
Spreading Depolarization as a Therapeutic Target in Severe Ischemic Stroke: Physiological and Pharmacological Strategies

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Electrocorticographic Monitoring
2.3. Data Analysis
2.4. Statistical Approaches
3. Results
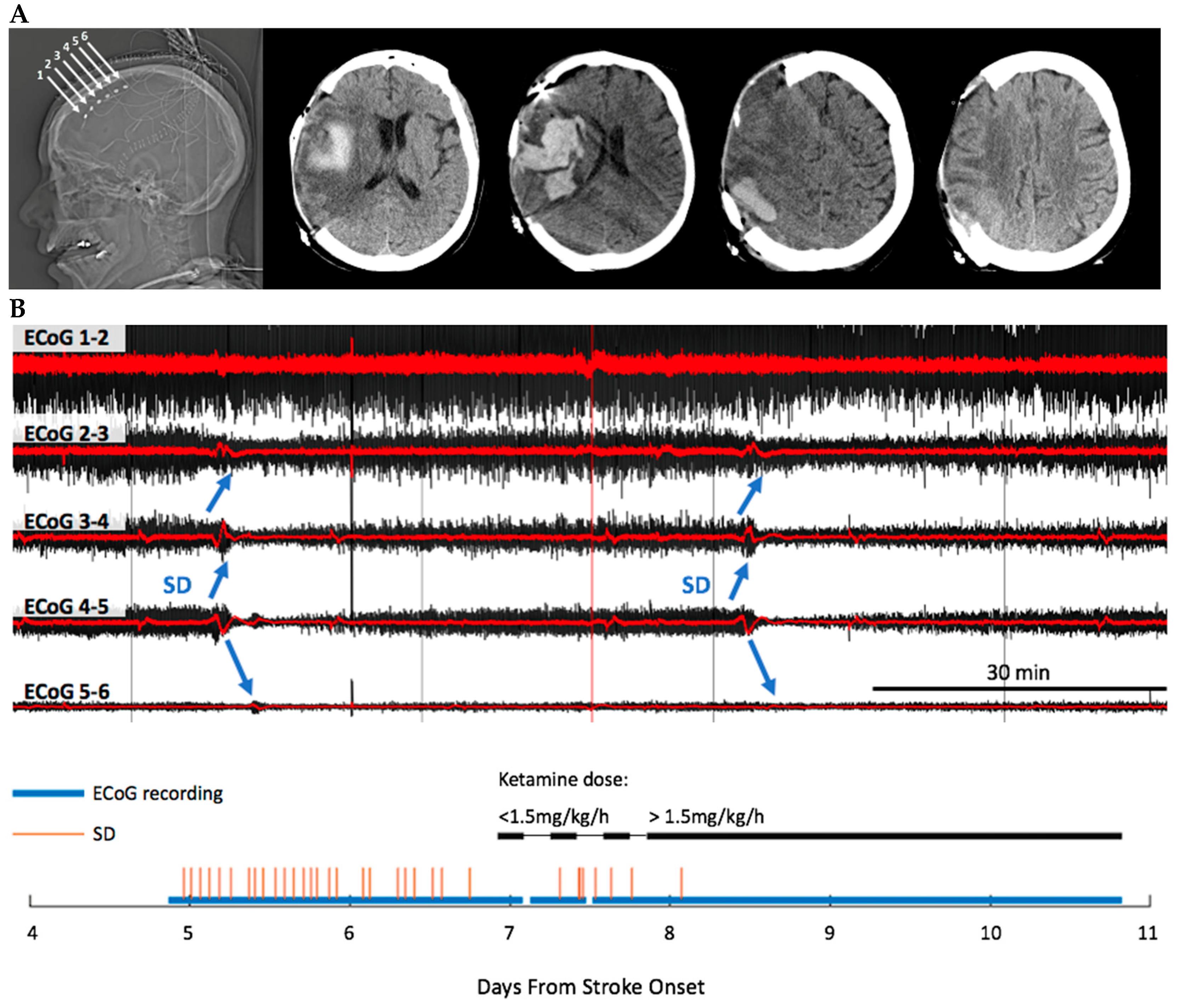
3.1. Recordings
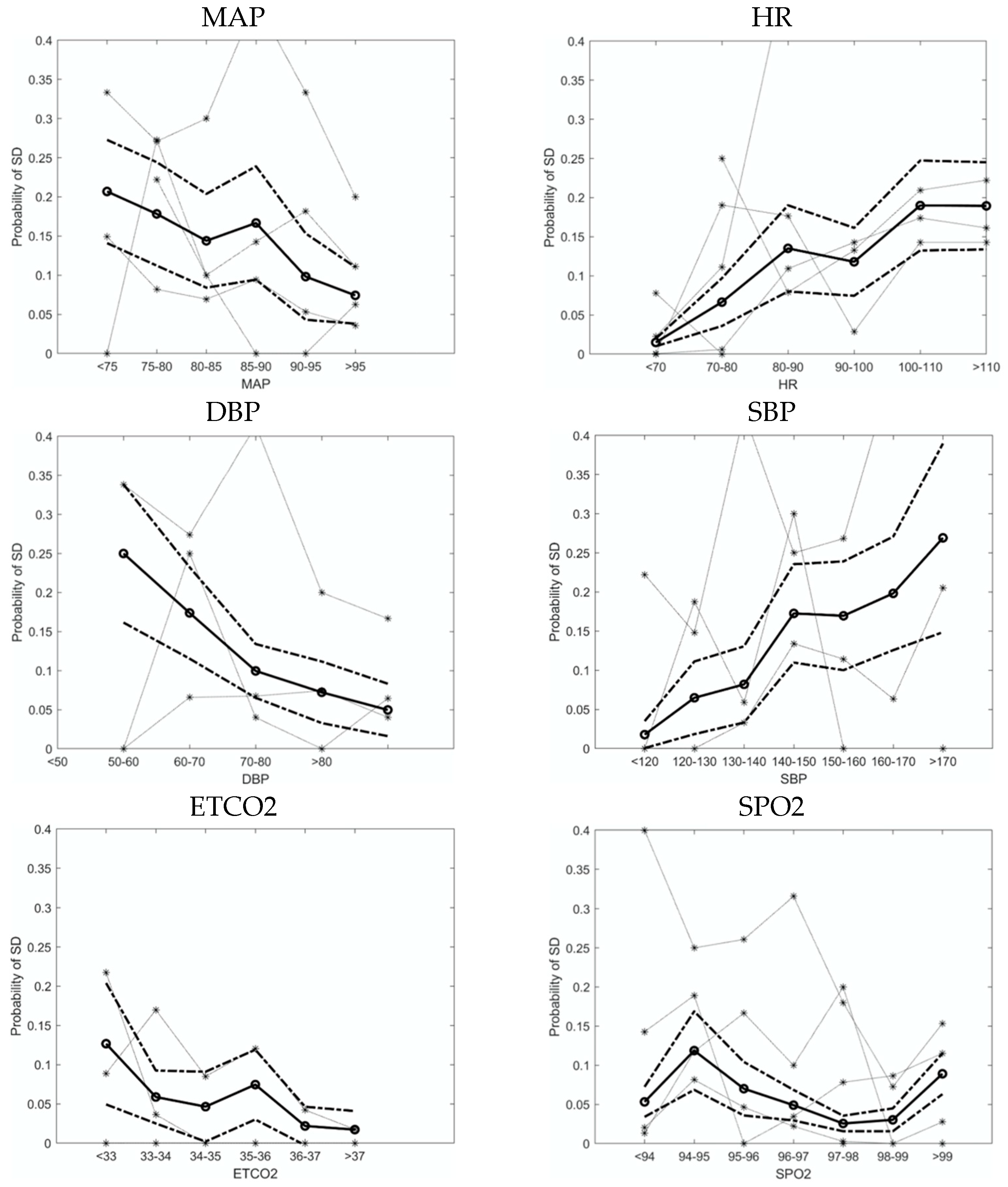
3.2. Physiological Variables Affect the Probability of SD
3.3. Risk Factors and Protective Factors for SD
3.4. Ketamine Exhibits Dose-Dependent Inhibition of SD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ropper, A.H.; Shafran, B. Brain edema after stroke. Clinical syndrome and intracranial pressure. Arch. Neurol. 1984, 41, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Hacke, W.; Schwab, S.; Horn, M.; Spranger, M.; De Georgia, M.; von Kummer, R. ‘Malignant’ middle cerebral artery territory infarction: Clinical course and prognostic signs. Arch. Neurol. 1996, 53, 309–315. [Google Scholar] [CrossRef]
- Wijdicks, E.F.; Sheth, K.N.; Carter, B.S.; Greer, D.M.; Kasner, S.E.; Kimberly, W.T.; Schwab, S.; Smith, E.E.; Tamargo, R.J.; Wintermark, M.; et al. Recommendations for the management of cerebral and cerebellar infarction with swelling: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2014, 45, 1222–1238. [Google Scholar] [CrossRef]
- Zha, A.M.; Sari, M.; Torbey, M.T. Recommendations for management of large hemispheric infarction. Curr. Opin. Crit. Care 2015, 21, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.M.; Morgan Jones, G.; Hawryluk, G.W.J.; Mailloux, P.; McLaughlin, D.; Papangelou, A.; Samuel, S.; Tokumaru, S.; Venkatasubramanian, C.; Zacko, C.; et al. Guidelines for the Acute Treatment of Cerebral Edema in Neurocritical Care Patients. Neurocrit. Care 2020, 32, 647–666. [Google Scholar] [CrossRef] [PubMed]
- Vahedi, K.; Hofmeijer, J.; Juettler, E.; Vicaut, E.; George, B.; Algra, A.; Amelink, G.J.; Schmiedeck, P.; Schwab, S.; Rothwell, P.M.; et al. Early decompressive surgery in malignant infarction of the middle cerebral artery: A pooled analysis of three randomised controlled trials. Lancet Neurol. 2007, 6, 215–222. [Google Scholar] [CrossRef]
- Jüttler, E.; Schwab, S.; Schmiedek, P.; Unterberg, A.; Hennerici, M.; Woitzik, J.; Witte, S.; Jenetzky, E.; Hacke, W.; DESTINY Study Group. Decompressive Surgery for the Treatment of Malignant Infarction of the Middle Cerebral Artery (DESTINY): A randomized, controlled trial. Stroke 2007, 38, 2518–2525. [Google Scholar] [CrossRef]
- Vahedi, K.; Vicaut, E.; Mateo, J.; Kurtz, A.; Orabi, M.; Guichard, J.P.; Boutron, C.; Couvreur, G.; Rouanet, F.; Touzé, E.; et al. Sequential-design, multicenter, randomized, controlled trial of early decompressive craniectomy in malignant middle cerebral artery infarction (DECIMAL Trial). Stroke 2007, 38, 2506–2517. [Google Scholar] [CrossRef]
- Maciel, C.B.; Sheth, K.N. Malignant MCA Stroke: An Update on Surgical Decompression and Future Directions. Curr. Atheroscler. Rep. 2015, 17, 40. [Google Scholar] [CrossRef]
- Das, S.; Mitchell, P.; Ross, N.; Whitfield, P.C. Decompressive Hemicraniectomy in the Treatment of Malignant Middle Cerebral Artery Infarction: A Meta-Analysis. World Neurosurg. 2019, 123, 8–16. [Google Scholar] [CrossRef]
- van Middelaar, T.; Nederkoorn, P.J.; van der Worp, H.B.; Stam, J.; Richard, E. Quality of life after surgical decompression for space-occupying middle cerebral artery infarction: Systematic review. Int. J. Stroke 2015, 10, 170–176. [Google Scholar] [CrossRef]
- Rahme, R.; Zuccarello, M.; Kleindorfer, D.; Adeoye, O.M.; Ringer, A.J. Decompressive hemicraniectomy for malignant middle cerebral artery territory infarction: Is life worth living? J. Neurosurg. 2012, 117, 749–754. [Google Scholar] [CrossRef]
- Mestre, H.; Du, T.; Sweeney, A.M.; Liu, G.; Samson, A.J.; Peng, W.; Mortensen, K.N.; Stæger, F.F.; Bork, P.A.R.; Bashford, L.; et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science 2020, 367, eaax7171. [Google Scholar] [CrossRef]
- Dreier, J.P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef]
- Hartings, J.A.; Shuttleworth, C.W.; Kirov, S.A.; Ayata, C.; Hinzman, J.M.; Foreman, B.; Andrew, R.D.; Boutelle, M.G.; Brennan, K.C.; Carlson, A.P.; et al. The continuum of spreading depolarizations in acute cortical lesion development: Examining Leão’s legacy. J. Cereb. Blood Flow Metab. 2017, 37, 1571–1594. [Google Scholar] [CrossRef]
- Leao, A.A.P. Spreading depression of activity in the cerebral cortex. J. Neurophysiol. 1944, 7, 359–390. [Google Scholar] [CrossRef]
- Strong, A.; Fabricius, M.; Boutelle, M.; Hibbins, S.; Hopwood, S.; Jones, R.; Parkin, M.; Lauritzen, M. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke 2002, 33, 2738–2743. [Google Scholar] [CrossRef]
- Dreier, J.P.; Woitzik, J.; Fabricius, M.; Bhatia, R.; Major, S.; Drenckhahn, C.; Lehmann, T.N.; Sarrafzadeh, A.; Willumsen, L.; Hartings, J.A.; et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain 2006, 129, 3224–3237. [Google Scholar] [CrossRef]
- Lauritzen, M.; Strong, A.J. Spreading depression of Leao’ and its emerging relevance to acute brain injury in humans. J. Cereb. Blood Flow Metab. 2017, 37, 1553–1570. [Google Scholar] [CrossRef]
- Dohmen, C.; Sakowitz, O.W.; Fabricius, M.; Bosche, B.; Reithmeier, T.; Ernestus, R.I.; Brinker, G.; Dreier, J.P.; Woitzik, J.; Strong, A.J.; et al. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann. Neurol. 2008, 63, 720–728. [Google Scholar] [CrossRef]
- Hinzman, J.M.; DiNapoli, V.A.; Mahoney, E.J.; Gerhardt, G.A.; Hartings, J.A. Spreading depolarizations mediate excitotoxicity in the development of acute cortical lesions. Exp. Neurol. 2015, 267, 243–253. [Google Scholar] [CrossRef]
- Hartings, J.A.; Rolli, M.L.; Lu, X.C.; Tortella, F.C. Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: Relation to infarct growth and neuroprotection. J. Neurosci. 2003, 23, 11602–11610. [Google Scholar] [CrossRef]
- Iijima, T.; Mies, G.; Hossmann, K.A. Repeated negative DC deflections in rat cortex following middle cerebral artery occlusion are abolished by MK-801: Effect on volume of ischemic injury. J. Cereb. Blood Flow Metab. 1992, 12, 727–733. [Google Scholar] [CrossRef]
- Gill, R.; Andiné, P.; Hillered, L.; Persson, L.; Hagberg, H. The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischaemia in the rat. J. Cereb. Blood Flow Metab. 1992, 12, 371–379. [Google Scholar] [CrossRef]
- Takano, K.; Latour, L.L.; Formato, J.E.; Carano, R.A.; Helmer, K.G.; Hasegawa, Y.; Sotak, C.H.; Fisher, M. The role of spreading depression in focal ischemia evaluated by diffusion mapping. Ann. Neurol. 1996, 39, 308–318. [Google Scholar] [CrossRef]
- Busch, E.; Gyngell, M.L.; Eis, M.; Hoehn-Berlage, M.; Hossmann, K.A. Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: Contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging. J. Cereb. Blood Flow Metab. 1996, 16, 1090–1099. [Google Scholar] [CrossRef]
- Back, T.; Ginsberg, M.D.; Dietrich, W.D.; Watson, B.D. Induction of spreading depression in the ischemic hemisphere following experimental middle cerebral artery occlusion: Effect on infarct morphology. J. Cereb. Blood Flow Metab. 1996, 16, 202–213. [Google Scholar] [CrossRef]
- Hartings, J.A.; Strong, A.J.; Fabricius, M.; Manning, A.; Bhatia, R.; Dreier, J.P.; Mazzeo, A.T.; Tortella, F.C.; Bullock, M.R.; Co-Operative Study of Brain Injury Depolarizations. Spreading depolarizations and late secondary insults after traumatic brain injury. J. Neurotrauma 2009, 26, 1857–1866. [Google Scholar] [CrossRef]
- Schumm, L.; Lemale, C.L.; Major, S.; Hecht, N.; Nieminen-Kelhä, M.; Zdunczyk, A.; Kowoll, C.M.; Martus, P.; Thiel, C.M.; Dreier, J.P.; et al. Physiological variables in association with spreading depolarizations in the late phase of ischemic stroke. J. Cereb. Blood Flow Metab. 2022, 42, 121–135. [Google Scholar] [CrossRef]
- Sakowitz, O.W.; Kiening, K.L.; Krajewski, K.L.; Sarrafzadeh, A.S.; Fabricius, M.; Strong, A.J.; Unterberg, A.W.; Dreier, J.P. Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke 2009, 40, e519–e522. [Google Scholar] [CrossRef] [Green Version]
- Hertle, D.N.; Dreier, J.P.; Woitzik, J.; Hartings, J.A.; Bullock, R.; Okonkwo, D.O.; Shutter, L.A.; Vidgeon, S.; Strong, A.J.; Kowoll, C.; et al. Effect of analgesics and sedatives on the occurrence of spreading depolarizations accompanying acute brain injury. Brain 2012, 135 Pt 8, 2390–2398. [Google Scholar] [CrossRef] [PubMed]
- Carlson, A.P.; Abbas, M.; Alunday, R.L.; Qeadan, F.; Shuttleworth, C.W. Spreading depolarization in acute brain injury inhibited by ketamine: A prospective, randomized, multiple crossover trial. J. Neurosurg. 2018. epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Hartings, J.A.; Ngwenya, L.B.; Carroll, C.P.; Foreman, B. Ketamine sedation for the suppression of spreading depolarizations. J. Neurosurg. 2018. epub ahead of print. [Google Scholar] [CrossRef]
- Helbok, R.; Hartings, J.A.; Schiefecker, A.; Balança, B.; Jewel, S.; Foreman, B.; Ercole, A.; Balu, R.; Ayata, C.; Ngwenya, L.; et al. What Should a Clinician Do When Spreading Depolarizations are Observed in a Patient? Neurocrit. Care 2020, 32, 306–310. [Google Scholar] [CrossRef]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; Shuttleworth, C.W.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J. Cereb. Blood Flow Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef]
- Owen, B.; Vangala, A.; Fritch, C.; Alsarah, A.A.; Jones, T.; Davis, H.; Shuttleworth, C.W.; Carlson, A.P. Cerebral Autoregulation Correlation with Outcomes and Spreading Depolarization in Aneurysmal Subarachnoid Hemorrhage. Stroke 2022. epub ahead of print. [Google Scholar] [CrossRef]
- von Bornstädt, D.; Houben, T.; Seidel, J.L.; Zheng, Y.; Dilekoz, E.; Qin, T.; Sandow, N.; Kura, S.; Eikermann-Haerter, K.; Endres, M.; et al. Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron 2015, 85, 1117–1131. [Google Scholar] [CrossRef]
- Carlson, A.P.; Davis, H.T.; Jones, T.; Brennan, K.C.; Torbey, M.; Ahmadian, R.; Qeadan, F.; Shuttleworth, C.W. Is the Human Touch Always Therapeutic? Patient Stimulation and Spreading Depolarization after Acute Neurological Injuries. Transl. Stroke Res. 2022. epub ahead of print. [Google Scholar] [CrossRef]
- Zhao, H.T.; Tuohy, M.C.; Chow, D.; Kozberg, M.G.; Kim, S.H.; Shaik, M.A.; Hillman, E.M.C. Neurovascular dynamics of repeated cortical spreading depolarizations after acute brain injury. Cell Rep. 2021, 37, 109794. [Google Scholar] [CrossRef]
- Østergaard, L.; Dreier, J.P.; Hadjikhani, N.; Jespersen, S.N.; Dirnagl, U.; Dalkara, T. Neurovascular coupling during cortical spreading depolarization and -depression. Stroke 2015, 46, 1392–1401. [Google Scholar] [CrossRef] [Green Version]
- Takano, T.; Tian, G.F.; Peng, W.; Lou, N.; Lovatt, D.; Hansen, A.J.; Kasischke, K.A.; Nedergaard, M. Cortical spreading depression causes and coincides with tissue hypoxia. Nat. Neurosci. 2007, 10, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Hinzman, J.M.; Wilson, J.A.; Mazzeo, A.T.; Bullock, M.R.; Hartings, J.A. Excitotoxicity and Metabolic Crisis Are Associated with Spreading Depolarizations in Severe Traumatic Brain Injury Patients. J. Neurotrauma 2016, 33, 1775–1783. [Google Scholar] [CrossRef]
- Aiba, I.; Shuttleworth, C.W. Sustained NMDA receptor activation by spreading depolarizations can initiate excitotoxic injury in metabolically compromised neurons. J. Physiol. 2012, 590, 5877–5893. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P.; Major, S.; Foreman, B.; Winkler, M.K.L.; Kang, E.J.; Milakara, D.; Lemale, C.L.; DiNapoli, V.; Hinzman, J.M.; Woitzik, J.; et al. Terminal spreading depolarization and electrical silence in death of human cerebral cortex. Ann. Neurol. 2018, 83, 295–310. [Google Scholar] [CrossRef]
- Nakamura, H.; Strong, A.J.; Dohmen, C.; Sakowitz, O.W.; Vollmar, S.; Sué, M.; Kracht, L.; Hashemi, P.; Bhatia, R.; Yoshimine, T.; et al. Spreading depolarizations cycle around and enlarge focal ischaemic brain lesions. Brain 2010, 133 Pt 7, 1994–2006. [Google Scholar] [CrossRef] [PubMed]
- Woitzik, J.; Hecht, N.; Pinczolits, A.; Sandow, N.; Major, S.; Winkler, M.K.; Weber-Carstens, S.; Dohmen, C.; Graf, R.; Strong, A.J.; et al. Propagation of cortical spreading depolarization in the human cortex after malignant stroke. Neurology 2013, 80, 1095–1102. [Google Scholar] [CrossRef]
- Kontos, H.A.; Wei, E.P.; Raper, A.J.; Patterson, J.L., Jr. Local mechanism of CO2 action of cat pial arterioles. Stroke 1977, 8, 226–229. [Google Scholar] [CrossRef]
- Heistad, D.D.; Kontos, H.A. Cerebral circulation. In Handbook of Physiology, Section 2: The Cardiovascular System; Shepherd, J.T., Abboud, F.M., Eds.; American Physiological Society: Bethesda, MD, USA, 1983; Volume 3, pp. 137–182. [Google Scholar]
- Curley, G.; Kavanagh, B.P.; Laffey, J.G. Hypocapnia and the injured brain: More harm than benefit. Crit. Care Med. 2010, 38, 1348–1359. [Google Scholar] [CrossRef]
- Coles, J.P.; Fryer, T.D.; Coleman, M.R.; Smielewski, P.; Gupta, A.K.; Minhas, P.S.; Aigbirhio, F.; Chatfield, D.A.; Williams, G.B.; Boniface, S.; et al. Hyperventilation following head injury: Effect on ischemic burden and cerebral oxidative metabolism. Crit. Care Med. 2007, 35, 568–578. [Google Scholar] [CrossRef]
- Brain Trauma Foundation; American Association of Neurological Surgeons; Congress of Neurological Surgeons; Joint Section on Neurotrauma and Critical Care; AANS/CNS; Bratton, S.L.; Chestnut, R.M.; Ghajar, J.; McConnell Hammond, F.F.; Harris, O.A.; et al. Guidelines for the management of severe traumatic brain injury. XIV. Hyperventilation. J. Neurotrauma 2007, 24 (Suppl. 1), S87–S90, Erratum in: J. Neurotrauma 2008, 25, 276–278. Multiple Author Names Added. [Google Scholar] [CrossRef]
- Gorelova, N.A.; Koroleva, V.I.; Amemori, T.; Pavlík, V.; Burĕs, J. Ketamine blockade of cortical spreading depression in rats. Electroencephalogr. Clin. Neurophysiol. 1987, 66, 440–447. [Google Scholar] [CrossRef]
- Hernándéz-Cáceres, J.; Macias-González, R.; Brozek, G.; Bures, J. Systemic ketamine blocks cortical spreading depression but does not delay the onset of terminal anoxic depolarization in rats. Brain Res. 1987, 437, 360–364. [Google Scholar] [CrossRef]
- Marrannes, R.; Willems, R.; De Prins, E.; Wauquier, A. Evidence for a role of the N-methyl-D-aspartate (NMDA) receptor in cortical spreading depression in the rat. Brain Res. 1988, 457, 226–240. [Google Scholar] [CrossRef]
- Reinhart, K.M.; Shuttleworth, C.W. Ketamine reduces deleterious consequences of spreading depolarizations. Exp. Neurol. 2018, 305, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, M.; Hansen, A.J. The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. J. Cereb. Blood Flow Metab. 1992, 12, 223–229. [Google Scholar] [CrossRef]
- Krüger, H.; Heinemann, U.; Luhmann, H.J. Effects of ionotropic glutamate receptor blockade and 5-HT1A receptor activation on spreading depression in rat neocortical slices. Neuroreport 1999, 10, 2651–2656. [Google Scholar] [CrossRef]
- Peeters, M.; Gunthorpe, M.J.; Strijbos, P.J.; Goldsmith, P.; Upton, N.; James, M.F. Effects of pan- and subtype-selective N-methyl-D-aspartate receptor antagonists on cortical spreading depression in the rat: Therapeutic potential for migraine. J. Pharmacol. Exp. Ther. 2007, 321, 564–572. [Google Scholar] [CrossRef]
- Jansen, N.A.; Schenke, M.; Voskuyl, R.A.; Thijs, R.D.; van den Maagdenberg, A.M.J.M.; Tolner, E.A. Apnea Associated with Brainstem Seizures in Cacna1aS218L Mice Is Caused by Medullary Spreading Depolarization. J. Neurosci. 2019, 39, 9633–9644. [Google Scholar] [CrossRef]
- Reinhart, K.M.; Humphrey, A.; Brennan, K.C.; Carlson, A.P.; Shuttleworth, C.W. Memantine Improves Recovery after Spreading Depolarization in Brain Slices and can be Considered for Future Clinical Trials. Neurocrit. Care 2021, 35 (Suppl. 2), 135–145. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Subject | Age | Sex | Stroke Territory | Comorbidities | Time to Craniectomy * | Duration of ECoG Monitoring | Total SD | Early mRS ** |
|---|---|---|---|---|---|---|---|---|
| 1 | 29 | F | Left MCA, left PCA | T2DM, Alcohol use | 27 h | 96 h | 40 | 4 |
| 2 | 24 | M | Multifocal hemorrhagic | Polysubstance use | 47 h | 149 h | 104 | 4 |
| 3 | 63 | F | Right MCA | Unknown | 69 h | 74 h | 20 | 4 |
| 4 | 64 | F | Right MCA | TIA, HTN, HLD, CD | 98 h | 234 h | 33 | 4 |
| 5 | 60 | M | Right ICA terminus | HTN, Hyperthyroid | 6 h | 91 h | 7 | 4 |
| A. Univariable Analysis | B. Multivariable Analysis | |||||
|---|---|---|---|---|---|---|
| Odds Ratio | 95% Confidence Interval | Odds Ratio | 95%Confidence Interval | |||
| Temp | 2.048 | 1.442 | 2.909 | 1.621 | 1.593 | 1.649 |
| WBC | 1.113 | 1.081 | 1.922 | 1.041 | 1.037 | 1.044 |
| HR | 1.021 | 1.013 | 1.03 | 1.010 | 1.009 | 1.011 |
| SBP | 1.015 | 1.003 | 1.027 | 1.001 | 1.000 | 1.001 |
| Glucose | 1.013 | 1.003 | 1.022 | 1.014 | 1.013 | 1.014 |
| PaO2 | 0.993 | 0.988 | 0.998 | 1.001 | 1.000 | 1.001 |
| SpO2 | 0.986 | 0.921 | 1.055 | -- | ||
| MAP | 0.975 | 0.96 | 0.991 | 0.975 | 0.960 | 0.991 |
| DBP | 0.958 | 0.941 | 0.991 | 0.986 | 0.984 | 0.989 |
| PaCO2 | 0.936 | 0.9 | 0.973 | 0.973 | 0.971 | 0.975 |
| ETCO2 | 0.772 | 0.655 | 0.91 | 0.943 | 0.940 | 0.946 |
| Lactate | 0.644 | 0.398 | 1.042 | -- | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chau, L.; Davis, H.T.; Jones, T.; Greene-Chandos, D.; Torbey, M.; Shuttleworth, C.W.; Carlson, A.P. Spreading Depolarization as a Therapeutic Target in Severe Ischemic Stroke: Physiological and Pharmacological Strategies. J. Pers. Med. 2022, 12, 1447. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm12091447
Chau L, Davis HT, Jones T, Greene-Chandos D, Torbey M, Shuttleworth CW, Carlson AP. Spreading Depolarization as a Therapeutic Target in Severe Ischemic Stroke: Physiological and Pharmacological Strategies. Journal of Personalized Medicine. 2022; 12(9):1447. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm12091447
Chicago/Turabian StyleChau, Lily, Herbert T. Davis, Thomas Jones, Diana Greene-Chandos, Michel Torbey, C. William Shuttleworth, and Andrew P. Carlson. 2022. "Spreading Depolarization as a Therapeutic Target in Severe Ischemic Stroke: Physiological and Pharmacological Strategies" Journal of Personalized Medicine 12, no. 9: 1447. https://0-doi-org.brum.beds.ac.uk/10.3390/jpm12091447