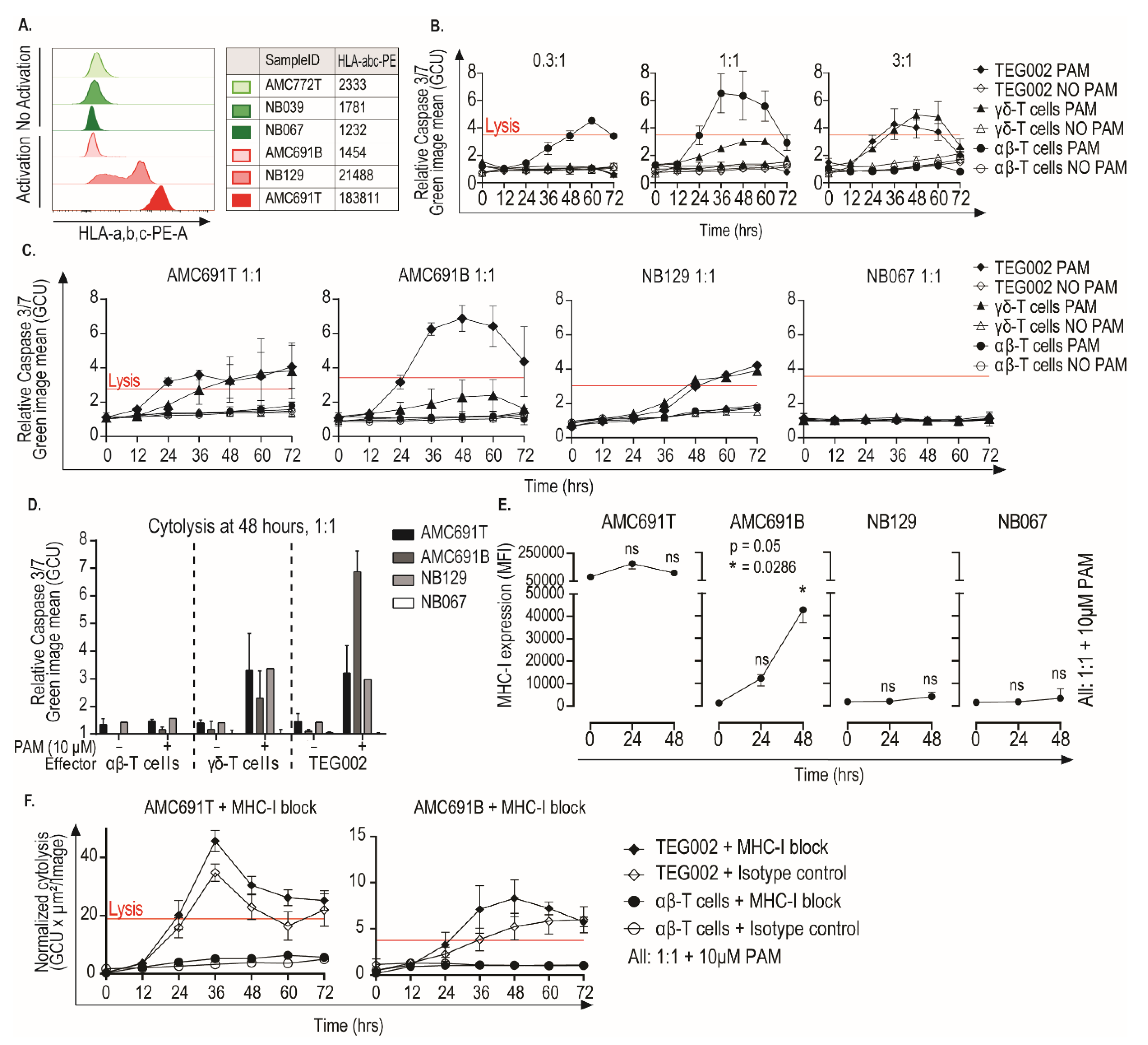
3. Results
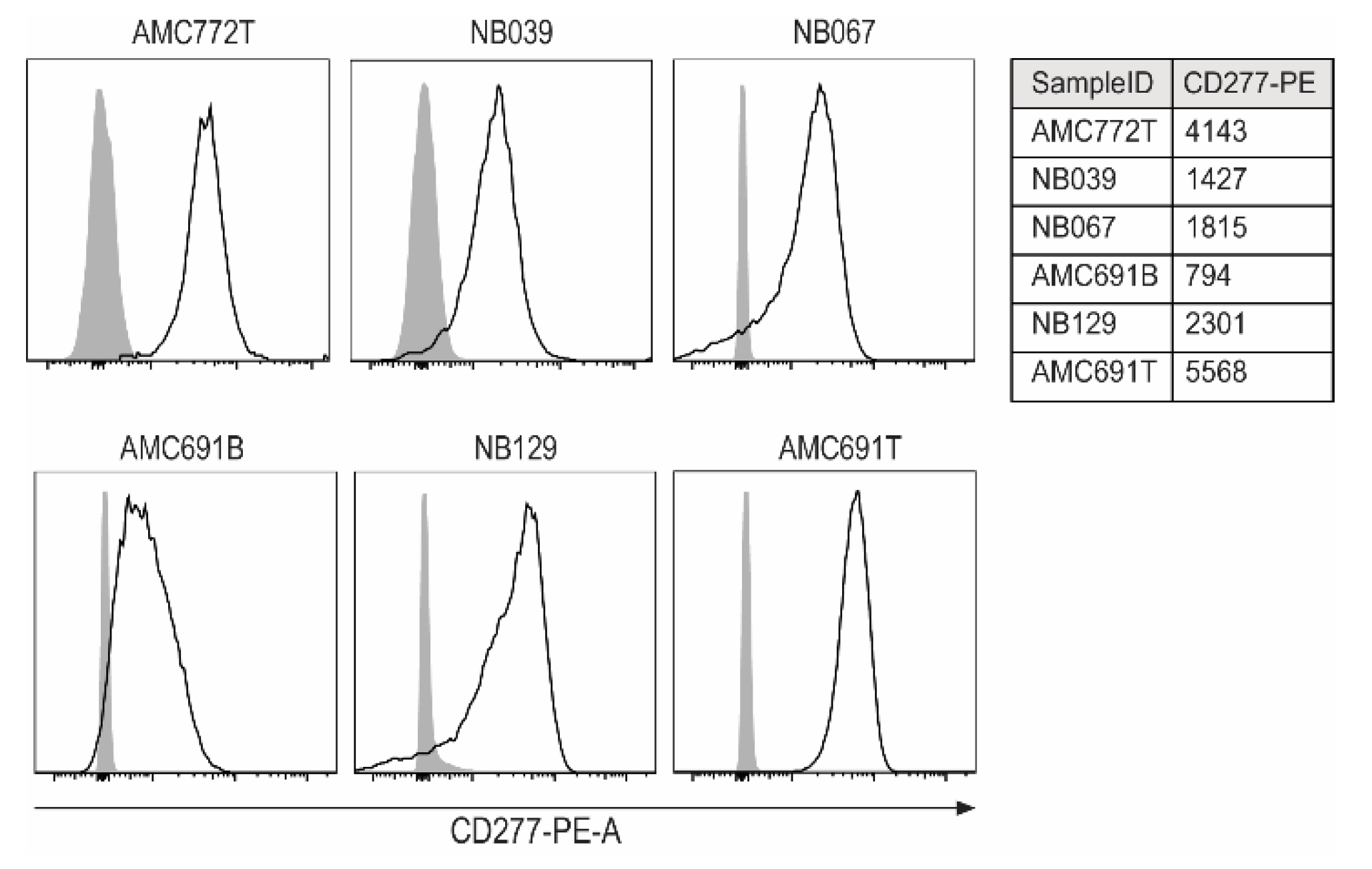
3.1. Expression of CD277 on Neuroblastoma Organoids
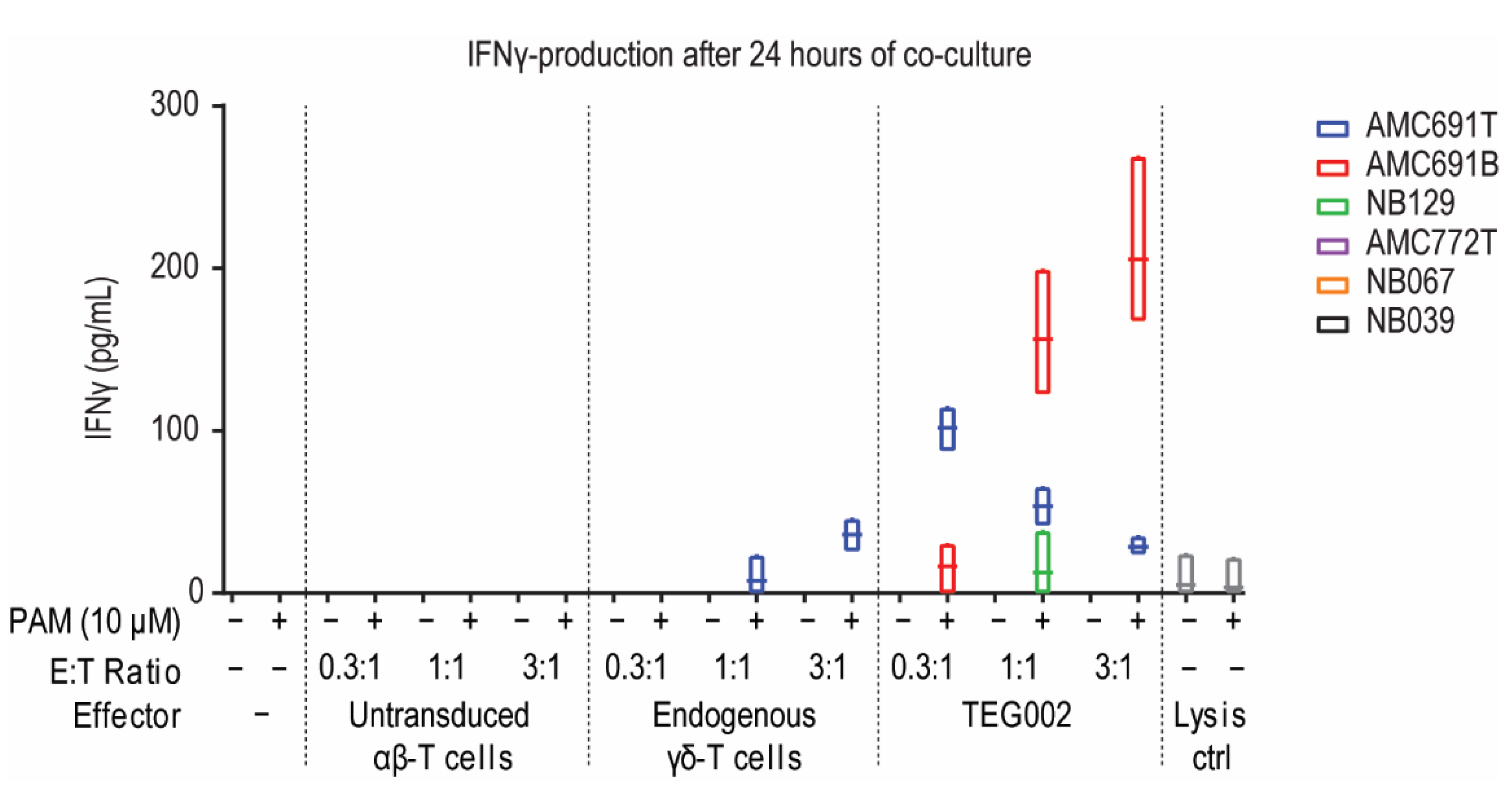
3.2. TEG002 Are Activated by 50% of Organoids in Co-Cultures
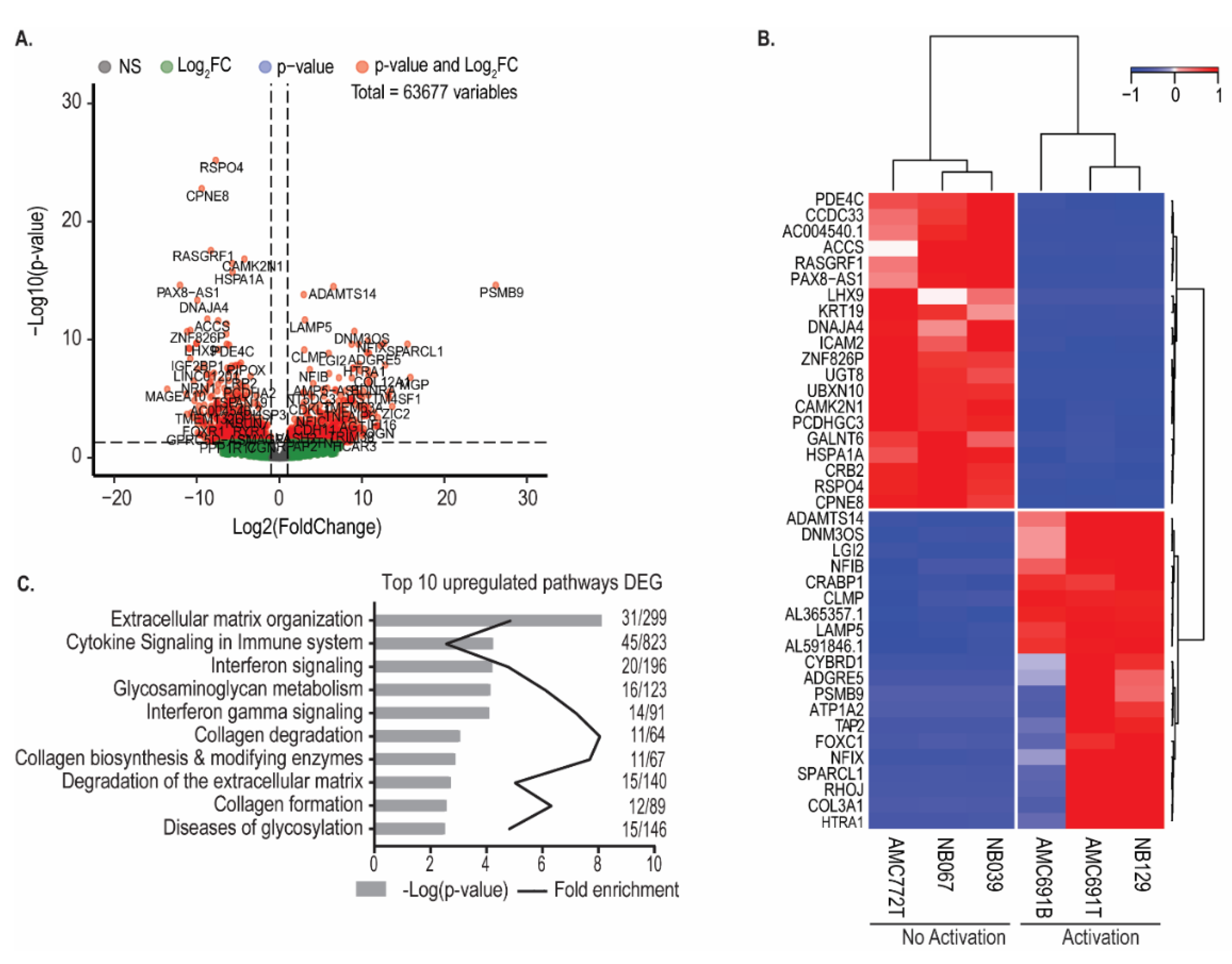
3.3. Neuroblastoma Organoids Susceptible to Recognition by TEG002 Have a Distinct Transcriptomic Profile
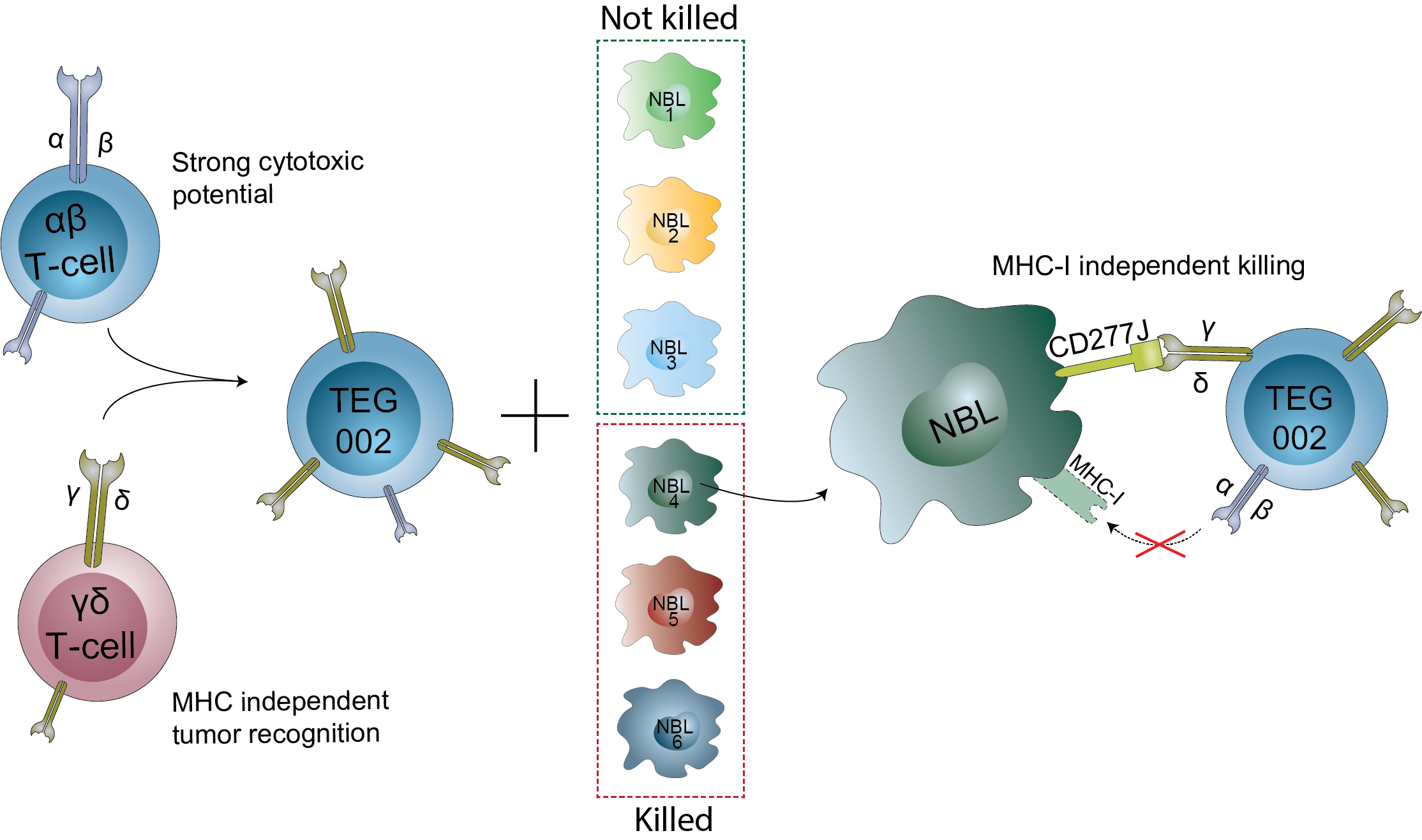
3.4. Neuroblastoma Organoids Are Killed by TEG002 Independent of MHC-I Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Chan, T.A.; Kroemer, G.; Wolchok, J.D.; López-Soto, A. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 2018, 10, eaat7807. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wienke, J.; Dierselhuis, M.P.; Tytgat, G.A.; Künkele, A.; Nierkens, S.; Molenaar, J.J. The immune landscape of neuroblastoma: Challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur. J. Cancer 2021, 144, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W. Neuroblastoma. Nat. Rev. Dis. Prim. 2016, 2, 16078. [Google Scholar] [CrossRef]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of Multiple Copies of the N-mycOncogene with Rapid Progression of Neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Shimada, H.; Ambros, I.M.; Dehner, L.P.; Hata, J.; Joshi, V.V.; Roald, B.; O Stram, D.; Gerbing, R.B.; Lukens, J.N.; Matthay, K.K.; et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer 1999, 86, 364–372. [Google Scholar] [CrossRef]
- Fisher, J.P.; Heuijerjans, J.; Yan, M.; Gustafsson, K.; Anderson, J. γδ T cells for cancer immunotherapy. OncoImmunology 2014, 3, e27572. [Google Scholar] [CrossRef] [Green Version]
- Scheper, W.; Gründer, C.; Kuball, J. Multifunctional γδ T cells and their receptors for targeted anticancer immunotherapy. OncoImmunology 2013, 2, e23974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebestyen, Z.; Scheper, W.; Vyborova, A.; Gu, S.; Rychnavska, Z.; Schiffler, M.; Cleven, A.; Chéneau, C.; van Noorden, M.; Peigné, C.-M.; et al. RhoB Mediates Phosphoantigen Recognition by Vγ9Vδ2 T Cell Receptor. Cell Rep. 2016, 15, 1973–1985. [Google Scholar] [CrossRef] [Green Version]
- Harly, C.; Guillaume, Y.; Nedellec, S.; Peigné, C.-M.; Mönkkönen, H.; Mönkkönen, J.; Li, J.; Kuball, J.; Adams, E.J.; Netzer, S.; et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human γδ T-cell subset. Blood 2012, 120, 2269–2279. [Google Scholar] [CrossRef] [Green Version]
- Yazdanifar, M.; Barbarito, G.; Bertaina, A.; Airoldi, I. γδ T Cells: The Ideal Tool for Cancer Immunotherapy. Cells 2020, 9, 1305. [Google Scholar] [CrossRef]
- Straetemans, T.; Gründer, C.; Heijhuurs, S.; Hol, S.; Slaper-Cortenbach, I.; Bönig, H.; Sebestyen, Z.; Kuball, J. Untouched GMP-Ready Purified Engineered Immune Cells to Treat Cancer. Clin. Cancer Res. 2015, 21, 3957–3968. [Google Scholar] [CrossRef] [Green Version]
- Gründer, C.; Van Dorp, S.; Hol, S.; Drent, E.; Straetemans, T.; Heijhuurs, S.; Scholten, K.; Scheper, W.; Sebestyen, Z.; Martens, A.; et al. γ9 and δ2CDR3 domains regulate functional avidity of T cells harboring γ9δ2TCRs. Blood 2012, 120, 5153–5162. [Google Scholar] [CrossRef] [Green Version]
- Johanna, I.; Straetemans, T.; Heijhuurs, S.; Aarts-Riemens, T.; Norell, H.; Bongiovanni, L.; De Bruin, A.; Sebestyen, Z.; Kuball, J. Evaluating in vivo efficacy–toxicity profile of TEG001 in humanized mice xenografts against primary human AML disease and healthy hematopoietic cells. J. Immunother. Cancer 2019, 7, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcu-Malina, V.; Heijhuurs, S.; Van Buuren, M.; Hartkamp, L.; Strand, S.; Sebestyen, Z.; Scholten, K.; Martens, A.; Kuball, J. Redirecting αβT cells against cancer cells by transfer of a broadly tumor-reactive γδT-cell receptor. Blood 2011, 118, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Amir, A.L.; Van Der Steen, D.M.; Van Loenen, M.M.; Hagedoorn, R.S.; De Boer, R.; Kester, M.D.; De Ru, A.H.; Lugthart, G.; van Kooten, C.; Hiemstra, P.; et al. PRAME-Specific Allo-HLA–Restricted T Cells with Potent Antitumor Reactivity Useful for Therapeutic T-Cell Receptor Gene Transfer. Clin. Cancer Res. 2011, 17, 5615–5625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Incucyte® Caspase-3/7 Green Dye for Apoptosis–Incucyte EU eShop. Available online: https://eu-shop.essenbioscience.com/products/caspase-3-7-green-apoptosis-assay-reagent (accessed on 30 March 2021).
- QIAGEN. RNeasy Mini Kit-QIAGEN Online Shop. Available online: https://www.qiagen.com/nl/products/discovery-and-translational-research/dna-rna-purification/rna-purification/total-rna/rneasy-mini-kit/?clear=true#orderinginformation (accessed on 11 January 2021).
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalised medicine for high-risk paediatric cancer patients—The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Bate-Eya, L.T.; Ebus, M.E.; Koster, J.; den Hartog, I.J.; Zwijnenburg, D.A.; Schild, L.; van der Ploeg, I.; Dolman, M.E.; Caron, H.N.; Versteeg, R.; et al. Newly-derived neuroblastoma cell lines propagated in serum-free media recapitulate the genotype and phenotype of primary neuroblastoma tumours. Eur. J. Cancer 2014, 50, 628–637. [Google Scholar] [CrossRef]
- Spel, L.; Boelens, J.J.; Van Der Steen, D.M.; Blokland, N.J.; Van Noesel, M.M.; Molenaar, J.J.; Heemskerk, M.H.; Boes, M.; Nierkens, S. Natural killer cells facilitate PRAME-specific T-cell reactivity against neuroblastoma. Oncotarget 2015, 6, 35770–35781. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.; Borowska, M.T.; Boughter, C.T.; Adams, E.J. Butyrophilin3A proteins and Vγ9Vδ2 T cell activation. Semin. Cell Dev. Biol. 2018, 84, 65–74. [Google Scholar] [CrossRef]
- Sebestyen, Z.; Prinz, I.; Déchanet-Merville, J.; Silva-Santos, B.; Kuball, J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov. 2020, 19, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Santos, B.; Serre, K.; Norell, H. γδ T cells in cancer. Nat. Rev. Immunol. 2015, 15, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Lawson, K.A.; Sousa, C.M.; Zhang, X.; Kim, E.; Akthar, R.; Caumanns, J.J.; Yao, Y.; Mikolajewicz, N.; Ross, C.; Brown, K.R.; et al. Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nat. Cell Biol. 2020, 586, 120–126. [Google Scholar] [CrossRef]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with γδ T cells: Many paths ahead of us. Cell. Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Smetak, M.; Schaefer-Eckart, K.; Kimmel, B.; Birkmann, J.; Einsele, H.; Kunzmann, V. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J. Transl. Med. 2014, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Pressey, J.G.; Adams, J.; Harkins, L.; Kelly, D.; You, Z.; Lamb, L.S. In vivo expansion and activation of γδ T cells as immunotherapy for refractory neuroblastoma. Medicine 2016, 95, e4909. [Google Scholar] [CrossRef]
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T Cell Therapy for Neuroblastoma. Front. Immunol. 2018, 9, 2380. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strijker, J.G.M.; Pscheid, R.; Drent, E.; van der Hoek, J.J.F.; Koopmans, B.; Ober, K.; van Hooff, S.R.; Kholosy, W.M.; Cornel, A.M.; Coomans, C.; et al. αβ-T Cells Engineered to Express γδ-T Cell Receptors Can Kill Neuroblastoma Organoids Independent of MHC-I Expression. J. Pers. Med. 2021, 11, 923. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11090923
Strijker JGM, Pscheid R, Drent E, van der Hoek JJF, Koopmans B, Ober K, van Hooff SR, Kholosy WM, Cornel AM, Coomans C, et al. αβ-T Cells Engineered to Express γδ-T Cell Receptors Can Kill Neuroblastoma Organoids Independent of MHC-I Expression. Journal of Personalized Medicine. 2021; 11(9):923. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11090923
Chicago/Turabian StyleStrijker, Josephine G. M., Ronja Pscheid, Esther Drent, Jessica J. F. van der Hoek, Bianca Koopmans, Kimberley Ober, Sander R. van Hooff, Waleed M. Kholosy, Annelisa M. Cornel, Chris Coomans, and et al. 2021. "αβ-T Cells Engineered to Express γδ-T Cell Receptors Can Kill Neuroblastoma Organoids Independent of MHC-I Expression" Journal of Personalized Medicine 11, no. 9: 923. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm11090923