Nitrogen-Based Heterocyclic Compounds: A Promising Class of Antiviral Agents against Chikungunya Virus

 , , , and
, , , and
Abstract
:1. Introduction
2. CHIKV Infection
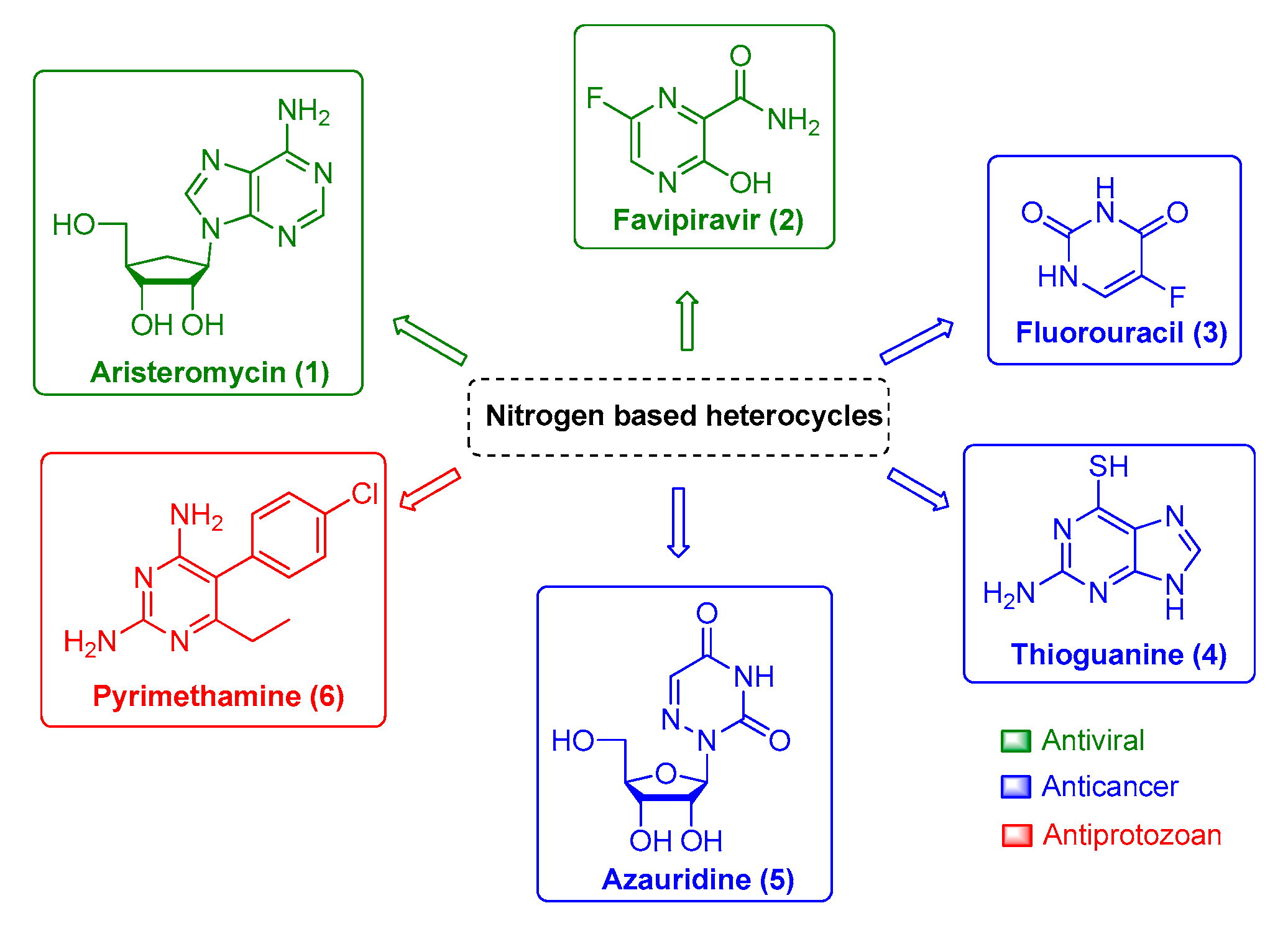
3. Overview of CHIKV Drugs and the Versatility of Nitrogen-Based Heterocycles Derivatives
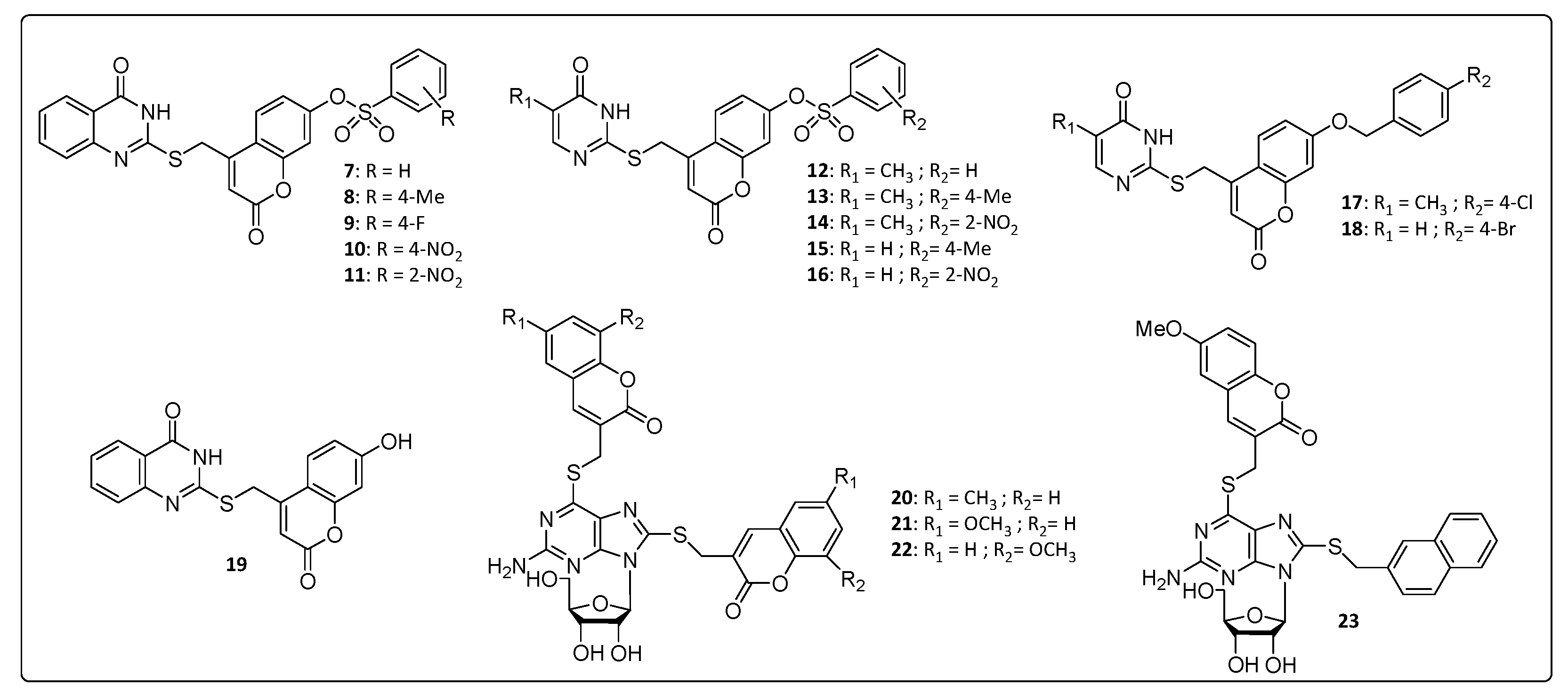
3.1. Nitrogen Heterocycle–Coumarin Hybrid Compounds
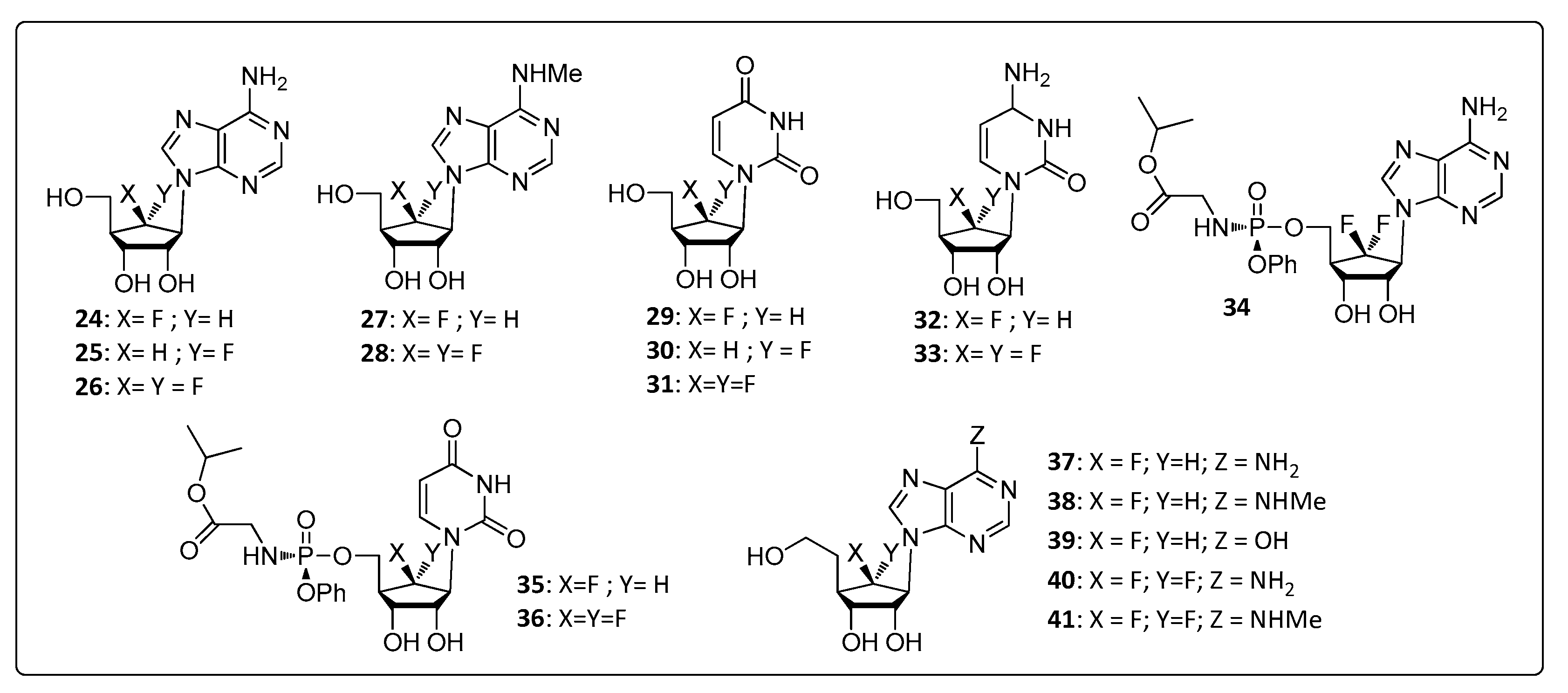
3.2. Aristeromycin Analogs
3.3. Ribonucleoside Derivatives
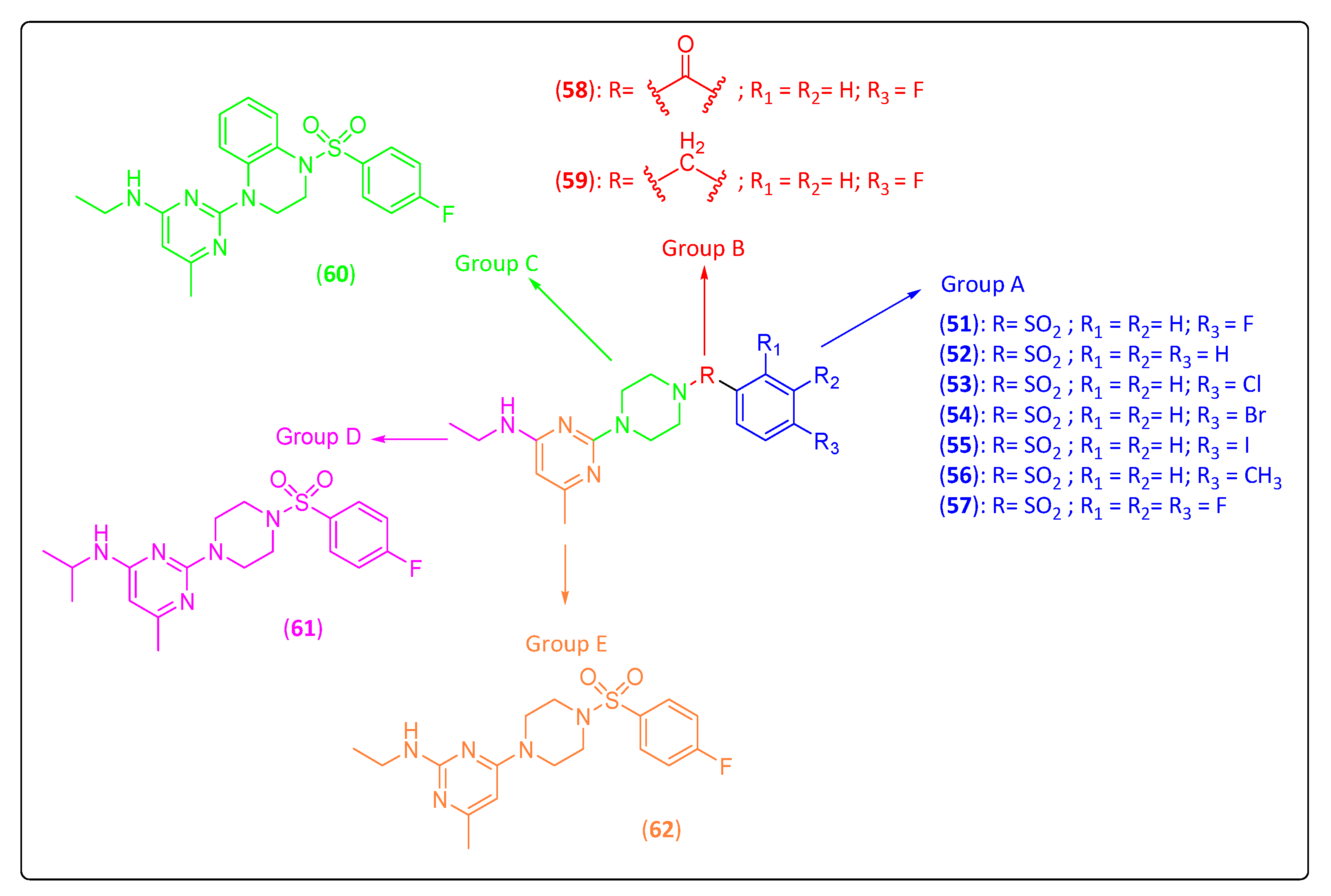
3.4. Piperazinyl–Pyrimidine Derivatives
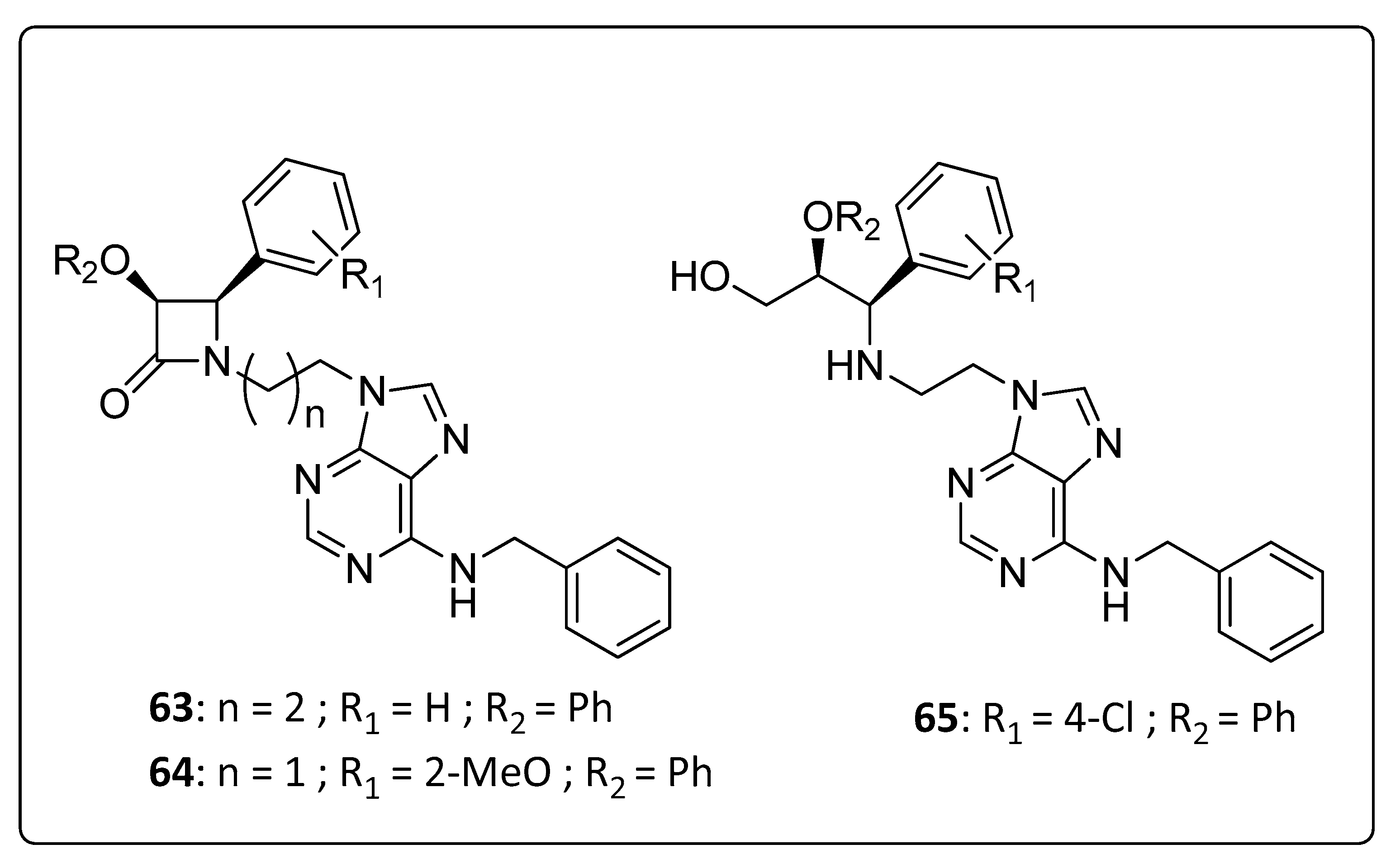
3.5. Purine-β-Lactam Hybrids
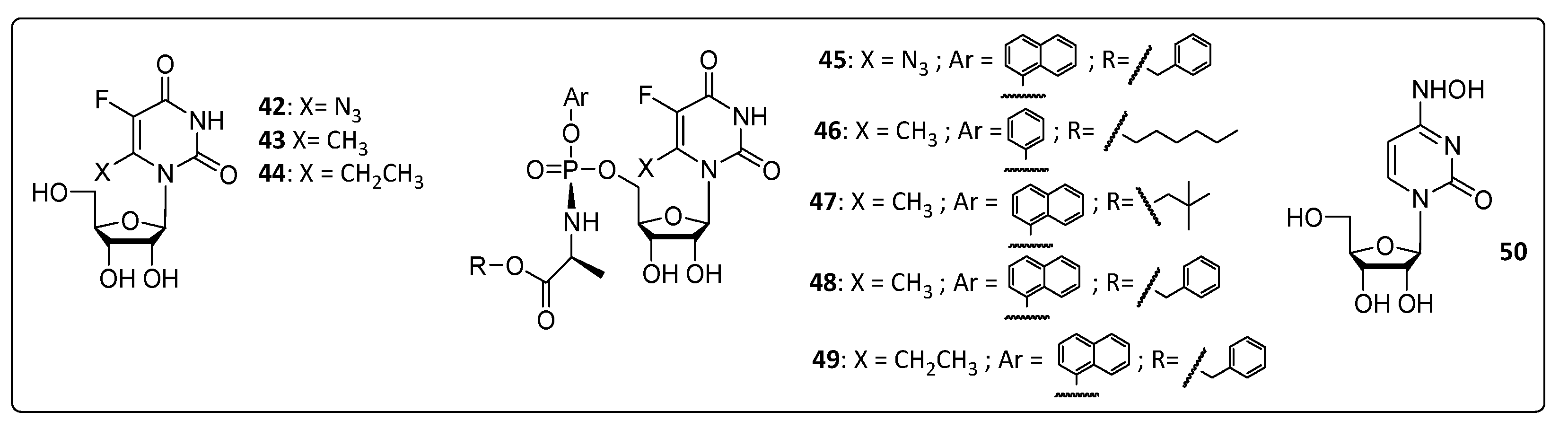
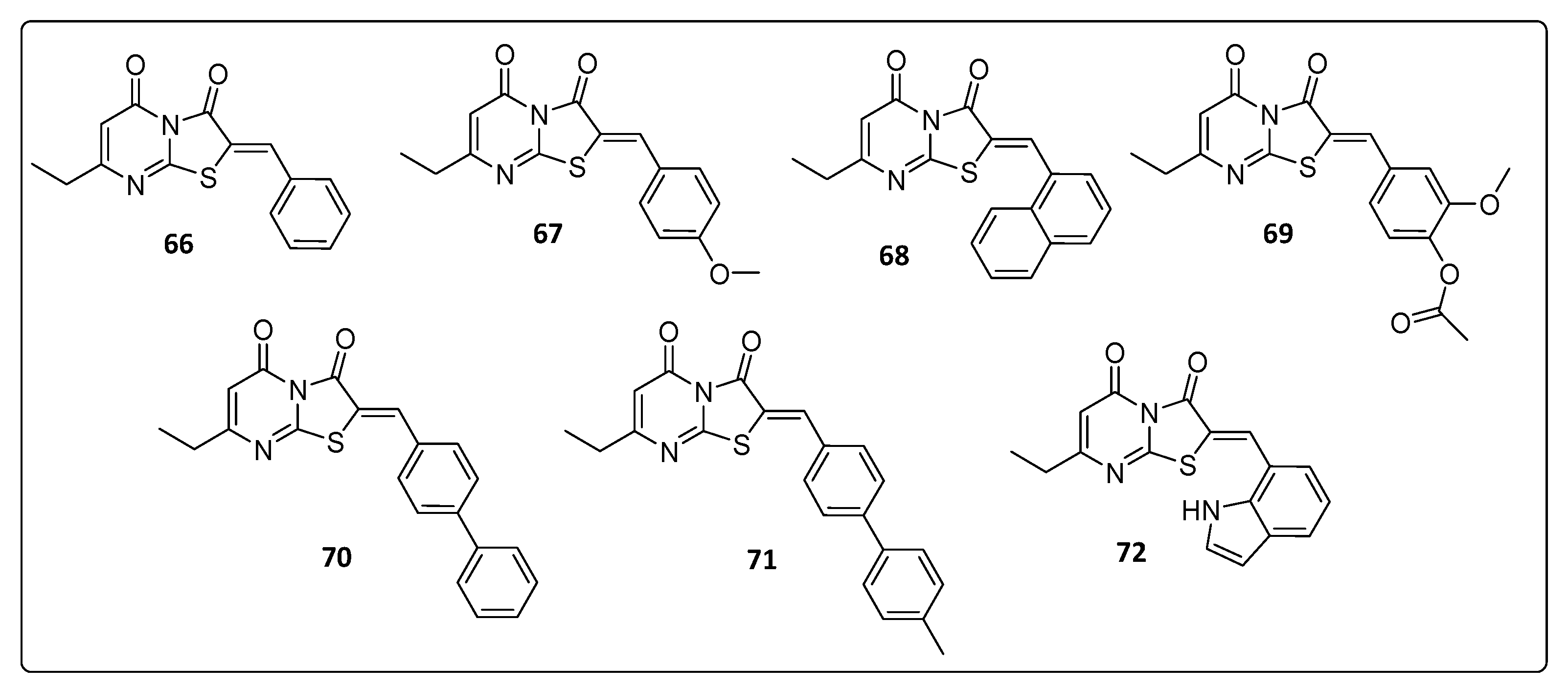
3.6. Thiazolopyrimidine and Triazolopyrimidine Derivatives
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burt, F.J.; Chen, W.; Miner, J.J.; Lenschow, D.J.; Merits, A.; Schnettler, E.; Kohl, A.; Rudd, P.A.; Taylor, A.; Herrero, L.J.; et al. Chikungunya virus: An update on the biology and pathogenesis of this emerging pathogen. Lancet Infect. Dis. 2017, 17, e107–e117. [Google Scholar] [CrossRef]
- Robinson, M.C. An Epidemic Of Virus Disease In Southern Province, Tanganyika Territory, In 1952-53. Trans. R. Soc. Trop. Med. Hyg. 1955, 49, 28–32. [Google Scholar] [CrossRef]
- Ghildiyal, R.; Gabrani, R. Antiviral therapeutics for chikungunya virus. Expert Opin. Ther. Pat. 2020, 30, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsetsarkin, K.A.; Chen, R.; Leal, G.; Forrester, N.; Higgs, S.; Huang, J.; Weaver, S.C. Chikungunya virus emergence is constrained in Asia by lineage-specific adaptive landscapes. Proc. Natl. Acad. Sci. USA 2011, 108, 7872–7877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeller, H.; Van Bortel, W.; Sudre, B. Chikungunya: Its history in Africa and Asia and its spread to new regions in 2013–2014. Proc. J. Infect. Dis. 2016, 214, S436–S440. [Google Scholar] [CrossRef] [PubMed]
- Centre, E.; Prevention, D. Chikungunya Worldwide Overview; ECDC: Solna, Sweden, 2020. [Google Scholar]
- Azevedo, R.D.S.D.S.; Oliveira, C.S.; Vasconcelos, P.F.D.C. Chikungunya risk for Brazil. Rev. Saude Publica 2015, 49. [Google Scholar] [CrossRef]
- da Cunha, R.V.; Trinta, K.S. Chikungunya virus: Clinical aspects and treatment. Mem. Inst. Oswaldo Cruz 2017, 112, 523–531. [Google Scholar] [CrossRef]
- Moesslacher, J.; Battisti, V.; Delang, L.; Neyts, J.; Abdelnabi, R.; Pürstinger, G.; Urban, E.; Langer, T. Identification of 2-(4-(Phenylsulfonyl)piperazine-1-yl)pyrimidine Analogues as Novel Inhibitors of Chikungunya Virus. ACS Med. Chem. Lett. 2020, 11, 906–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albulescu, I.C.; White-Scholten, L.; Tas, A.; Hoornweg, T.E.; Ferla, S.; Kovacikova, K.; Smit, J.M.; Brancale, A.; Snijder, E.J.; van Hemert, M.J. Suramin inhibits chikungunya virus replication by interacting with virions and blocking the early steps of infection. Viruses 2020, 12, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delang, L.; Li, C.; Tas, A.; Quérat, G.; Albulescu, I.C.; De Burghgraeve, T.; Segura Guerrero, N.A.; Gigante, A.; Piorkowski, G.; Decroly, E.; et al. The viral capping enzyme nsP1: A novel target for the inhibition of chikungunya virus infection. Sci. Rep. 2016, 6, 31819. [Google Scholar] [CrossRef]
- da Silva-Júnior, E.F.; Leoncini, G.O.; Rodrigues, É.E.S.; Aquino, T.M.; Araújo-Júnior, J.X. The medicinal chemistry of Chikungunya virus. Bioorg. Med. Chem. 2017, 25, 4219–4244. [Google Scholar] [CrossRef] [PubMed]
- Hitakarun, A.; Khongwichit, S.; Wikan, N.; Roytrakul, S.; Yoksan, S.; Rajakam, S.; Davidson, A.D.; Smith, D.R. Evaluation of the antiviral activity of orlistat (tetrahydrolipstatin) against dengue virus, Japanese encephalitis virus, Zika virus and chikungunya virus. Sci. Rep. 2020, 10, 1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirne-Santos, C.C.; de Barros, C.S.; Nogueira, C.C.R.; Azevedo, R.C.; Yamamoto, K.A.; Meira, G.L.S.; de Vasconcelos, Z.F.M.; Ratcliffe, N.A.; Teixeira, V.L.; Schmidt-Chanasit, J.; et al. Inhibition by marine algae of chikungunya virus isolated from patients in a recent disease outbreak in rio de janeiro. Front. Microbiol. 2019, 10, 2426. [Google Scholar] [CrossRef] [PubMed]
- Solignat, M.; Gay, B.; Higgs, S.; Briant, L.; Devaux, C. Replication cycle of chikungunya: A re-emerging arbovirus. Virology 2009, 393, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- Powers, A.M. Vaccine and therapeutic options to control chikungunya virus. Clin. Microbiol. Rev. 2018, 31, e00104-16. [Google Scholar] [CrossRef] [Green Version]
- Feibelman, K.M.; Fuller, B.P.; Li, L.; LaBarbera, D.V.; Geiss, B.J. Identification of small molecule inhibitors of the Chikungunya virus nsP1 RNA capping enzyme. Antivir. Res. 2018, 154, 124–131. [Google Scholar] [CrossRef]
- Gigante, A.; Gómez-SanJuan, A.; Delang, L.; Li, C.; Bueno, O.; Gamo, A.M.; Priego, E.M.; Camarasa, M.J.; Jochmans, D.; Leyssen, P.; et al. Antiviral activity of [1,2,3]triazolo[4,5-d]pyrimidin-7(6H)-ones against chikungunya virus targeting the viral capping nsP1. Antivir. Res. 2017, 144, 216–222. [Google Scholar] [CrossRef]
- Utt, A.; Das, P.K.; Varjak, M.; Lulla, V.; Lulla, A.; Merits, A. Mutations Conferring a Noncytotoxic Phenotype on Chikungunya Virus Replicons Compromise Enzymatic Properties of Nonstructural Protein 2. J. Virol. 2015, 89, 3145–3162. [Google Scholar] [CrossRef] [Green Version]
- Bhakat, S.; Soliman, M.E.S. Chikungunya virus (CHIKV) inhibitors from natural sources: A medicinal chemistry perspective. J. Nat. Med. 2015, 69, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Campagnola, G.; McDonald, S.; Beaucourt, S.; Vignuzzi, M.; Peersen, O.B. Structure-Function Relationships Underlying the Replication Fidelity of Viral RNA-Dependent RNA Polymerases. J. Virol. 2015, 89, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacikova, K.; Morren, B.M.; Tas, A.; Albulescu, I.C.; Van Rijswijk, R.; Jarhad, D.B.; Shin, Y.S.; Jang, M.H.; Kim, G.; Lee, H.W.; et al. 6′-β-fluoro-homoaristeromycin and 6′-fluoro-homoneplanocin A are potent inhibitors of Chikungunya virus replication through their direct effect on viral nonstructural protein 1. Antimicrob. Agents Chemother. 2020, 64, e02532-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekins, S.; Lane, T.R.; Madrid, P.B. Tilorone: A Broad-Spectrum Antiviral Invented in the USA and Commercialized in Russia and beyond. Pharm. Res. 2020, 37, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, A.C.; Reis, P.A.; de Freitas, C.S.; Sacramento, C.Q.; Hoelz, L.V.B.; Bastos, M.M.; Mattos, M.; Rocha, N.; de Azevedo Quintanilha, I.G.; da Silva Gouveia Pedrosa, C.; et al. Beyond members of the Flaviviridae family, sofosbuvir also inhibits chikungunya virus replication. Antimicrob. Agents Chemother. 2019, 63, e01389-18. [Google Scholar] [CrossRef] [Green Version]
- Briolant, S.; Garin, D.; Scaramozzino, N.; Jouan, A.; Crance, J.M. In vitro inhibition of Chikungunya and Semliki Forest viruses replication by antiviral compounds: Synergistic effect of interferon-α and ribavirin combination. Antivir. Res. 2004, 61, 111–117. [Google Scholar] [CrossRef]
- Ozden, S.; Lucas-Hourani, M.; Ceccaldi, P.E.; Basak, A.; Valentine, M.; Benjannet, S.; Hamelin, J.; Jacob, Y.; Mamchaoui, K.; Mouly, V.; et al. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors: Impairment of the maturation of the E2 surface glycoprotein. J. Biol. Chem. 2008, 283, 21899–21908. [Google Scholar] [CrossRef] [Green Version]
- Delogu, I.; Pastorino, B.; Baronti, C.; Nougairède, A.; Bonnet, E.; de Lamballerie, X. In vitro antiviral activity of arbidol against Chikungunya virus and characteristics of a selected resistant mutant. Antivir. Res. 2011, 90, 99–107. [Google Scholar] [CrossRef]
- Pohjala, L.; Utt, A.; Varjak, M.; Lulla, A.; Merits, A.; Ahola, T.; Tammela, P. Inhibitors of alphavirus entry and replication identified with a stable Chikungunya replicon cell line and virus-based assays. PLoS ONE 2011, 6, e28923. [Google Scholar] [CrossRef]
- Kaur, P.; Chu, J.J.H. Chikungunya virus: An update on antiviral development and challenges. Drug Discov. Today 2013, 18, 969–983. [Google Scholar] [CrossRef]
- Gigante, A.; Canela, M.D.; Delang, L.; Priego, E.M.; Camarasa, M.J.; Querat, G.; Neyts, J.; Leyssen, P.; Pérez-Pérez, M.J. Identification of [1,2,3]triazolo[4,5-d ]pyrimidin-7(6H)-ones as novel inhibitors of chikungunya virus replication. J. Med. Chem. 2014, 57, 4000–4008. [Google Scholar] [CrossRef]
- Gómez-Sanjuan, A.; Gamo, A.M.; Delang, L.; Pérez-Sánchez, A.; Amrun, S.N.; Abdelnabi, R.; Jacobs, S.; Priego, E.M.; Camarasa, M.J.; Jochmans, D.; et al. Inhibition of the Replication of Different Strains of Chikungunya Virus by 3-Aryl-[1,2,3]triazolo[4,5- d] pyrimidin-7(6 H)-ones. ACS Infect. Dis. 2018, 4, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Tănase, C.I.; Drăghici, C.; Hanganu, A.; Pintilie, L.; Maganu, M.; Volobueva, A.; Sinegubova, E.; Zarubaev, V.V.; Neyts, J.; Jochmans, D.; et al. New HSV-1 anti-viral 10-homocarbocyclic nucleoside analogs with an optically active substituted bicyclo[2.2.1]heptane fragment as a glycoside moiety. Molecules 2019, 24, 2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwu, J.R.; Kapoor, M.; Tsay, S.C.; Lin, C.C.; Hwang, K.C.; Horng, J.C.; Chen, I.C.; Shieh, F.K.; Leyssen, P.; Neyts, J. Benzouracil-coumarin-arene conjugates as inhibiting agents for chikungunya virus. Antivir. Res. 2015, 118, 103–109. [Google Scholar] [CrossRef]
- Shin, Y.S.; Jarhad, D.B.; Jang, M.H.; Kovacikova, K.; Kim, G.; Yoon, J.S.; Kim, H.R.; Hyun, Y.E.; Tipnis, A.S.; Chang, T.S.; et al. Identification of 6′-β-fluoro-homoaristeromycin as a potent inhibitor of chikungunya virus replication. Eur. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.S.; Kim, G.; Jarhad, D.B.; Kim, H.R.; Shin, Y.S.; Qu, S.; Sahu, P.K.; Kim, H.O.; Lee, H.W.; Wang, S.B.; et al. Design, Synthesis, and Anti-RNA Virus Activity of 6′-Fluorinated-Aristeromycin Analogues. J. Med. Chem. 2019, 62, 6346–6362. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, M.; Ferla, S.; Brancale, A.; McGuigan, C. Synthesis and Biological Evaluation of 6-Substituted-5-Fluorouridine ProTides. Bioorg. Med. Chem. 2018, 26, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Ehteshami, M.; Tao, S.; Zandi, K.; Hsiao, H.M.; Jiang, Y.; Hammond, E.; Amblard, F.; Russell, O.O.; Merits, A.; Schinazi, R.F. Characterization of β-D-N4-hydroxycytidine as a novel inhibitor of Chikungunya virus. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fares, M.; McCosker, P.M.; Alsherbiny, M.A.; Willis, A.C.; Clark, T.; Neyts, J.; Jochmans, D.; Keller, P.A. Regioselective convergent synthesis of 2-arylidene thiazolo[3,2-: A] pyrimidines as potential anti-chikungunya agents. RSC Adv. 2020, 10, 5191–5195. [Google Scholar] [CrossRef] [Green Version]
- Goh, V.S.L.; Mok, C.K.; Chu, J.J.H. Antiviral natural products for arbovirus infections. Molecules 2020, 25, 2796. [Google Scholar] [CrossRef]
- Jain, J.; Kumar, A.; Narayanan, V.; Ramaswamy, R.S.; Sathiyarajeswaran, P.; Shree Devi, M.S.; Kannan, M.; Sunil, S. Antiviral activity of ethanolic extract of Nilavembu Kudineer against dengue and chikungunya virus through in vitro evaluation. J. Ayurveda Integr. Med. 2020, 11, 329–335. [Google Scholar] [CrossRef]
- Mishra, S.; Pandey, A.; Manvati, S. Coumarin: An emerging antiviral agent. Heliyon 2020, 6, e03217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louvet, M.S.; Gault, G.; Lefebvre, S.; Popowycz, F.; Boulven, M.; Besse, S.; Benoit, E.; Lattard, V.; Grancher, D. Comparative inhibitory effect of prenylated coumarins, ferulenol and ferprenin, contained in the “poisonous chemotype” of Ferula communis on mammal liver microsomal VKORC1 activity. Phytochemistry 2015, 118, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Menezes, J.C.J.M.D.S.; Diederich, M. Translational role of natural coumarins and their derivatives as anticancer agents. Future Med. Chem. 2019, 11, 1057–1082. [Google Scholar] [CrossRef] [PubMed]
- Osman, H.; Yusufzai, S.K.; Khan, M.S.; Abd Razik, B.M.; Sulaiman, O.; Mohamad, S.; Gansau, J.A.; Ezzat, M.O.; Parumasivam, T.; Hassan, M.Z. New thiazolyl-coumarin hybrids: Design, synthesis, characterization, X-ray crystal structure, antibacterial and antiviral evaluation. J. Mol. Struct. 2018, 1166, 147–154. [Google Scholar] [CrossRef]
- Márquez, N.; Sancho, R.; Bedoya, L.M.; Alcamí, J.; López-Pérez, J.L.; San Feliciano, A.; Fiebich, B.L.; Muñoz, E. Mesuol, a natural occurring 4-phenylcoumarin, inhibits HIV-1 replication by targeting the NF-κB pathway. Antivir. Res. 2005, 66, 137–145. [Google Scholar] [CrossRef]
- Peng, H.K.; Chen, W.C.; Lee, J.C.; Yang, S.Y.; Tzeng, C.C.; Lin, Y.T.; Yang, S.C. Novel anilinocoumarin derivatives as agents against hepatitis C virus by the induction of IFN-mediated antiviral responses. Org. Biomol. Chem. 2013, 11, 1858–1866. [Google Scholar] [CrossRef]
- Yusufzai, S.K.; Osman, H.; Khan, M.S.; Abd Razik, B.M.; Ezzat, M.O.; Mohamad, S.; Sulaiman, O.; Gansau, J.A.; Parumasivam, T. 4-Thiazolidinone coumarin derivatives as two-component NS2B/NS3 DENV flavivirus serine protease inhibitors: Synthesis, molecular docking, biological evaluation and structure–activity relationship studies. Chem. Cent. J. 2018, 12, 69. [Google Scholar] [CrossRef]
- Hwu, J.R.; Singha, R.; Hong, S.C.; Chang, Y.H.; Das, A.R.; Vliegen, I.; De Clercq, E.; Neyts, J. Synthesis of new benzimidazole-coumarin conjugates as anti-hepatitis C virus agents. Antivir. Res. 2008, 77, 157–162. [Google Scholar] [CrossRef]
- Neyts, J.; De Clercq, E.; Singha, R.; Chang, Y.H.; Das, A.R.; Chakraborty, S.K.; Hong, S.C.; Tsay, S.C.; Hsu, M.H.; Hwu, J.R. Structure-activity relationship of new anti-hepatitis C virus agents: Heterobicycle-coumarin conjugates. J. Med. Chem. 2009, 52, 1486–1490. [Google Scholar] [CrossRef]
- Hwu, J.R.; Lin, S.Y.; Tsay, S.C.; De Clercq, E.; Leyssen, P.; Neyts, J. Coumarin-purine ribofuranoside conjugates as new agents against hepatitis c virus. J. Med. Chem. 2011, 54, 2114–2126. [Google Scholar] [CrossRef]
- Rashamuse, T.J.; Klein, R.; Kaye, P.T. Synthesis of Baylis-Hillman-derived phosphonated 3-(benzylaminomethyl) coumarins. Synth. Commun. 2010, 40, 3683–3690. [Google Scholar] [CrossRef]
- Thaisrivongs, S.; Tomich, P.K.; Watenpaugh, K.D.; Chong, K.T.; Howe, J.W.; Yang, C.P.; Strohbach, J.W.; Turner, S.R.; McGrath, J.P.; Bohanon, M.J.; et al. Structure-Based Design of HIV Protease Inhibitors: 4-Hydroxycoumarins and 4-Hydroxy-2-pyrones as Non-peptidic Inhibitors. J. Med. Chem. 1994, 37, 3200–3204. [Google Scholar] [CrossRef] [PubMed]
- Hwu, J.R.; Huang, W.C.; Lin, S.Y.; Tan, K.T.; Hu, Y.C.; Shieh, F.K.; Bachurin, S.O.; Ustyugov, A.; Tsay, S.C. Chikungunya virus inhibition by synthetic coumarin–guanosine conjugates. Eur. J. Med. Chem. 2019, 166, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; O’Rear, J.; Stollar, V. Mutagenesis of the Sindbis virus nsP1 protein: Effects on methyltransferase activity and viral infectivity. Virology 1996. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E. In search of selective antiviral chemotherapy. Clin. Microbiol. Rev. 1997, 10, 674–693. [Google Scholar] [CrossRef]
- Balzarini, J.; Aquaro, S.; Hassan-Abdallah, A.; Daluge, S.M.; Perno, C.F.; McGuigan, C. Improved antiviral activity of the aryloxymethoxyalaninyl phosphoramidate (APA) prodrug of abacavir (ABC) is due to the formation of markedly increased carbovir 5′-triphosphate metabolite levels. FEBS Lett. 2004, 573, 38–44. [Google Scholar] [CrossRef]
- Veinberg, G.; Shestakova, I.; Vorona, M.; Kanepe, I.; Lukevics, E. Synthesis of antitumor 6-alkylidenepenicillanate sulfones and related 3-alkylidene-2-azetidinones. Bioorg. Med. Chem. Lett. 2004, 14, 147–150. [Google Scholar] [CrossRef]
- Nivsarkar, M.; Thavaselvam, D.; Prasanna, S.; Sharma, M.; Kaushik, M.P. Design, synthesis and biological evaluation of novel bicyclic β-lactams as potential antimalarials. Bioorg. Med. Chem. Lett. 2005, 15, 1371–1373. [Google Scholar] [CrossRef]
- Sperka, T.; Pitlik, J.; Bagossi, P.; Tözsér, J. Beta-lactam compounds as apparently uncompetitive inhibitors of HIV-1 protease. Bioorg. Med. Chem. Lett. 2005, 15, 3086–3090. [Google Scholar] [CrossRef]
- Kværnø, L.; Werder, M.; Hauser, H.; Carreira, E.M. Synthesis and in vitro evaluation of inhibitors of intestinal cholesterol absorption. J. Med. Chem. 2005, 48, 6035–6053. [Google Scholar] [CrossRef]
- D’hooghe, M.; Mollet, K.; De Vreese, R.; Jonckers, T.H.M.; Dams, G.; De Kimpe, N. Design, Synthesis, and Antiviral Evaluation of Purine-β-lactam and Purine-aminopropanol Hybrids. J. Med. Chem. 2012, 55, 5637–5641. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Compound | CC50 (μM) | EC50 (μM) | SI | LogP |
|---|---|---|---|---|---|
| Hwu (2015) [34] | 7 | 178 | 19.1 | 9.3 | 1.91 |
| 8 | 117 | 10.2 | 11.5 | 2.13 | |
| 9 | 30 | 18.4 | 1.6 | ND | |
| 10 | 117 | 54.5 | 2.2 | ND | |
| 11 | 144 | 17.2 | 8.8 | −1.07 | |
| 12 | 126 | 58 | 2.2 | ND | |
| 13 | 114 | 26.4 | 4.3 | 0.611 | |
| 14 | 107 | 19.0 | 5.6 | −2.60 | |
| 15 | 60.2 | 23.1 | 2.6 | 0.467 | |
| 16 | 75.2 | 13.0 | 5.8 | −2.74 | |
| 17 | 104 | 45.1 | 2.3 | ND | |
| 18 | 13.8 | 4.6 | 3.0 | ND | |
| 19 | >284 | 192 | >1.5 | 1.32 | |
| Hwu (2019) [54] | 20 | 81.3 | 40.9 | 1.98 | 3.22 |
| 21 | >212 | 9.9 | >21.7 | 3.01 | |
| 22 | 96.5 | 10.3 | 9.37 | 2.97 | |
| 23 | >227 | 13.9 | >16.3 | 3.77 | |
| Standard: 3-[(methylthio)-methyl]coumarin | ND | >485 | ND | - |
| Reference | Compound | SAH Hydrolase IC50 (μM) | CC50 (μM) | EC50 (μM) | SI |
|---|---|---|---|---|---|
| Yoon (2019) [36] | Standard (1) | 1.32 | 6.3 | 0.8 | 7.9 |
| 24 | 0.37 | >100 | >100 | ND | |
| 25 | 9.70 | 1.32 | 0.53 | 2.49 | |
| 26 | 1.06 | >1.25 | 0.13 | >9.6 | |
| 27 | 4.39 | >100 | >100 | ND | |
| 28 | 0.76 | >100 | >100 | ND | |
| 29 | >100 | >100 | >100 | ND | |
| 30 | >100 | >100 | >100 | ND | |
| 31 | >100 | >100 | >100 | ND | |
| 32 | >100 | >100 | >100 | ND | |
| 33 | >100 | >100 | >100 | ND | |
| 34 | >100 | >12.5 | 1.95 | >6.4 | |
| 35 | >100 | >100 | >100 | ND | |
| 36 | >100 | >100 | >100 | ND | |
| Shin (2020) [35] | 37 | 0.36 | >250 | 0.12 | ND |
| 38 | 0.37 | >100 | >100 | ND | |
| 39 | >100 | >100 | >100 | ND | |
| 40 | 2.03 | >25 | >100 | ND | |
| 41 | 3.05 | >100 | >100 | ND |
| Reference | Compound | CC50 (μM) | EC50 (μM) |
|---|---|---|---|
| Slusarczyk (2018) [37] | Standard (5) | >10 | 0.468 |
| 42 | 47.02 | >200 | |
| 43 | 9.04 | >200 | |
| 44 | >200 | >200 | |
| 45 | 81.80 | >200 | |
| 46 | >200 | >200 | |
| 47 | 81.29 | >200 | |
| 48 | >200 | >200 | |
| 49 | 53.10 | >50 | |
| Ehteshami (2016) [38] | 50 | 30.6 c 7.7 d 2.5 e | 0.2 a,b |
| Standard (2) | ND | 0.3 a 0.6 b |
| Compound | CC50 (µM) | EC50 (µM) |
|---|---|---|
| Standard (51) | 122 ± 24 | 8.7 ± 1 |
| 52 | 156 ± 35 | 14 ± 3 |
| 53 | 51 ± 19 | 5 ± 0.4 |
| 54 | 44 ± 19 | 7.1 ± 0.01 |
| 55 | 19 ± 2 | 5.9 ± 0.2 |
| 56 | 95 ± 74 | 15 ± 2 |
| 57 | 181 ± 0.4 | 10 ± 0.2 |
| 58 | >260 | 4.0 ± 1 |
| 59 | 69 ± 7 | 2.5 ± 1 |
| 60 | 106 ± 69 | 16 ± 1 |
| 61 | 66.4 ± 6 | 9.5 ± 2 |
| 62 | 59 ± 18 | 3.2 ± 0.2 |
| Compound | CC50 (μM) | EC50 (μM) | SI |
|---|---|---|---|
| 63 | >98.36 | 17.11 | >5.75 |
| 64 | 58.97 | 13.01 | 4.53 |
| 65 | 71.20 | 11.51 | 6.19 |
| Compound | % Inhibition at 20 μg/mL | EC50 (μΜ) |
|---|---|---|
| 66 | 19 | ND |
| 67 | 4.3 | ND |
| 68 | 28 | ND |
| 69 | 5.5 | ND |
| 70 | 5.0 | ND |
| 71 | 58 | 42 |
| 72 | 34 | ND |
| Reference | Compound | SI | CC50 (µM) | EC50 (µM) | |||||
|---|---|---|---|---|---|---|---|---|---|
| CHIKV 899 | CHIKV Venturini | CHIKV Congo 95 | CHIKV St. Martin | CHIKV OPY | CHIKV nsP1-P34S | ||||
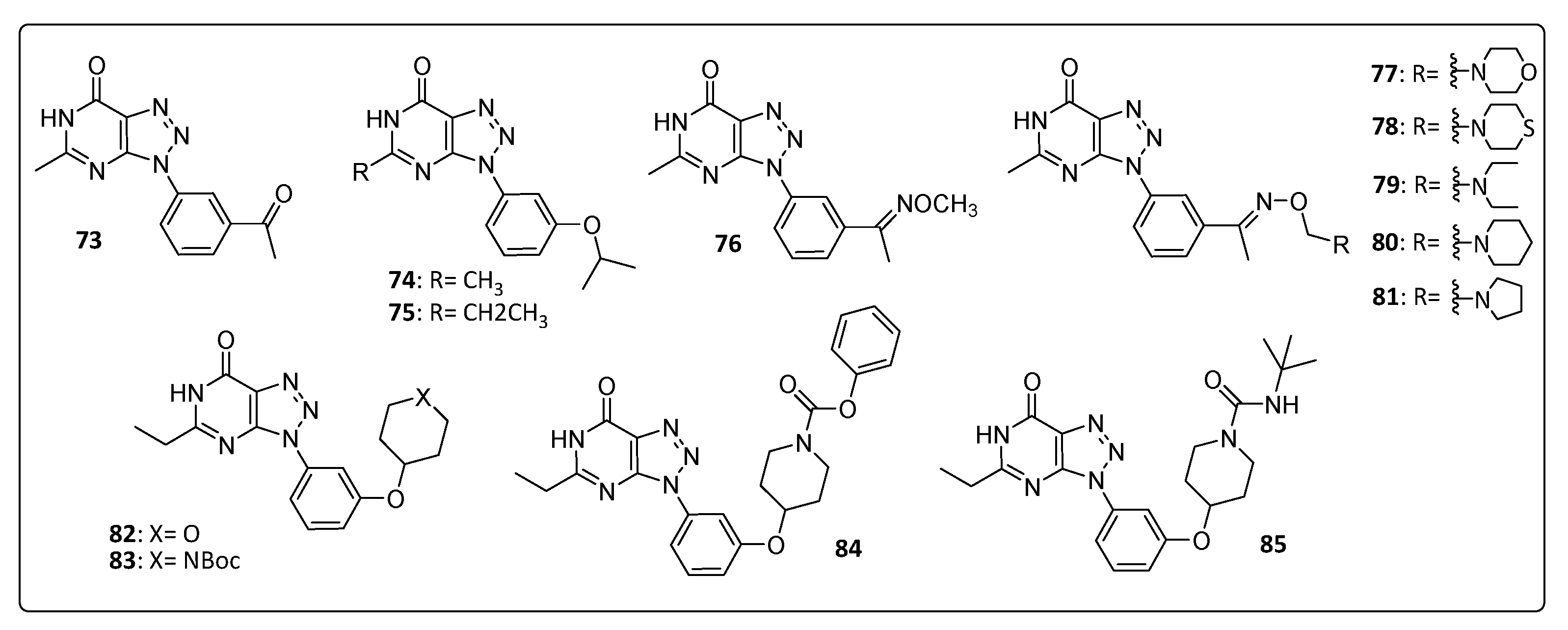
| Gigante (2014) [31] | 73 | 39 ± 5 | >743 | 19 ± 2 | 26 ± 2 | 6.35 ± 0.05 | 23.9 ± 0.5 | 28 | 116 ± 60 |
| 74 | ND | >704 | 12 ± 4 | ND | ND | ND | ND | ND | |
| 75 | 64 | >668 [31] 167 ± 94 [32] | 3 ± 1 | 1.43 ± 0.01 | 0.75 ± 0.35 | 2.9 ± 0.05 | 2.6 ± 0.5 | >167 | |
| Gigante (2017) [19] | 76 | 101 ± 73 | 3.6 ± 1.3 | 5.2 ± 0.2 | 1.1 ± 0.1 | 4.2 ± 0.8 | ND | ND | ND |
| 77 | >729 | 12 ± 1 | 6.7 | 2.5 | ND | ND | ND | ND | |
| 78 | 162 ± 1 | 5.2 ± 0.5 | ND | ND | ND | ND | ND | ND | |
| 79 | 165 ± 8 | 16 ± 6 | ND | ND | ND | ND | ND | ND | |
| 80 | 95 ± 40 | 6.9 ± 2 | 9.6 ± 1.2 | 3.0 ± 0.5 | 14 ± 3.5 | ND | ND | ND | |
| 81 | 166 ± 26 | 16 ± 1.8 | ND | ND | ND | ND | ND | ND | |
| Gómez-Sanjuan (2018) [32] | 82 | 70 | 84 ± 19 | 1.2 ± 0.01 | 1.65 ± 0.5 | 0.3 ± 0.0 | 2 ± 0.3 | 5.2 | 65 ± 40 |
| 83 | 18 | 59 ± 23 | 3.2 ± 1.0 | 2.6 ± 1.1 | 1 ± 0.1 | ND | 11.5 | 35 ± 29 | |
| 84 | >147 | >220 | 1.5 ± 0.3 | 1.2 ± 0.2 | 0.35 ± 0.5 | 1.2 ± 0.5 | 3.4 ± 1.2 | ND | |
| 85 | 23 | 201 ± 26 | 8.9 ± 6.1 | 2.9 ± 0.4 | 1.0 ± 0.4 | 3 ± 2.5 | 5.4 ± 2.4 | 23 ± 14 | |
| Standard: Chloroquine | 8.1 | 89 ± 28 | 11 ± 7 | ND | ND | ND | ND | ND | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santana, A.C.; Silva Filho, R.C.; Menezes, J.C.J.M.D.S.; Allonso, D.; Campos, V.R. Nitrogen-Based Heterocyclic Compounds: A Promising Class of Antiviral Agents against Chikungunya Virus. Life 2021, 11, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/life11010016
Santana AC, Silva Filho RC, Menezes JCJMDS, Allonso D, Campos VR. Nitrogen-Based Heterocyclic Compounds: A Promising Class of Antiviral Agents against Chikungunya Virus. Life. 2021; 11(1):16. https://0-doi-org.brum.beds.ac.uk/10.3390/life11010016
Chicago/Turabian StyleSantana, Andreza C., Ronaldo C. Silva Filho, José C. J. M. D. S. Menezes, Diego Allonso, and Vinícius R. Campos. 2021. "Nitrogen-Based Heterocyclic Compounds: A Promising Class of Antiviral Agents against Chikungunya Virus" Life 11, no. 1: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/life11010016