
Interaction between Neurons and the Oligodendroglial Lineage in Multiple Sclerosis and Its Preclinical Models

 ,
, 
Abstract
:1. Introduction
2. The Organization and Functioning of the Myelinated Axon
3. Neuronal Activity as a Mediator of Myelin Plasticity
4. Oligodendrocyte Lineage and Neuron: A Story of Mutual Dependence
5. Altered Myelin and Axonal Damage: Lessons from the Animal Models
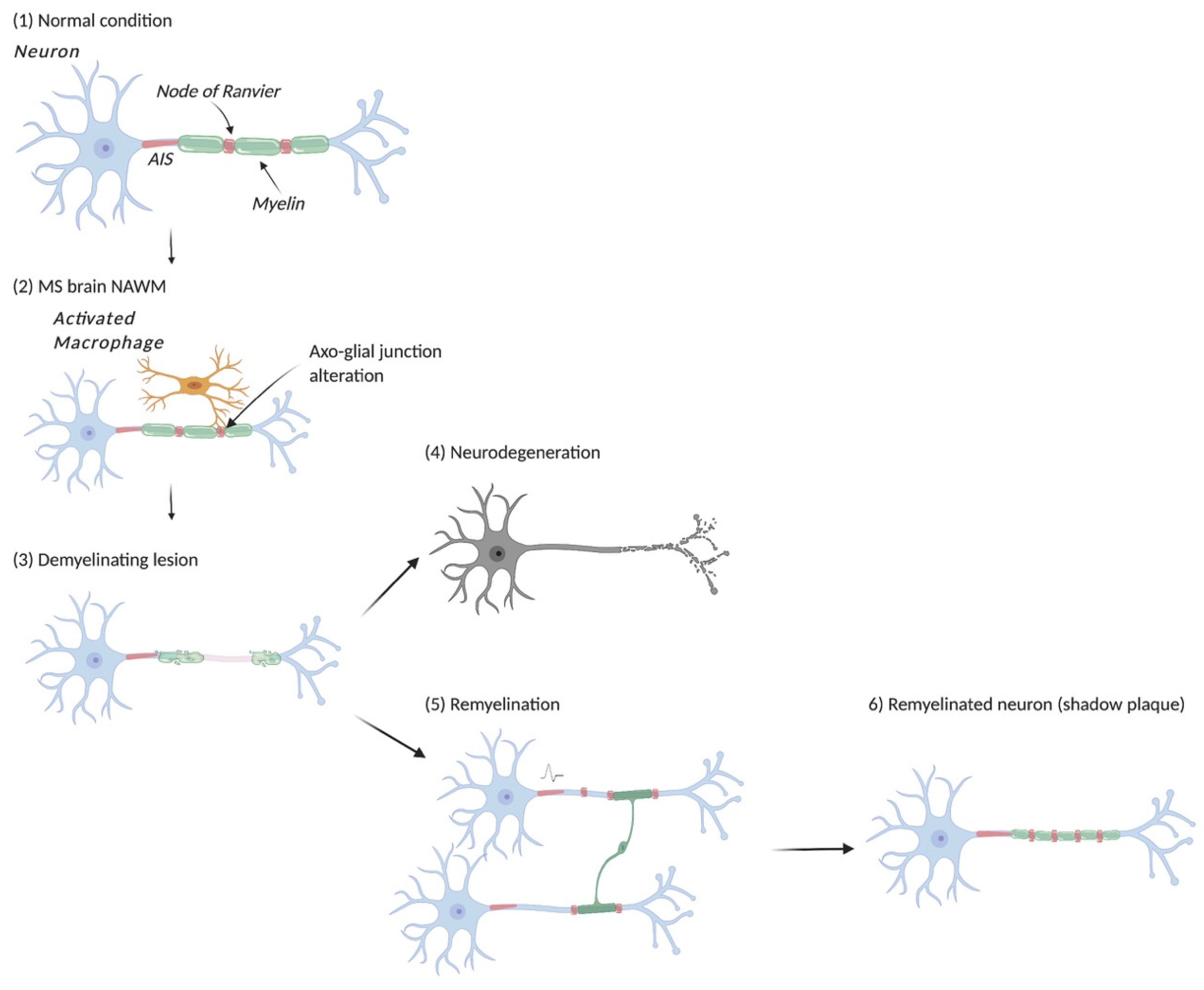
6. Myelin Insult and Neuronal Damage in Multiple Sclerosis
7. Endogenous Remyelination Occurs in MS and Its Animal Models
8. Remyelination Failure in MS
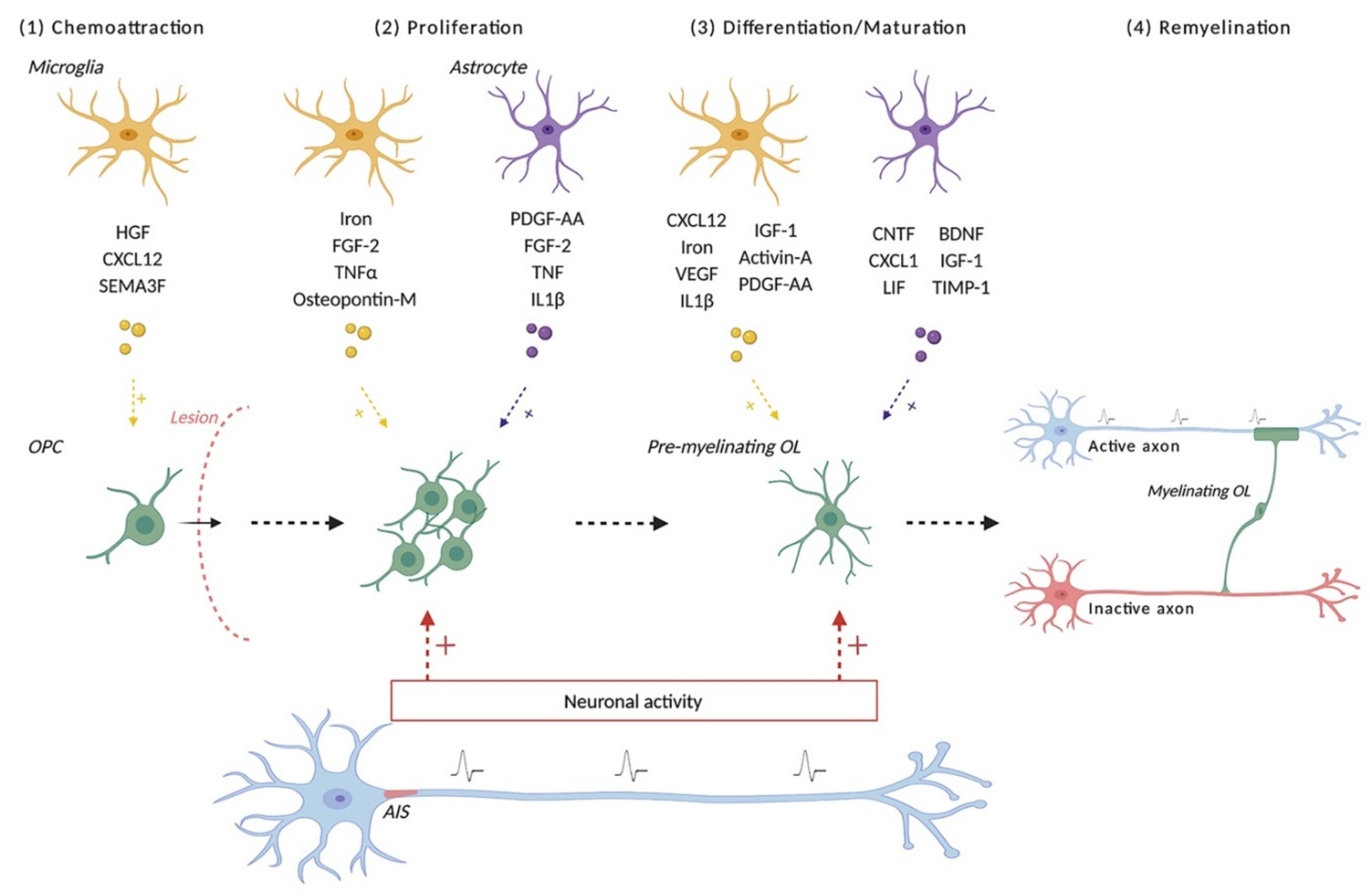
9. After a Demyelinating Insult, What Are the Required Steps for New Myelin Sheath Formation?
10. Other Cellular Contributors to Successful Remyelination
11. Myelin Repair: From Animal Models to Human Translation
11.1. The Challenges of Myelin Repair Strategies.
11.2. Repurposing Existing Drugs for Their Remyelinating Potential
11.3. Strategies Promoting OPC Differentiation
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zalc, B.; Goujet, D.; Colman, D. The Origin of the Myelination Program in Vertebrates. Curr. Biol. 2008, 18, R511–R512. [Google Scholar] [CrossRef] [Green Version]
- Zalc, B.; Colman, D.R. Origins of Vertebrate Success. Science 2000, 288, 271–272. [Google Scholar] [CrossRef]
- Rushton, W.A.H. A Theory of the Effects of Fibre Size in Medullated Nerve. J. Physiol. 1951, 115, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G.; Bennett, M.V.L. Relative Conduction Velocities of Small Myelinated and Non-Myelinated Fibres in the Central Nervous System. Nat. New Biol. 1972, 238, 217–219. [Google Scholar] [CrossRef]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic Oligodendrocytes Maintain Myelin and Long-Term Axonal Integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Leach, M.K.; Redmond, S.A.; Chong, S.Y.C.; Mellon, S.H.; Tuck, S.J.; Feng, Z.-Q.; Corey, J.M.; Chan, J.R. A Culture System to Study Oligodendrocyte Myelination Processes Using Engineered Nanofibers. Nat. Methods 2012, 9, 917–922. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, S.; Warrington, A.; Bansal, R. The Oligodendrocyte and Its Many Cellular Processes. Trends Cell Biol. 1993, 3, 191–197. [Google Scholar] [CrossRef]
- Czopka, T.; ffrench-Constant, C.; Lyons, D.A. Individual Oligodendrocytes Have Only a Few Hours in Which to Generate New Myelin Sheaths In Vivo. Dev. Cell 2013, 25, 599–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumas, L.; Heitz-Marchaland, C.; Fouquet, S.; Suter, U.; Livet, J.; Moreau-Fauvarque, C.; Chédotal, A. Multicolor Analysis of Oligodendrocyte Morphology, Interactions, and Development with Brainbow: Multicolor Imaging of Myelination. Glia 2015, 63, 699–717. [Google Scholar] [CrossRef]
- Matthews, M.A.; Duncan, D. A Quantitative Study of Morphological Changes Accompanying the Initiation and Progress of Myelin Production in the Dorsal Funiculus of the Rat Spinal Cord. J. Comp. Neurol. 1971, 142, 1–22. [Google Scholar] [CrossRef]
- Bacmeister, C.M.; Barr, H.J.; McClain, C.R.; Thornton, M.A.; Nettles, D.; Welle, C.G.; Hughes, E.G. Motor Learning Promotes Remyelination via New and Surviving Oligodendrocytes. Nat. Neurosci. 2020, 23, 819–831. [Google Scholar] [CrossRef]
- Bechler, M.E.; Byrne, L.; ffrench-Constant, C. CNS Myelin Sheath Lengths Are an Intrinsic Property of Oligodendrocytes. Curr. Biol. 2015, 25, 2411–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubetzki, C.; Demerens, C.; Anglade, P.; Villarroya, H.; Frankfurter, A.; Lee, V.M.; Zalc, B. Even in Culture, Oligodendrocytes Myelinate Solely Axons. Proc. Natl. Acad. Sci. USA 1993, 90, 6820–6824. [Google Scholar] [CrossRef] [Green Version]
- Zalc, B. The Acquisition of Myelin: An Evolutionary Perspective. Brain Res. 2016, 1641, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goebbels, S.; Wieser, G.L.; Pieper, A.; Spitzer, S.; Weege, B.; Yan, K.; Edgar, J.M.; Yagensky, O.; Wichert, S.P.; Agarwal, A.; et al. A Neuronal PI(3,4,5)P3-Dependent Program of Oligodendrocyte Precursor Recruitment and Myelination. Nat. Neurosci. 2017, 20, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, C.; Remahl, S.; Persson, H.; Bjartmar, C. Myelinated Nerve Fibres in the CNS. Prog. Neurobiol. 1993, 40, 319–384. [Google Scholar] [CrossRef]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.; et al. Oligodendroglia Metabolically Support Axons and Contribute to Neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Raff, M.C. Axonal Control of Oligodendrocyte Development. J. Cell Biol. 1999, 147, 1123–1128. [Google Scholar] [CrossRef]
- Demerens, C.; Stankoff, B.; Logak, M.; Anglade, P.; Allinquant, B.; Couraud, F.; Zalc, B.; Lubetzki, C. Induction of Myelination in the Central Nervous System by Electrical Activity. Proc. Natl. Acad. Sci. USA 1996, 93, 9887–9892. [Google Scholar] [CrossRef] [Green Version]
- Gautier, H.O.B.; Evans, K.A.; Volbracht, K.; James, R.; Sitnikov, S.; Lundgaard, I.; James, F.; Lao-Peregrin, C.; Reynolds, R.; Franklin, R.J.M.; et al. Neuronal Activity Regulates Remyelination via Glutamate Signalling to Oligodendrocyte Progenitors. Nat. Commun. 2015, 6, 8518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, E.M.; Purger, D.; Mount, C.W.; Goldstein, A.K.; Lin, G.L.; Wood, L.S.; Inema, I.; Miller, S.E.; Bieri, G.; Zuchero, J.B.; et al. Neuronal Activity Promotes Oligodendrogenesis and Adaptive Myelination in the Mammalian Brain. Science 2014, 344, 1252304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hines, J.H.; Ravanelli, A.M.; Schwindt, R.; Scott, E.K.; Appel, B. Neuronal Activity Biases Axon Selection for Myelination in Vivo. Nat. Neurosci. 2015, 18, 683–689. [Google Scholar] [CrossRef] [Green Version]
- Káradóttir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA Receptors Are Expressed in Oligodendrocytes and Activated in Ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef] [Green Version]
- Kukley, M.; Nishiyama, A.; Dietrich, D. The Fate of Synaptic Input to NG2 Glial Cells: Neurons Specifically Downregulate Transmitter Release onto Differentiating Oligodendroglial Cells. J. Neurosci. 2010, 30, 8320–8331. [Google Scholar] [CrossRef]
- Mensch, S.; Baraban, M.; Almeida, R.; Czopka, T.; Ausborn, J.; El Manira, A.; Lyons, D.A. Synaptic Vesicle Release Regulates Myelin Sheath Number of Individual Oligodendrocytes in Vivo. Nat. Neurosci. 2015, 18, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple Sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple Sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Brück, W.; Rodriguez, M.; Lassmann, H. Distinct Patterns of Multiple Sclerosis Pathology Indicates Heterogeneity in Pathogenesis. Brain Pathol. 1996, 6, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M. Regenerating CNS Myelin—from Mechanisms to Experimental Medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef]
- Lubetzki, C.; Zalc, B.; Williams, A.; Stadelmann, C.; Stankoff, B. Remyelination in Multiple Sclerosis: From Basic Science to Clinical Translation. Lancet Neurol. 2020, 19, 678–688. [Google Scholar] [CrossRef]
- Neumann, B.; Foerster, S.; Zhao, C.; Bodini, B.; Reich, D.S.; Bergles, D.E.; Káradóttir, R.T.; Lubetzki, C.; Lairson, L.L.; Zalc, B.; et al. Problems and Pitfalls of Identifying Remyelination in Multiple Sclerosis. Cell Stem Cell 2020, 26, 617–619. [Google Scholar] [CrossRef]
- Bodini, B.; Veronese, M.; García-Lorenzo, D.; Battaglini, M.; Poirion, E.; Chardain, A.; Freeman, L.; Louapre, C.; Tchikviladze, M.; Papeix, C.; et al. Dynamic I Maging of I Ndividual R Emyelination P Rofiles in M Ultiple S Clerosis. Ann. Neurol. 2016, 79, 726–738. [Google Scholar] [CrossRef] [Green Version]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Bruck, W.; Lucchinetti, C.; Lassmann, H. Remyelination Is Extensive in a Subset of Multiple Sclerosis Patients. Brain 2006, 129, 3165–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, M.; Antel, J.; Brück, W.; Stadelmann, C. Extensive Cortical Remyelination in Patients with Chronic Multiple Sclerosis. Brain Pathol. 2007, 17, 129–138. [Google Scholar] [CrossRef]
- Lubetzki, C.; Sol-Foulon, N.; Desmazières, A. Nodes of Ranvier during Development and Repair in the CNS. Nat. Rev. Neurol. 2020, 16, 426–439. [Google Scholar] [CrossRef]
- Poliak, S.; Peles, E. The Local Differentiation of Myelinated Axons at Nodes of Ranvier. Nat. Rev. Neurosci. 2003, 4, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Arancibia-Carcamo, I.L.; Attwell, D. The Node of Ranvier in CNS Pathology. Acta Neuropathol. 2014, 128, 161–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakiri, Y.; Káradóttir, R.; Cossell, L.; Attwell, D. Morphological and Electrical Properties of Oligodendrocytes in the White Matter of the Corpus Callosum and Cerebellum: Oligodendrocyte Electrical Properties. J. Physiol. 2011, 589, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Hartline, D.K.; Colman, D.R. Rapid Conduction and the Evolution of Giant Axons and Myelinated Fibers. Curr. Biol. 2007, 17, R29–R35. [Google Scholar] [CrossRef] [Green Version]
- Dubessy, A.-L.; Mazuir, E.; Rappeneau, Q.; Ou, S.; Abi Ghanem, C.; Piquand, K.; Aigrot, M.-S.; Thétiot, M.; Desmazières, A.; Chan, E.; et al. Role of a Contactin Multi-Molecular Complex Secreted by Oligodendrocytes in Nodal Protein Clustering in the CNS. Glia 2019, 67, 2248–2263. [Google Scholar] [CrossRef] [Green Version]
- Thetiot, M.; Freeman, S.A.; Roux, T.; Dubessy, A.; Aigrot, M.; Rappeneau, Q.; Lejeune, F.; Tailleur, J.; Sol-Foulon, N.; Lubetzki, C.; et al. An Alternative Mechanism of Early Nodal Clustering and Myelination Onset in GABAergic Neurons of the Central Nervous System. Glia 2020, 68, 1891–1909. [Google Scholar] [CrossRef] [PubMed]
- Charles, P. Re-Expression of PSA-NCAM by Demyelinated Axons: An Inhibitor of Remyelination in Multiple Sclerosis? Brain 2002, 125, 1972–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elazar, N.; Vainshtein, A.; Rechav, K.; Tsoory, M.; Eshed-Eisenbach, Y.; Peles, E. Coordinated Internodal and Paranodal Adhesion Controls Accurate Myelination by Oligodendrocytes. J. Cell Biol. 2019, 218, 2887–2895. [Google Scholar] [CrossRef] [Green Version]
- Klingseisen, A.; Ristoiu, A.-M.; Kegel, L.; Sherman, D.L.; Rubio-Brotons, M.; Almeida, R.G.; Koudelka, S.; Benito-Kwiecinski, S.K.; Poole, R.J.; Brophy, P.J.; et al. Oligodendrocyte Neurofascin Independently Regulates Both Myelin Targeting and Sheath Growth in the CNS. Dev. Cell 2019, 51, 730–744.e6. [Google Scholar] [CrossRef] [Green Version]
- Fields, R.D. White Matter in Learning, Cognition and Psychiatric Disorders. Trends Neurosci. 2008, 31, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, T. Neuron-Oligodendrocyte Interaction in Neuroinflammation. Clin. Exp. Neuroimmunol. 2015, 6, 232–244. [Google Scholar] [CrossRef]
- Simons, M.; Trajkovic, K. Neuron-Glia Communication in the Control of Oligodendrocyte Function and Myelin Biogenesis. J. Cell Sci. 2006, 119, 4381–4389. [Google Scholar] [CrossRef] [Green Version]
- Karimian, M.; Dibenedetto, D.; Moerel, M.; Burwick, T.; Westra, R.L.; De Weerd, P.; Senden, M. Effects of Synaptic and Myelin Plasticity on Learning in a Network of Kuramoto Phase Oscillators. Chaos 2019, 29, 083122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bells, S.; Lefebvre, J.; Longoni, G.; Narayanan, S.; Arnold, D.L.; Yeh, E.A.; Mabbott, D.J. White Matter Plasticity and Maturation in Human Cognition. Glia 2019, 67, 2020–2037. [Google Scholar] [CrossRef] [PubMed]
- Oluich, L.-J.; Stratton, J.A.S.; Lulu Xing, Y.; Ng, S.W.; Cate, H.S.; Sah, P.; Windels, F.; Kilpatrick, T.J.; Merson, T.D. Targeted Ablation of Oligodendrocytes Induces Axonal Pathology Independent of Overt Demyelination. J. Neurosci. 2012, 32, 8317–8330. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Nave, K.-A. Myelin Dynamics: Protecting and Shaping Neuronal Functions. Curr. Opin. Neurobiol. 2017, 47, 104–112. [Google Scholar] [CrossRef]
- Snaidero, N.; Simons, M. Myelination at a Glance. J. Cell Sci. 2014, 127, 2999–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Ohayon, D.; McKenzie, I.A.; Sinclair-Wilson, A.; Wright, J.L.; Fudge, A.D.; Emery, B.; Li, H.; Richardson, W.D. Rapid Production of New Oligodendrocytes Is Required in the Earliest Stages of Motor-Skill Learning. Nat. Neurosci. 2016, 19, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Gallo, V.; Zhou, J.; McBain, C.; Wright, P.; Knutson, P.; Armstrong, R. Oligodendrocyte Progenitor Cell Proliferation and Lineage Progression Are Regulated by Glutamate Receptor-Mediated K+ Channel Block. J. Neurosci. 1996, 16, 2659–2670. [Google Scholar] [CrossRef]
- Nagy, B.; Hovhannisyan, A.; Barzan, R.; Chen, T.-J.; Kukley, M. Different Patterns of Neuronal Activity Trigger Distinct Responses of Oligodendrocyte Precursor Cells in the Corpus Callosum. PLoS Biol. 2017, 15, e2001993. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Lee, P.R.; Fields, R.D. Control of Local Protein Synthesis and Initial Events in Myelination by Action Potentials. Science 2011, 333, 1647–1651. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Eisen, A.M.; McBain, C.J.; Gallo, V. A Role for Glutamate and Its Receptors in the Regulation of Oligodendrocyte Development in Cerebellar Tissue Slices. Development 1998, 125, 2901–2914. [Google Scholar]
- Stevens, B.; Porta, S.; Haak, L.L.; Gallo, V.; Fields, R.D. Adenosine: A Neuron-Glial Transmitter Promoting Myelination in the CNS in Response to Action Potentials. Neuron 2002, 36, 855–868. [Google Scholar] [CrossRef] [Green Version]
- Ronzano, R.; Thetiot, M.; Lubetzki, C.; Desmazieres, A. Myelin Plasticity and Repair: Neuro-Glial Choir Sets the Tuning. Front. Cell. Neurosci. 2020, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Baraban, M.; Koudelka, S.; Lyons, D.A. Ca2+ Activity Signatures of Myelin Sheath Formation and Growth in Vivo. Nat. Neurosci. 2018, 21, 19–23. [Google Scholar] [CrossRef]
- Ortiz, F.C.; Habermacher, C.; Graciarena, M.; Houry, P.-Y.; Nishiyama, A.; Oumesmar, B.N.; Angulo, M.C. Neuronal Activity in Vivo Enhances Functional Myelin Repair. JCI Insight 2019, 4, e123434. [Google Scholar] [CrossRef] [Green Version]
- Trapp, B.D.; Nishiyama, A.; Cheng, D.; Macklin, W. Differentiation and Death of Premyelinating Oligodendrocytes in Developing Rodent Brain. J. Cell Biol. 1997, 137, 459–468. [Google Scholar] [CrossRef]
- Wilkins, A.; Majed, H.; Layfield, R.; Compston, A.; Chandran, S. Oligodendrocytes Promote Neuronal Survival and Axonal Length by Distinct Intracellular Mechanisms: A Novel Role for Oligodendrocyte-Derived Glial Cell Line-Derived Neurotrophic Factor. J. Neurosci. 2003, 23, 4967–4974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.; Nave, K.-A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2016, 8, a020479. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetanova, I.D.; Nave, K.-A. The Role of Myelin and Oligodendrocytes in Axonal Energy Metabolism. Curr. Opin. Neurobiol. 2013, 23, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Richter, N.; Fan, Z.; Siemonsmeier, G.; Pivneva, T.; Jordan, P.; Steinhäuser, C.; Semtner, M.; Nolte, C.; Kettenmann, H. Oligodendrocytes in the Mouse Corpus Callosum Maintain Axonal Function by Delivery of Glucose. Cell Rep. 2018, 22, 2383–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.; Meschkat, M.; Ruhwedel, T.; Trevisiol, A.; Tzvetanova, I.D.; Battefeld, A.; Kusch, K.; Kole, M.H.P.; Strenzke, N.; Möbius, W.; et al. A Role of Oligodendrocytes in Information Processing. Nat. Commun. 2020, 11, 5497. [Google Scholar] [CrossRef]
- Montag, D.; Giese, K.P.; Bartsch, U.; Martini, R.; Lang, Y.; Blüthmann, H.; Karthigasan, J.; Kirschner, D.A.; Wintergerst, E.S.; Nave, K.-A.; et al. Mice Deficient for the Glycoprotein Show Subtle Abnormalities in Myelin. Neuron 1994, 13, 229–246. [Google Scholar] [CrossRef]
- Griffiths, I. Axonal Swellings and Degeneration in Mice Lacking the Major Proteolipid of Myelin. Science 1998, 280, 1610–1613. [Google Scholar] [CrossRef]
- Garbern, J.Y.; Yool, D.A.; Moore, G.J.; Wilds, I.B.; Faulk, M.W.; Klugmann, M.; Nave, K.-A.; Sistermans, E.A.; van der Knaap, M.S.; Bird, T.D.; et al. Patients Lacking the Major CNS Myelin Protein, Proteolipid Protein 1, Develop Length-Dependent Axonal Degeneration in the Absence of Demyelination and Inflammation. Brain 2002, 125, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Delaney, K.H.; Kwiecien, J.M.; Wegiel, J.; Wisniewski, H.M.; Percy, D.H.; Fletch, A.L. Familial Dysmyelination in a Long Evans Rat Mutant. Lab. Anim. Sci. 1995, 45, 547–553. [Google Scholar]
- Rosenbluth, J. Central Myelin in the Mouse Mutant Shiverer. J. Comp. Neurol. 1980, 194, 639–648. [Google Scholar] [CrossRef]
- Smith, C.M.; Cooksey, E.; Duncan, I.D. Myelin Loss Does Not Lead to Axonal Degeneration in a Long-Lived Model of Chronic Demyelination. J. Neurosci. 2013, 33, 2718–2727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charcot, J.M. Histologie de La Sclerose En Plaques. Gaz. Hop. 1868, 41, 554–566. [Google Scholar]
- Zalc, B. One Hundred and Fifty Years Ago Charcot Reported Multiple Sclerosis as a New Neurological Disease. Brain 2018, 141, 3482–3488. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of Multiple Sclerosis: 2017 Revisions of the McDonald Criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An Updated Histological Classification System for Multiple Sclerosis Lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of Multiple Sclerosis Lesions: Implications for the Pathogenesis of Demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Bramow, S.; Frischer, J.M.; Lassmann, H.; Koch-Henriksen, N.; Lucchinetti, C.F.; Sørensen, P.S.; Laursen, H. Demyelination versus Remyelination in Progressive Multiple Sclerosis. Brain 2010, 133, 2983–2998. [Google Scholar] [CrossRef] [Green Version]
- Coman, I.; Aigrot, M.S.; Seilhean, D.; Reynolds, R.; Girault, J.A.; Zalc, B.; Lubetzki, C. Nodal, Paranodal and Juxtaparanodal Axonal Proteins during Demyelination and Remyelination in Multiple Sclerosis. Brain 2006, 129, 3186–3195. [Google Scholar] [CrossRef] [PubMed]
- Craner, M.J.; Newcombe, J.; Black, J.A.; Hartle, C.; Cuzner, M.L.; Waxman, S.G. Molecular Changes in Neurons in Multiple Sclerosis: Altered Axonal Expression of Nav1.2 and Nav1.6 Sodium Channels and Na+/Ca2+ Exchanger. Proc. Natl. Acad. Sci. USA 2004, 101, 8168–8173. [Google Scholar] [CrossRef] [Green Version]
- Howell, O.W.; Palser, A.; Polito, A.; Melrose, S.; Zonta, B.; Scheiermann, C.; Vora, A.J.; Brophy, P.J.; Reynolds, R. Disruption of Neurofascin Localization Reveals Early Changes Preceding Demyelination and Remyelination in Multiple Sclerosis. Brain 2006, 129, 3173–3185. [Google Scholar] [CrossRef]
- Ronzano, R.; Roux, T.; Thetiot, M.; Aigrot, M.S.; Richard, L.; Lejeune, F.X.; Mazuir, E.; Vallat, J.M.; Lubetzki, C.; Desmazières, A. Microglia-Neuron Communication at Nodes of Ranvier Depends on Neuronal Activity through Potassium Release and Contributes to Myelin Repair. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nikić, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Brück, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A Reversible Form of Axon Damage in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef]
- Lubetzki, C.; Zalc, B. Remyelination: A Brief History. Lancet Neurol. 2020, 19, 649. [Google Scholar] [CrossRef]
- Goldschmidt, T.; Antel, J.; König, F.B.; Brück, W.; Kuhlmann, T. Remyelination Capacity of the MS Brain Decreases with Disease Chronicity. Neurology 2009, 72, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Patani, R.; Balaratnam, M.; Vora, A.; Reynolds, R. Remyelination Can Be Extensive in Multiple Sclerosis despite a Long Disease Course. Neuropathol. Appl. Neurobiol. 2007, 33, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Barnard, R.O.; Revesz, T.; Kwon, E.E.; Sharer, L.; Cho, E.S. Multiple Sclerosis. Pathology of Recurrent Lesions. Brain 1993, 116 Pt 3, 681–693. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Frisén, J.; Lyons, D.A. Revisiting Remyelination: Towards a Consensus on the Regeneration of CNS Myelin. Semin. Cell Dev. Biol. 2020, S1084952120301579. [Google Scholar] [CrossRef]
- Levine, J.M.; Reynolds, R. Activation and Proliferation of Endogenous Oligodendrocyte Precursor Cells during Ethidium Bromide-Induced Demyelination. Exp. Neurol. 1999, 160, 333–347. [Google Scholar] [CrossRef]
- Fancy, S.P.J.; Zhao, C.; Franklin, R.J.M. Increased Expression of Nkx2.2 and Olig2 Identifies Reactive Oligodendrocyte Progenitor Cells Responding to Demyelination in the Adult CNS. Mol. Cell. Neurosci. 2004, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Kuhlmann, T.; Antel, J.P. Cells of the Oligodendroglial Lineage, Myelination, and Remyelination. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2011, 1812, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Snaidero, N.; Schifferer, M.; Mezydlo, A.; Zalc, B.; Kerschensteiner, M.; Misgeld, T. Myelin Replacement Triggered by Single-Cell Demyelination in Mouse Cortex. Nat. Commun. 2020, 11, 4901. [Google Scholar] [CrossRef]
- Neely, S.A.; Williamson, J.M.; Klingseisen, A.; Zoupi, L.; Early, J.J.; Williams, A.; Lyons, D.A. New Oligodendrocytes Exhibit More Abundant and Accurate Myelin Regeneration than Those That Survive Demyelination. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nait-Oumesmar, B.; Picard-Riera, N.; Kerninon, C.; Decker, L.; Seilhean, D.; Hoglinger, G.U.; Hirsch, E.C.; Reynolds, R.; Baron-Van Evercooren, A. Activation of the Subventricular Zone in Multiple Sclerosis: Evidence for Early Glial Progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 4694–4699. [Google Scholar] [CrossRef] [Green Version]
- Remaud, S.; Ortiz, F.C.; Perret-Jeanneret, M.; Aigrot, M.-S.; Gothié, J.-D.; Fekete, C.; Kvárta-Papp, Z.; Gereben, B.; Langui, D.; Lubetzki, C.; et al. Transient Hypothyroidism Favors Oligodendrocyte Generation Providing Functional Remyelination in the Adult Mouse Brain. eLife 2017, 6, e29996. [Google Scholar] [CrossRef]
- Xing, Y.L.; Röth, P.T.; Stratton, J.A.S.; Chuang, B.H.A.; Danne, J.; Ellis, S.L.; Ng, S.W.; Kilpatrick, T.J.; Merson, T.D. Adult Neural Precursor Cells from the Subventricular Zone Contribute Significantly to Oligodendrocyte Regeneration and Remyelination. J. Neurosci. 2014, 34, 14128–14146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubetzki, C.; Gansmüller, A.; Lachapelle, F.; Lombrail, P.; Gumpel, M. Myelination by Oligodendrocytes Isolated from 4-6-Week-Old Rat Central Nervous System and Transplanted into Newborn Shiverer Brain. J. Neurol. Sci. 1988, 88, 161–175. [Google Scholar] [CrossRef]
- Yeung, M.S.Y.; Zdunek, S.; Bergmann, O.; Bernard, S.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Brundin, L.; et al. Dynamics of Oligodendrocyte Generation and Myelination in the Human Brain. Cell 2014, 159, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Yeung, M.S.Y.; Djelloul, M.; Steiner, E.; Bernard, S.; Salehpour, M.; Possnert, G.; Brundin, L.; Frisén, J. Dynamics of Oligodendrocyte Generation in Multiple Sclerosis. Nature 2019, 566, 538–542. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. A Quantitative Analysis of Oligodendrocytes in Multiple Sclerosis Lesions. A Study of 113 Cases. Brain 1999, 122 Pt 12, 2279–2295. [Google Scholar] [CrossRef]
- Brown, R.A.; Narayanan, S.; Arnold, D.L. Imaging of Repeated Episodes of Demyelination and Remyelination in Multiple Sclerosis. Neuroimage Clin. 2014, 6, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.J.M. Why Does Remyelination Fail in Multiple Sclerosis? Nat. Rev. Neurosci. 2002, 3, 705–714. [Google Scholar] [CrossRef]
- Ruckh, J.M.; Zhao, J.-W.; Shadrach, J.L.; van Wijngaarden, P.; Rao, T.N.; Wagers, A.J.; Franklin, R.J.M. Rejuvenation of Regeneration in the Aging Central Nervous System. Cell Stem Cell 2012, 10, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J.M. The Age-Related Decrease in CNS Remyelination Efficiency Is Attributable to an Impairment of Both Oligodendrocyte Progenitor Recruitment and Differentiation. J. Neurosci. 2002, 22, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.; Sati, P.; Schindler, M.; Leibovitch, E.C.; Ohayon, J.; Wu, T.; Meani, A.; Filippi, M.; Jacobson, S.; Cortese, I.C.M.; et al. Persistent 7-Tesla Phase Rim Predicts Poor Outcome in New Multiple Sclerosis Patient Lesions. J. Clin. Investig. 2016, 126, 2597–2609. [Google Scholar] [CrossRef]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Höftberger, R.; Berger, T.; Auff, E.; Leutmezer, F.; Trattnig, S.; Lassmann, H.; et al. Slow Expansion of Multiple Sclerosis Iron Rim Lesions: Pathology and 7 T Magnetic Resonance Imaging. Acta Neuropathol. 2017, 133, 25–42. [Google Scholar] [CrossRef] [Green Version]
- Luchetti, S.; Fransen, N.L.; van Eden, C.G.; Ramaglia, V.; Mason, M.; Huitinga, I. Progressive Multiple Sclerosis Patients Show Substantial Lesion Activity That Correlates with Clinical Disease Severity and Sex: A Retrospective Autopsy Cohort Analysis. Acta Neuropathol. 2018, 135, 511–528. [Google Scholar] [CrossRef] [Green Version]
- Kornek, B.; Lassmann, H. Neuropathology of Multiple Sclerosis—New Concepts. Brain Res. Bull. 2003, 61, 321–326. [Google Scholar] [CrossRef]
- Mei, F.; Lehmann-Horn, K.; Shen, Y.-A.A.; Rankin, K.A.; Stebbins, K.J.; Lorrain, D.S.; Pekarek, K.; Sagan, S.A.; Xiao, L.; Teuscher, C.; et al. Accelerated Remyelination during Inflammatory Demyelination Prevents Axonal Loss and Improves Functional Recovery. eLife 2019, 5, e18246. [Google Scholar] [CrossRef]
- Piaton, G.; Aigrot, M.-S.; Williams, A.; Moyon, S.; Tepavcevic, V.; Moutkine, I.; Gras, J.; Matho, K.S.; Schmitt, A.; Soellner, H.; et al. Class 3 Semaphorins Influence Oligodendrocyte Precursor Recruitment and Remyelination in Adult Central Nervous System. Brain 2011, 134, 1156–1167. [Google Scholar] [CrossRef] [Green Version]
- Tepavčević, V.; Kerninon, C.; Aigrot, M.S.; Meppiel, E.; Mozafari, S.; Arnould-Laurent, R.; Ravassard, P.; Kennedy, T.E.; Nait-Oumesmar, B.; Lubetzki, C. Early Netrin-1 Expression Impairs Central Nervous System Remyelination: Netrin-1 and CNS Remyelination. Ann. Neurol. 2014, 76, 252–268. [Google Scholar] [CrossRef]
- Ishibashi, T.; Dakin, K.A.; Stevens, B.; Lee, P.R.; Kozlov, S.V.; Stewart, C.L.; Fields, R.D. Astrocytes Promote Myelination in Response to Electrical Impulses. Neuron 2006, 49, 823–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankoff, B.; Aigrot, M.-S.; Noël, F.; Wattilliaux, A.; Zalc, B.; Lubetzki, C. Ciliary Neurotrophic Factor (CNTF) Enhances Myelin Formation: A Novel Role for CNTF and CNTF-Related Molecules. J. Neurosci. 2002, 22, 9221–9227. [Google Scholar] [CrossRef] [PubMed]
- Craner, M.J. Abnormal Sodium Channel Distribution in Optic Nerve Axons in a Model of Inflammatory Demyelination. Brain 2003, 126, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Orthmann-Murphy, J.; Call, C.L.; Molina-Castro, G.C.; Hsieh, Y.C.; Rasband, M.N.; Calabresi, P.A.; Bergles, D.E. Remyelination Alters the Pattern of Myelin in the Cerebral Cortex. eLife 2020, 9, e56621. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.A.; Desmazières, A.; Fricker, D.; Lubetzki, C.; Sol-Foulon, N. Mechanisms of Sodium Channel Clustering and Its Influence on Axonal Impulse Conduction. Cell. Mol. Life Sci. 2016, 73, 723–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, M.R.; Cho, M.-H.; Ullian, E.M.; Isom, L.L.; Levinson, S.R.; Barres, B.A. Differential Control of Clustering of the Sodium Channels Nav1.2 and Nav1.6 at Developing CNS Nodes of Ranvier. Neuron 2001, 30, 105–119. [Google Scholar] [CrossRef] [Green Version]
- John, G.R.; Shankar, S.L.; Shafit-Zagardo, B.; Massimi, A.; Lee, S.C.; Raine, C.S.; Brosnan, C.F. Multiple Sclerosis: Re-Expression of a Developmental Pathway That Restricts Oligodendrocyte Maturation. Nat. Med. 2002, 8, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the Brain Extracellular Matrix: A New Target for Remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Kuboyama, K.; Tanga, N.; Suzuki, R.; Fujikawa, A.; Noda, M. Protamine Neutralizes Chondroitin Sulfate Proteoglycan-Mediated Inhibition of Oligodendrocyte Differentiation. PLoS ONE 2017, 12, e0189164. [Google Scholar] [CrossRef]
- Luo, F.; Tran, A.P.; Xin, L.; Sanapala, C.; Lang, B.T.; Silver, J.; Yang, Y. Modulation of Proteoglycan Receptor PTPσ Enhances MMP-2 Activity to Promote Recovery from Multiple Sclerosis. Nat. Commun. 2018, 9, 4126. [Google Scholar] [CrossRef]
- Pekny, M.; Wilhelmsson, U.; Pekna, M. The Dual Role of Astrocyte Activation and Reactive Gliosis. Neurosci. Lett. 2014, 565, 30–38. [Google Scholar] [CrossRef]
- Franklin, R.J.; Crang, A.J.; Blakemore, W.F. Transplanted Type-1 Astrocytes Facilitate Repair of Demyelinating Lesions by Host Oligodendrocytes in Adult Rat Spinal Cord. J. Neurocytol. 1991, 20, 420–430. [Google Scholar] [CrossRef]
- Neumann, H.; Kotter, M.R.; Franklin, R.J.M. Debris Clearance by Microglia: An Essential Link between Degeneration and Regeneration. Brain 2008, 132, 288–295. [Google Scholar] [CrossRef]
- Kotter, M.R.; Zhao, C.; van Rooijen, N.; Franklin, R.J.M. Macrophage-Depletion Induced Impairment of Experimental CNS Remyelination Is Associated with a Reduced Oligodendrocyte Progenitor Cell Response and Altered Growth Factor Expression. Neurobiol. Dis. 2005, 18, 166–175. [Google Scholar] [CrossRef]
- Bauer, J.; Sminia, T.; Wouterlood, F.G.; Dijkstra, C.D. Phagocytic Activity of Macrophages and Microglial Cells during the Course of Acute and Chronic Relapsing Experimental Autoimmune Encephalomyelitis. J. Neurosci. Res. 1994, 38, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.F.; Davies, C.L.; Miron, V.E. Microglia: Origins, Homeostasis, and Roles in Myelin Repair. Curr. Opin. Neurobiol. 2017, 47, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, A.F.; Miron, V.E. The Pro-Remyelination Properties of Microglia in the Central Nervous System. Nat. Rev. Neurol. 2019, 15, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.-W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 Microglia and Macrophages Drive Oligodendrocyte Differentiation during CNS Remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miron, V.E. Microglia-Driven Regulation of Oligodendrocyte Lineage Cells, Myelination, and Remyelination. J. Leukoc. Biol. 2017, 101, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Dziembowska, M.; Tham, T.N.; Lau, P.; Vitry, S.; Lazarini, F.; Dubois-Dalcq, M. A Role for CXCR4 Signaling in Survival and Migration of Neural and Oligodendrocyte Precursors. Glia 2005, 50, 258–269. [Google Scholar] [CrossRef]
- Connor, J.R.; Menzies, S.L. Relationship of Iron to Oligodendrocytes and Myelination. Glia 1996, 17, 83–93. [Google Scholar] [CrossRef]
- Shields, S.A.; Gilson, J.M.; Blakemore, W.F.; Franklin, R.J. Remyelination Occurs as Extensively but More Slowly in Old Rats Compared to Young Rats Following Gliotoxin-Induced CNS Demyelination. Glia 1999, 28, 77–83. [Google Scholar] [CrossRef]
- Wolswijk, G. Chronic Stage Multiple Sclerosis Lesions Contain a Relatively Quiescent Population of Oligodendrocyte Precursor Cells. J. Neurosci. 1998, 18, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannoni, G.; Bermel, R.; Phillips, T.; Rudick, R. A Brief History of NEDA. Mult. Scler. Relat. Disord. 2018, 20, 228–230. [Google Scholar] [CrossRef]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of Neurodegeneration and Axonal Dysfunction in Multiple Sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Dallenga, T.; Winkler, A.; Roemer, S.; Maruschak, B.; Siebert, H.; Brück, W.; Stadelmann, C. Relationship of Acute Axonal Damage, Wallerian Degeneration, and Clinical Disability in Multiple Sclerosis. J. Neuroinflamm. 2017, 14, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooijmans, C.R.; Hlavica, M.; Schuler, F.A.F.; Good, N.; Good, A.; Baumgartner, L.; Galeno, G.; Schneider, M.P.; Jung, T.; de Vries, R.; et al. Remyelination Promoting Therapies in Multiple Sclerosis Animal Models: A Systematic Review and Meta-Analysis. Sci. Rep. 2019, 9, 822. [Google Scholar] [CrossRef] [Green Version]
- Bodini, B.; Stankoff, B. Imaging Central Nervous System Demyelination and Remyelination by Positron-Emission Tomography. Brain Plast. 2016, 2, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Mei, F.; Fancy, S.P.J.; Shen, Y.-A.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar Arrays as a High-Throughput Screening Platform for Therapeutics in Multiple Sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; He, Y.; Fan, S.; Sun, B. Clemastine Rescues Behavioral Changes and Enhances Remyelination in the Cuprizone Mouse Model of Demyelination. Neurosci. Bull. 2015, 31, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine Fumarate as a Remyelinating Therapy for Multiple Sclerosis (ReBUILD): A Randomised, Controlled, Double-Blind, Crossover Trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef] [Green Version]
- Schwartzbach, C.J.; Grove, R.A.; Brown, R.; Tompson, D.; Then Bergh, F.; Arnold, D.L. Lesion Remyelinating Activity of GSK239512 versus Placebo in Patients with Relapsing-Remitting Multiple Sclerosis: A Randomised, Single-Blind, Phase II Study. J. Neurol. 2017, 264, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A Regenerative Approach to the Treatment of Multiple Sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Bongarzone, E.R.; Howard, S.G.; Schonmann, V.; Campagnoni, A.T. Identification of the Dopamine D3 Receptor in Oligodendrocyte Precursors: Potential Role in Regulating Differentiation and Myelin Formation. J. Neurosci. 1998, 18, 5344–5353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xu, H.; Jiang, W.; Xiao, L.; Yan, B.; He, J.; Wang, Y.; Bi, X.; Li, X.; Kong, J. Quetiapine Alleviates the Cuprizone-Induced White Matter Pathology in the Brain of C57BL/6 Mouse. Schizophr. Res. 2008, 106, 182–191. [Google Scholar] [CrossRef]
- Gonzalez, G.A.; Hofer, M.P.; Syed, Y.A.; Amaral, A.I.; Rundle, J.; Rahman, S.; Zhao, C.; Kotter, M.R.N. Tamoxifen Accelerates the Repair of Demyelinated Lesions in the Central Nervous System. Sci. Rep. 2016, 6, 31599. [Google Scholar] [CrossRef]
- Chan, J.R.; Phillips, L.J.; Glaser, M. Glucocorticoids and Progestins Signal the Initiation and Enhance the Rate of Myelin Formation. Proc. Natl. Acad. Sci. USA 1998, 95, 10459–10464. [Google Scholar] [CrossRef] [Green Version]
- Najm, F.J.; Madhavan, M.; Zaremba, A.; Shick, E.; Karl, R.T.; Factor, D.C.; Miller, T.E.; Nevin, Z.S.; Kantor, C.; Sargent, A.; et al. Drug-Based Modulation of Endogenous Stem Cells Promotes Functional Remyelination in Vivo. Nature 2015, 522, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Neumann, B.; Baror, R.; Zhao, C.; Segel, M.; Dietmann, S.; Rawji, K.S.; Foerster, S.; McClain, C.R.; Chalut, K.; van Wijngaarden, P.; et al. Metformin Restores CNS Remyelination Capacity by Rejuvenating Aged Stem Cells. Cell Stem Cell 2019, 25, 473–485.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göttle, P.; Manousi, A.; Kremer, D.; Reiche, L.; Hartung, H.-P.; Küry, P. Teriflunomide Promotes Oligodendroglial Differentiation and Myelination. J. Neuroinflamm. 2018, 15, 76. [Google Scholar] [CrossRef] [Green Version]
- Mannioui, A.; Vauzanges, Q.; Fini, J.B.; Henriet, E.; Sekizar, S.; Azoyan, L.; Thomas, J.L.; Pasquier, D.D.; Giovannangeli, C.; Demeneix, B.; et al. The Xenopus Tadpole: An in Vivo Model to Screen Drugs Favoring Remyelination. Mult. Scler. 2018, 24, 1421–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miron, V.E.; Rajasekharan, S.; Jarjour, A.A.; Zamvil, S.S.; Kennedy, T.E.; Antel, J.P. Simvastatin Regulates Oligodendroglial Process Dynamics and Survival. Glia 2007, 55, 130–143. [Google Scholar] [CrossRef]
- Chataway, J.; Schuerer, N.; Alsanousi, A.; Chan, D.; MacManus, D.; Hunter, K.; Anderson, V.; Bangham, C.R.M.; Clegg, S.; Nielsen, C.; et al. Effect of High-Dose Simvastatin on Brain Atrophy and Disability in Secondary Progressive Multiple Sclerosis (MS-STAT): A Randomised, Placebo-Controlled, Phase 2 Trial. Lancet 2014, 383, 2213–2221. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, P.S.; Lycke, J.; Erälinna, J.-P.; Edland, A.; Wu, X.; Frederiksen, J.L.; Oturai, A.; Malmeström, C.; Stenager, E.; Sellebjerg, F.; et al. Simvastatin as Add-on Therapy to Interferon Beta-1a for Relapsing-Remitting Multiple Sclerosis (SIMCOMBIN Study): A Placebo-Controlled Randomised Phase 4 Trial. Lancet Neurol. 2011, 10, 691–701. [Google Scholar] [CrossRef]
- Shen, S.; Sandoval, J.; Swiss, V.A.; Li, J.; Dupree, J.; Franklin, R.J.M.; Casaccia-Bonnefil, P. Age-Dependent Epigenetic Control of Differentiation Inhibitors Is Critical for Remyelination Efficiency. Nat. Neurosci. 2008, 11, 1024–1034. [Google Scholar] [CrossRef] [Green Version]
- Moreau-Fauvarque, C.; Kumanogoh, A.; Camand, E.; Jaillard, C.; Barbin, G.; Boquet, I.; Love, C.; Jones, E.Y.; Kikutani, H.; Lubetzki, C.; et al. The Transmembrane Semaphorin Sema4D/CD100, an Inhibitor of Axonal Growth, Is Expressed on Oligodendrocytes and Upregulated after CNS Lesion. J. Neurosci. 2003, 23, 9229–9239. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.S.; Jonason, A.; Reilly, C.; Veeraraghavan, J.; Fisher, T.; Doherty, M.; Klimatcheva, E.; Mallow, C.; Cornelius, C.; Leonard, J.E.; et al. SEMA4D Compromises Blood–Brain Barrier, Activates Microglia, and Inhibits Remyelination in Neurodegenerative Disease. Neurobiol. Dis. 2015, 73, 254–268. [Google Scholar] [CrossRef] [Green Version]
- LaGanke, C.; Samkoff, L.; Edwards, K.; Jung Henson, L.; Repovic, P.; Lynch, S.; Stone, L.; Mattson, D.; Galluzzi, A.; Fisher, T.L.; et al. Safety/Tolerability of the Anti-Semaphorin 4D Antibody VX15/2503 in a Randomized Phase 1 Trial. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derfuss, T.; Curtin, F.; Guebelin, C.; Bridel, C.; Rasenack, M.; Matthey, A.; Du Pasquier, R.; Schluep, M.; Desmeules, J.; Lang, A.B.; et al. A Phase IIa Randomised Clinical Study of GNbAC1, a Humanised Monoclonal Antibody against the Envelope Protein of Multiple Sclerosis-Associated Endogenous Retrovirus in Multiple Sclerosis Patients. Mult. Scler. 2015, 21, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Mi, S.; Hu, B.; Hahm, K.; Luo, Y.; Kam Hui, E.S.; Yuan, Q.; Wong, W.M.; Wang, L.; Su, H.; Chu, T.-H.; et al. LINGO-1 Antagonist Promotes Spinal Cord Remyelination and Axonal Integrity in MOG-Induced Experimental Autoimmune Encephalomyelitis. Nat. Med. 2007, 13, 1228–1233. [Google Scholar] [CrossRef]
- Kremer, D.; Schichel, T.; Förster, M.; Tzekova, N.; Bernard, C.; van der Valk, P.; van Horssen, J.; Hartung, H.-P.; Perron, H.; Küry, P. Human Endogenous Retrovirus Type W Envelope Protein Inhibits Oligodendroglial Precursor Cell Differentiation: Retroviruses and Myelin Repair. Ann. Neurol. 2013, 74, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Cadavid, D.; Balcer, L.; Galetta, S.; Aktas, O.; Ziemssen, T.; Vanopdenbosch, L.; Frederiksen, J.; Skeen, M.; Jaffe, G.J.; Butzkueven, H.; et al. Safety and Efficacy of Opicinumab in Acute Optic Neuritis (RENEW): A Randomised, Placebo-Controlled, Phase 2 Trial. Lancet Neurol. 2017, 16, 189–199. [Google Scholar] [CrossRef]
- Cadavid, D.; Mellion, M.; Hupperts, R.; Edwards, K.R.; Calabresi, P.A.; Drulović, J.; Giovannoni, G.; Hartung, H.-P.; Arnold, D.L.; Fisher, E.; et al. Safety and Efficacy of Opicinumab in Patients with Relapsing Multiple Sclerosis (SYNERGY): A Randomised, Placebo-Controlled, Phase 2 Trial. Lancet Neurol. 2019, 18, 845–856. [Google Scholar] [CrossRef]
- Martin, E.; Aigrot, M.-S.; Grenningloh, R.; Stankoff, B.; Lubetzki, C.; Boschert, U.; Zalc, B. Bruton’s Tyrosine Kinase Inhibition Promotes Myelin Repair. Brain Plast. 2020, 5, 123–133. [Google Scholar] [CrossRef]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S. Placebo-Controlled Trial of an Oral BTK Inhibitor in Multiple Sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef]
- Chandraratna, R.A.; Noelle, R.J.; Nowak, E.C. Treatment with Retinoid X Receptor Agonist IRX4204 Ameliorates Experimental Autoimmune Encephalomyelitis. Am. J. Transl. Res. 2016, 8, 1016–1026. [Google Scholar] [PubMed]
- Ljunggren-Rose, Å.; Natarajan, C.; Matta, P.; Pandey, A.; Upender, I.; Sriram, S. Anacardic Acid Induces IL-33 and Promotes Remyelination in CNS. Proc. Natl. Acad. Sci. USA 2020, 117, 21527–21535. [Google Scholar] [CrossRef]
- Melero-Jerez, C.; Fernández-Gómez, B.; Lebrón-Galán, R.; Ortega, M.C.; Sánchez-de Lara, I.; Ojalvo, A.C.; Clemente, D.; Castro, F. Myeloid-derived Suppressor Cells Support Remyelination in a Murine Model of Multiple Sclerosis by Promoting Oligodendrocyte Precursor Cell Survival, Proliferation, and Differentiation. Glia 2020, 69, 905–924. [Google Scholar] [CrossRef]
- Matías-Guiu, J.; Matías-Guiu, J.A.; Montero-Escribano, P.; Barcia, J.A.; Canales-Aguirre, A.A.; Mateos-Diaz, J.C.; Gómez-Pinedo, U. Particles Containing Cells as a Strategy to Promote Remyelination in Patients With Multiple Sclerosis. Front. Neurol. 2020, 11, 638. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug Name | Mode of Action | Preclinical Studies | Clinical Studies | ||||
|---|---|---|---|---|---|---|---|
| Model | Effect | Study | Population | Outcome | Results | ||
| ACTH | Corticotropin hormone | Cell culture | OPC differentiation/OL maturation | Phase IV (NCT02446886) | RMS | MWF over 12 months | Completed; results pending |
| Bexarotene/IRX4204 | Retinoid X receptor γ agonist | EAE | OPC differentiation/reduced EAE severity | Phase IIa ISRCTN14265371 | RMS | MTR; VEP latency | Improvement in MTR in a lesion subset; reduction in VEP latency ; serious side effects |
| Clemastine fumarate | H1 and M1/M3 receptors antagonist | Micropillar array screen; LPC; cuprizone | OPC differentiation/OL maturation/reduced EAE severity | Phase II ReBUILD trial Phase II Recover Trial | Chronic optic neuritis Acute optic neuritis | P100 VEP latency P100 VEP latency; RNFL thickness; EDSS; MWF | Reduced P100 VEP latency in chronic optic neuropathy without clinical improvement Ongoing |
| Domperidone | D2/D3 dopamine receptor antagonist | LPC | OL maturation | Phase II NCT02308137 Phase II NCT02493049 | SPMS RRMS | T25WF MRI lesions; EDSS | Completed; results pending Completed; results pending |
| GSK239512 | H3 receptor antagonist | Cuprizone | OPC differentiation/OL maturation | Phase II NCT01772199 | RRMS | MTR activity | Small improvement in lesion remyelination |
| Opicinumab | mAb against LINGO 1 | EAE; LPC | OPC differentiation/OL maturation | Phase IIa RENEW trial Phase IIb SYNERGY trial Phase II AFFINITY trial | Acute optic neuritis RMS RMS | P100 VEP latency; RNFL thickness; MRI lesions EDSS progression ODRS; MTR | Minor reduction in P100 VEP latency No improvement vs. placebo No improvement vs. placebo |
| Quetiapine | Antipsychotic drug | Cuprizone | OL maturation; inhibition of activated microglia | Phase I/II NCT020087631 | RMS | Tolerance; EDSS | Completed; results pending |
| Temelimab/GNbAC1 | mAb against HERV envelope protein | In vitro OPC cultures | OPC differentiation | Phase IIa CHANGE-MS trial Phase IIb ANGEL-MS trial | RMS RRMS | MRI lesions; morphometry | Significant reduction on cortical and thalamic atrophy; no decrease in new lesions |
| SAR442168 Evobrutinib/M2951 | BTK inhibitor | LPC demyelinized cultures | 1.7× improved remyelination comparing to placebo | Phase III HERCULES trial Phase III PERSEUS trial 2 PHASE III GEMINI1 and GEMINI2 Phase II NCT02975349 | SPMS PPMS RMS RMS | CDP;AAR; NfL MRI, ARR, EDSS | Ongoing Reduction in enhancing lesions and ARR, no effect on EDSS, transaminase elevation |
| Simvastatin | HMG-CoA reductase inhibitor | EAE | OPC differentiation/prevent EAE | Phase II MS-STAT trial Phase III MS-STAT2 trial Phase IV SIMCOMBIN trial | SPMS SPMS RRMS | AAR EDSS Multiparameter clinical outcomes ARR; MRI | 43% reduction of brain atrophy Ongoing No additional benefit to IFN treatment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pantazou, V.; Roux, T.; Oliveira Moreira, V.; Lubetzki, C.; Desmazières, A. Interaction between Neurons and the Oligodendroglial Lineage in Multiple Sclerosis and Its Preclinical Models. Life 2021, 11, 231. https://0-doi-org.brum.beds.ac.uk/10.3390/life11030231
Pantazou V, Roux T, Oliveira Moreira V, Lubetzki C, Desmazières A. Interaction between Neurons and the Oligodendroglial Lineage in Multiple Sclerosis and Its Preclinical Models. Life. 2021; 11(3):231. https://0-doi-org.brum.beds.ac.uk/10.3390/life11030231
Chicago/Turabian StylePantazou, Vasiliki, Thomas Roux, Vanessa Oliveira Moreira, Catherine Lubetzki, and Anne Desmazières. 2021. "Interaction between Neurons and the Oligodendroglial Lineage in Multiple Sclerosis and Its Preclinical Models" Life 11, no. 3: 231. https://0-doi-org.brum.beds.ac.uk/10.3390/life11030231