
Inflammation in Metabolic and Cardiovascular Disorders—Role of Oxidative Stress


1
Cardioimmunology Group, Medical Clinic II, University Heart Center Lübeck, 23538 Lübeck, Germany
2
University Heart Center Lübeck, Medical Clinic II, University Hospital, 23538 Lübeck, Germany
3
DZHK (German Research Centre for Cardiovascular Research), Partner Site Hamburg/Lübeck/Kiel, 23562 Lübeck, Germany
*
Author to whom correspondence should be addressed.
Life 2021, 11(7), 672; https://0-doi-org.brum.beds.ac.uk/10.3390/life11070672
Submission received: 9 June 2021
/
Revised: 30 June 2021
/
Accepted: 30 June 2021
/
Published: 9 July 2021
(This article belongs to the Special Issue New Molecules and Mechanisms to Fight the Onset and Progression Cardiovascular Diseases—the Role of Oxidative Stress)
{kind=link}
{kind=link}
Abstract
:Cardiovascular diseases (CVD) constitute the main cause of death worldwide. Both inflammation and oxidative stress have been reported to be involved in the progress of CVD. It is well known that generation of oxidative stress during the course of CVD is involved in tissue damage and inflammation, causing deleterious effects such as hypertension, dysfunctional metabolism, endothelial dysfunction, stroke, and myocardial infarction. Remarkably, natural antioxidant strategies have been increasingly discovered and are subject to current scientific investigations. Here, we addressed the activation of immune cells in the context of ROS production, as well as how their interaction with other cellular players and further (immune) mediators contribute to metabolic and cardiovascular disorders. We also highlight how a dysregulated complement system contributes to immune imbalance and tissue damage in the context of increases oxidative stress. Additionally, modulation of hypothalamic oxidative stress is discussed, which may offer novel treatment strategies for type-2 diabetes and obesity. Together, we provide new perspectives on therapy strategies for CVD caused by oxidative stress, with a focus on oxidative stress.
1. Introduction
Several cardiovascular diseases (CVD), including atherosclerosis, myocardial infarction (MI), and heart failure, are related to low-grade inflammation. A balanced interaction between ongoing immune responses and metabolic regulation were demonstrated to be decisive homeostatic mechanism, as dysregulation of this balance may lead to various chronic metabolic disorders, particularly obesity, type 2 diabetes, and subsequent CVD [1]. Different players of inflammation such as several types of immune cells and mediators of the innate immune system are major contributors to the pathogenesis of these conditions. Furthermore, oxidative stress has been reported as a driver of metabolic and CVD: An excessive generation of reactive oxygen species (ROS) can tilt the equilibrium between protective and harmful effects, resulting in oxidative stress and subsequent release of proinflammatory cytokines, endothelial dysfunction, and reduction of nitrogen monoxide (NO) utilization. Thus, the present review describes and summarizes the miscellaneous principles of inflammation contributing to metabolic disorders and CVD, which are associated with oxidative stress.
2. Innate Immune Activation Contributes to Metabolic Disorders
2.1. Macrophages, Dendritic Cells, and Further Immune Cells Contributing to Inflammation in Metabolic Disorders
To understand the intersection between metabolism and inflammation, one should consider the functional links of cells featuring metabolic or immune properties, such as macrophages, dendritic cells (DCs), and adipocytes [1]. Macrophages and DCs provide an early defense barrier against external pathogens [2]. In addition, both cell types respond to “danger signals” and cytokines, such as metabolic reprogramming in response to hypoxia or nutrient alterations [3]. Two different inflammatory phenotypes are known for macrophages: the M1-like macrophages are activated classically, typically by interferon (IFN)-γ or lipopolysaccharides (LPS), while M2-like macrophages are activated alternatively by exposure to several cytokines such as interleukin (IL)-4, IL-10, or IL-13. M1 macrophages secrete proinflammatory factors such as tumor necrosis factor-α (TNF-α), IL1β, and inducible nitric oxide synthase (iNOS)/NO [2,3,4,5,6]. By contrast, M2 macrophages have been linked to diminished levels of NO and TNF-α, as well as the generation of anti-inflammatory cytokines such as IL-10 [2]. During obesity, M1-like macrophage numbers elevate due to augmented levels of free fatty acids (FFAs), cholesterol, LPS, and hypoxia [7], contributing to adipose tissue (AT) inflammation and inhibition of insulin signaling. In contrast, anti-inflammatory M2-like macrophages dominate in lean humans and mice [8].
DCs are antigen-presenting cells of the mammalian immune system. They present internalized antigen material to T cells of the immune system. Thereby, they constitute an intersection point between the innate and the adaptive immune system [9]. DCs can be divided into plasmacytoid DCs (pDCs) and conventional DCs (cDCs), distinguished by morphology and function. Resting DCs have been demonstrated to display a catabolic metabolism, steadily decomposing nutrients for energy generation [10]. Thereby, tricarboxylic acid (TCA) cycle fueled via fatty acid β-oxidation (FAO) and glutaminolysis, drive activation of oxidative phosphorylation (OXPHOS), which is regulated by AMP-activated protein kinase (AMPK) [2,3,11,12,13]. Besides glucose, intracellular glycogen is used by resting DCs to support basal glycolytic demands, driving mitochondrial respiration [14]. Mature DCs perceive and respond to environmental stimuli. In secondary lymphoid tissues, DCs present antigens to T cells, promoting T cell proliferation as well as differentiation. Mice deficient of Flt3l (Fms-related tyrosine kinase 3 ligand) show a lack of DCs as well as reduced amounts of natural killer cells and regulatory T cells and B cells, while insulin sensitivity in diet-induced obesity is improved. This finding points out the pivotal role of DCs in the regulation of systemic metabolism [15]. Accordingly, CD11+DCs were identified in AT during insulin resistance inducing differentiation of Th17 in metabolic processes [16,17]. Interestingly, DCs have recently been shown to promote hypertension by mediating fluid retention and renal oxidative stress [18].
Eosinophils are cells of the innate immune system [19]. It is known that ROS can induce the death of eosinophils. Siglec-8 connection has been shown to enhance IL-5 in a ROS-dependent manner, inducing ERK phosphorylation, leading to eosinophil death [20]. Importantly, a synergistic effect of type 2 innate lymphoid cells (ILC2), eosinophils, and M2 macrophages controls metabolic homeostasis in visceral adipose tissue (VAT). Increased interaction of ILC2 with IL-33 in VAT leads to accumulation of eosinophils and goes together with a reduction of eosinophils in the bone marrow and spleen [21]. However, eosinophils are also related to the development of CVD [22]. Overexpression of eosinophil-specific chemokines, such as eotaxin, in smooth muscle cells of vessels have been demonstrated to stimulate TNF-α, interferon-α, and interferon-β production, promoting atherosclerosis, by regulating macrophages to stimulate the formation of foam cells [23,24,25].
2.2. Different Types of Adipose Tissue
AT is classically regarded as a tissue that stores surplus nutrients. During the last two decades of research, it has been found that AT is also a key regulator of body metabolism and homeostasis, as it secretes a variety of adipokines [26] (Figure 1). Subcutaneous and visceral adipose tissues display hyperplasia and hypertrophy following nutritional overload, resulting in adipokine dysregulation, hypoxia, and subsequent low-grade inflammation, which in turn fuels the infiltration and activation of immune cells into AT [27].
There are two main types of AT: WAT which consists of unilocular adipocytes that store energy and regulate metabolic homeostasis by the secretion of adipokines, and brown adipose tissue (BAT), which is shaped of mitochondria-rich multilocular adipocytes whose prior function is energy dissipation by thermogenesis [28]. Accumulation of WAT cells, local intrusion of immune cells, and increased levels of pro-inflammatory cytokines lead to peripheral insulin resistance in obesity [29]. In comparison to WAT, BAT of high-fat diet (HFD)-treated mice has shown a lower of immune cell-enriched mRNA expression and macrophage infiltration, indicating that BAT “resists” obesity-induced inflammation [30]. However, similar to WAT, BAT from mice with a sufficiently sustained obesogenic diet finally exhibited high mRNA levels of inflammation markers, such as TNF-α [31,32].
BAT is enriched by multilocular lipid droplets, high vascularization, and abundant mitochondria. As mentioned, BAT severs pivotally in thermoregulation through lipid oxidation-mediated heat generation [33]. Evidence suggests that the majority of WAT depots can switch phenotypically to brown fat under particular conditions, such as bariatric surgery, severe burns, cancer cachexia, cold exposure, and drug ingredients [34,35,36,37,38,39,40].
2.3. Platelets, Diabetes, and Its Sequel
Oxidative stress is of decisive importance for the development and maintenance of microvascular and macrovascular diabetes complications [41]. Under the metabolic environment of type 2 diabetes mellitus (T2DM), activated platelets can release chemokines such as CXCL4, CXCL5, and CCL5 on vascular cell surfaces, triggering atherogenesis [42,43,44,45]. Furthermore, several platelet receptors, e.g., P-Selectin, P2Y1 receptor, protease-activated receptors (PARs), and glycoprotein Ib-IX-V complex mediate the formation and activation of platelet–leukocyte microparticle complexes, fueling inflammation and subsequent atherosclerosis [46,47]. Due to different antioxidant enzymes, ROS expression is kept low in healthy patients. However, diabetic patients’ platelets display a reduced antioxidant capacity, and hence they are unable to adequately scavenge ROS [48], which in turn causes altered platelet function, dysregulated calcium homeostasis, and activation of protein kinase C (PKC). PKC acts as a mediator to promote platelet aggregation [49,50,51]. In particular, platelet CD40 ligand (CD40L) interacts with endothelial CD40, which fuels the production of chemokines, such as monocyte chemotactic protein 1 (MCP1) and IL-8, but also the expression of adhesion molecules such as vascular cell adhesion molecule 1 (VCAM-1) and E-selectin, which initiates an inflammatory response at the vessel wall. Furthermore, soluble CD40L (sCD40L) can be secreted by activated platelets express, inducing endothelial surface expression of P-selectin and secretion of IL-6 [52]. Interestingly, sCD40L mediates stimulation-induced platelet release of ROS and nitrogen species by activation of mitogen activated protein kinase (MAPK) and Akt signaling pathway, further amplifying platelet activation and indicating a crucial role of sCD40L and oxidative stress in mediating platelet-dependent inflammatory and thrombotic responses [53].
In vitro studies have shown several functional abnormalities in platelets of diabetes patients, such as hypersensitivity of platelets to aggregants and hyposensitivity of antiaggregants, indicating promotion of atherosclerosis by increasing platelet activity at site of vascular injury [54]. In diabetes patients, the prothrombotic tendency and platelet reactivity is accompanied by excessive oxidative stress and increased lipid peroxidation due to enhanced free radical activity [55,56]. Overall, platelet dysfunction leads to expanding risk of CVD in patients with T2DM.
2.4. Platelets and the Fat Tissue
In the process of adipose tissue hyperplasia, the release of certain adipokines such as IL-6, TNF-α, leptin, and resistin is increased, leading to endothelial dysfunction, followed by increase of vasoconstrictors, expression of adhesion factors, and decrease of vasodilator molecules [57,58,59]. The reduction of vasodilator molecules contributes to prothrombotic conditions, including increased concentrations of plasminogen activator inhibitor 1 and fibrinogen, leading to platelet activation/aggregation. This interaction between AT and platelets is of importance for the regulation of metabolic pathways.
Tozawa et al. [60] created an adipose-derived mesenchymal stem/stromal cell line (ASCL) that was cultured in megakaryocyte induction media. ASCL differentiated into megakaryocytes after 8 days, and platelets (ASCL-PLTs) were released after an additional 4 days. ASCL-PLTs resembled peripheral blood platelets and may have a further function as mesenchymal stem cells (MSCs). ASCL-PLTs do not need gene transfer or exogenous growth factors and display the same in vivo kinetics after application into irradiated immunodeficient mice.
3. Oxidative Stress as a Mediator of Tissue Damage and Promotor of Inflammation
The imbalance between the formation of ROS and ROS-degrading antioxidant systems can lead to CVD. This imbalance results in reduced endothelial nitric oxide synthase (eNOS), enhanced mitochondrial superoxide bioavailability, and subsequent exacerbation of hypertension [61]. Overproduction of ROS leads to NO degradation, and uncoupling of eNOS also contributes to a reduction in NO production and increased release of RNS. This mechanism is a crucial mediator of endothelial dysfunction and increases the occurrence of coronary artery disease (CAD) [62].
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is a well-known source of superoxide in vascular disease and an important mediator of oxidative stress. Interestingly, McCann et al. [63] have shown that endothelin-1-induced stroke in rat is associated to activation of two catalytic subunits of NADPH, Nox2, and Nox4. Blockade of these subunits might be helpful for treatment of stroke associated brain injury.
NO is an important vasodilator that has a protective effect on blood vessels. Oxidative stress and inflammation are the main driving forces of endothelial dysfunction. Several oxidase systems such as NADPH oxidase, xanthine oxidase, cyclooxygenase, lipoxygenase, myeloperoxidase, cytochrome P450 monooxygenase, uncoupled NOS, and peroxidase can cause NO inactivation, which represents an important mechanism that leads to endothelial dysfunction by increasing the level of superoxide dismutase [64,65]. Under a long-term oxidative stress environment, with accumulation of ROS, cell structures and functions may be damaged, inducing somatic mutations and tumorigenic transformation in several tissues [66,67]. For instance, increased ROS in prostate may cause somatic DNA mutations, promoting genetic instability, cell cycle arrest, and senescence, consequently causing the development of prostate cancer [67].
4. Complement Activation and Oxidative Stress
The complement system is an imperative part of the innate immune system. Excessive activation of host cells or inadequate control of complement activation can lead to immune dysregulation, which intensifies the vicious circle between complement, inflammatory cells, and tissue damage, and aggravates diverse clinical complications [68] (Figure 2). When granulocytes are exposed to activated complement (C), endothelial damage is induced. This damage is mainly mediated by oxygen radicals generated by granulocytes [69]. The indirect effects of anaphylatoxin and the direct effects of membrane attack complex C5b-9 can regulate white blood cell response, change vascular homeostasis, and lead to cell activation and early myocardial ischemia/reperfusion injury [70]. Activated C5b-9 modifies the production of ROS and Ca2+ Flux, aggravating ischemia/reperfusion injury [71]. Following ischemia-reperfusion injury, inhibition of C5b-9 reduces the expression of inflammatory factors [72]. In addition, overexpression of C5a can accelerate the development of atherosclerosis in ApoE−/− mice due to increasement of inflammatory activation, macrophage recruitment, and foam cell formation [73]. Beyond C5b-9, C3a, C4a, and C5a act on smooth muscle to promote vasodilatation [74,75]. Furthermore, C5a can promote neutrophil aggregation, chemotaxis, formation of ROS, and arachidonic acid metabolites. C5a regulates the adhesion of neutrophils to the endothelium by release of platelet activating factor [76,77]. Levels of C3 can predict prognosis of heart failure and is negatively correlated with cardiac remodeling [78]. Notably, C3 has been indicated as an important marker of insulin resistance in aged population [79]. AT can activate the alternative complement pathway in T2DM, which causes a low-grade inflammation [80]. The receptors of C3a (C3aR) and C5a (C5aR1 and C5aR2) have been reported to be expressed in adipocytes [81]. Therefore, in addition to the production of complement components, obesity is also a potential target of complement action. After HFD feeding, the expression of C3aR in WAT is increased. Both macrophages and WAT express large amounts of C3aR [82]. Onat et al. found that the increased level of complement C3 is related to the enhanced likelihood of CAD [83]. Furthermore, Complement C3 and C3a have separate roles in pathways leading to CVD. C3a was independently associated with aggravate atherosclerosis in heavy smokers and further promote CVD. On the contrary, in heavy smokers, C3 was exclusively associated with atherothrombosis rather than atherosclerosis [81].
5. Oxidative Stress Causes Hypothalamic Dysfunction in Metabolic Disease
There is growing evidence that obesity causes pervasive changes to the energy balance centers of the hypothalamus, perpetuating hyperphagia and metabolic disbalance. Mechanistically, HFD has been shown to promote an accumulation of toxic lipid species, thereby inducing an elevation of markers of inflammation, endoplasmic reticulum (ER) stress, and oxidative stress, which in turn leads to the loss of central leptin sensitivity, loss of insulin sensitivity, and promotion of subsequent obesity [84]. Interestingly, Kjaergaard et al. have investigated the effects of maternal intake of chocolate and soft drink (S) on hypothalamic homeostasis in rats. It has been demonstrated that hypothalamic oxidative stress is detectable prior to the inflammatory response in offspring exposed to maternal S. Furthermore, both maternal and postnatal S promoted hypothalamic inflammation prior to weight gain and diminished peripheral glucose homeostasis, pointing to a causative mechanism [85]. Indeed, hypothalamic inflammation and oxidative stress have been demonstrated to begin as early as after 1 week of HFD treatment [86]. Excessive levels of ROS were shown to alter various cellular components such as DNA, proteins, and lipids, leading to neuronal damage, thus activating several cellular inflammatory pathways [86]. In particular, neuronal oxidative stress may promote the activation of the c-Jun N-terminal kinase (JNK) and nuclear factor κB (NFκB) pathways [1,87], which are well known to induce suppressor of cytokine signaling 3 (SOCS3) and PTP1B (protein tyrosine phosphatase 1B) expression, thereby promoting central leptin resistance by inhibition of signal transducer and activator of transcription 3 (STAT3) phosphorylation as a part of the leptin post-receptor signaling [88]. Fascinatingly, the anorectic drug phenylpropanolamine has been able to restore hypothalamic STAT3 phosphorylation and decrease body weight by increasing levels of antioxidants [89]. In accordance, several beneficial effects on metabolic syndrome have been attributed to polyphenols, in part via modulation of hypothalamic inflammation and oxidative stress [90]. Thus, these insights could point to a novel new direction to research on hypothalamic energy regulation by considering the mechanisms by which oxidative stress regulates energy homeostasis, which may offer novel treatment strategies for obesity and T2DM.
6. Translational Implications
To develop novel therapy strategies for infections and cardiovascular diseases caused by oxidative stress, antioxidant therapy, reducing the ROS generation system and improving the antioxidant mechanism, receives major consideration. New ROS scavengers that target mitochondrial ROS are currently being studied, for example, mitochondrial-targeted antioxidant mitoquinone. It reduces the formation of free radicals without affecting the oxidative phosphorylation of mitochondria. In mice models, mitoquinone was shown to diminish the content of macrophages, reduce cell proliferation in atherosclerotic plaques, and constrain multiple features of metabolic syndrome [91,92,93]. Edaravone is another oxygen radical scavenger and an inhibitor of lipid peroxidation with potent antioxidant effects. Edaravone has been shown to reduce ischemia and reperfusion-associated vascular endothelial cell injury, diminishing neuronal death, brain edema, and associated neurological deficits [94]. In a large meta-analysis, antioxidant treatments (such as vitamin C and selenium) showed benefit in vitro and in animal models of CVD but failed to improve outcomes in a human context. Concentrations of antioxidant agents used in in vitro studies may have been much higher than possible oral ingestion in humans. Therefore, better antioxidant strategies need to be developed [95].
In addition, activation of NADPH oxidases promotes the production of ROS from other sources. Therefore, NADPH is recognized as another therapeutic target. NADPH oxidase (Nox) inhibitors have been developed and were shown to be effective in atherosclerosis in mice models [96].
Treatment of diabetic animals with superoxide dismutase/catalase mimics can prevent the oxidative inactivation of aortic prostacyclin synthase caused by diabetes [97]. Transgenic antioxidant enzyme expression or a combination of antioxidant compounds to inhibit hyperglycemia-induced ROS production in diabetic mice can prevent the development of experimental diabetic neuropathy, cardiomyopathy, nephropathy, and retinopathy [98,99,100,101,102]. Thus, preventive treatment of excessive superoxide caused by diabetes could be a future target for the prevention of diabetes complications [41].
7. Conclusions
CVDs remain a leading cause of morbidity and mortality. Particularly in the western world, diabetes, smoking, lack of regular exercise, and hypertension lead to CVD. To date, a growing amount of evidence has demonstrated that the chronic CVD process is related to inflammation and oxidative stress, which involves many cells, including their interactions, such as dendritic cells, platelets, eosinophils, and adipocytes. Furthermore, factors promoting inflammation such as the complement system are central to this process. Chronic low-grade inflammation in metabolic disorders, such as obesity and insulin resistance, is associated with an excessive production of ROS, resulting in endothelial cell dysfunction and release of further pro-inflammatory cytokines, which ultimately aggravates the development of ROS-mediated inflammation and consecutively cardiovascular diseases. Thus, understanding the relationship between oxidative stress, inflammation, and cardiovascular diseases will offer ways to control inflammation and oxidative stress, and is a promising direction for developing treatment strategies of affected patients.
Author Contributions
Y.S. wrote the manuscript. E.R. and H.M.N. critically revised the manuscript and wrote pars of the manuscript. H.F.L. conceptualized and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Projektnr. 374031971—TRR 240 and the DZHK (German Research Centre for Cardiovascular Research), partner site Hamburg/Lübeck/Kiel, 23562 Lübeck, Germany to HFL. And the ERA-CVD grant to HFL and HN (ERAPERMED2020-245).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Sarah Gekeler, Anke Constantz, and Jakob von Esebeck for perfect technical assistance. This work was supported by the Volkswagen Foundation (Lichtenberg program) and the German Research Council (KFO 274—Platelets—basic mechanisms and clinical implications; SFB/Transregio 240). HN is supported by the Clinician Scientist Programme of the DZHK (German Research Centre for Cardiovascular Research), partner site Hamburg/Lübeck/Kiel.
Conflicts of Interest
The authors declare that no conflict of interest exists.
References
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Fujisaka, S.; Usui, I.; Bukhari, A.; Ikutani, M.; Oya, T.; Kanatani, Y.; Tsuneyama, K.; Nagai, Y.; Takatsu, K.; Urakaze, M.; et al. Regulatory Mechanisms for Adipose Tissue M1 and M2 Macrophages in Diet-Induced Obese Mice. Diabetes 2009, 58, 2574–2582. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Choi, Y.J.; Joung, S.M.; Lee, B.H.; Jung, Y.-S.; Lee, J.Y. Hypoxic stress up-regulates the expression of Toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology 2010, 129, 516–524. [Google Scholar] [CrossRef]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Liu, Y.-J. Development of Dendritic-Cell Lineages. Immunity 2007, 26, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like Receptor–Induced Changes in Glycolytic Metabolism Regulate Dendritic Cell Activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Khouili, S.C.; Priego, E.; Heras-Murillo, I.; Sancho, D. Metabolic Control of Dendritic Cell Functions: Digesting Information. Front. Immunol. 2019, 10, 775. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.J.; Everts, B. Dendritic cell metabolism. Nat. Rev. Immunol. 2015, 15, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.G.; O’Neill, L.A.J. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. 2017, 591, 2992–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thwe, P.M.; Pelgrom, L.R.; Cooper, R.; Beauchamp, S.; Reisz, J.A.; D’Alessandro, A.; Everts, B.; Amiel, E. Cell-Intrinsic Glycogen Metabolism Supports Early Glycolytic Reprogramming Required for Dendritic Cell Immune Responses. Cell Metab. 2017, 26, 558–567.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanovic-Racic, M.; Yang, X.; Turner, M.S.; Mantell, B.S.; Stolz, D.B.; Sumpter, T.L.; Sipula, I.J.; Dedousis, N.; Scott, D.K.; Morel, P.A.; et al. Dendritic Cells Promote Macrophage Infiltration and Comprise a Substantial Proportion of Obesity-Associated Increases in CD11c+ Cells in Adipose Tissue and Liver. Diabetes 2012, 61, 2330–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertola, A.; Ciucci, T.; Rousseau, D.; Bourlier, V.; Duffaut, C.; Bonnafous, S.; Blin-Wakkach, C.; Anty, R.; Iannelli, A.; Gugenheim, J.; et al. Identification of Adipose Tissue Dendritic Cells Correlated With Obesity-Associated Insulin-Resistance and Inducing Th17 Responses in Mice and Patients. Diabetes 2012, 61, 2238–2247. [Google Scholar] [CrossRef] [Green Version]
- Macdougall, C.E.; Longhi, M.P. Adipose tissue dendritic cells in steady-state. Immunology 2019, 156, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Rudemiller, N.P.; Privratsky, J.R.; Ren, J.; Wen, Y.; Griffiths, R.; Crowley, S.D. Classical Dendritic Cells Mediate Hypertension by Promoting Renal Oxidative Stress and Fluid Retention. Hypertension 2020, 75, 131–138. [Google Scholar] [CrossRef]
- Weller, P.F.; Spencer, L.A. Functions of tissue-resident eosinophils. Nat. Rev. Immunol. 2017, 17, 746–760. [Google Scholar] [CrossRef]
- Kano, G.; Almanan, M.; Bochner, B.S.; Zimmermann, N. Mechanism of Siglec-8-mediated cell death in IL-5-activated eosinophils: Role for reactive oxygen species-enhanced MEK/ERK activation. J. Allergy Clin. Immunol. 2013, 132, 437–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molofsky, A.B.; Nussbaum, J.C.; Liang, H.-E.; Van Dyken, S.J.; Cheng, L.E.; Mohapatra, A.; Chawla, A.; Locksley, R.M. Innate Lymphoid Type 2 Cells Sustain Visceral Adipose Tissue Eosinophils and Alternatively Activated Macrophages. J. Exp. Med. 2013, 210, 535–549. [Google Scholar] [CrossRef]
- Niccoli, G.; Montone, R.A.; Sabato, V.; Crea, F. Role of Allergic Inflammatory Cells in Coronary Artery Disease. Circulation 2018, 138, 1736–1748. [Google Scholar] [CrossRef]
- Haley, K.J.; Lilly, C.M.; Yang, J.H.; Feng, Y.; Kennedy, S.P.; Turi, T.G.; Thompson, J.F.; Sukhova, G.H.; Libby, P.; Lee, R.T. Overexpression of Eotaxin and the CCR3 Receptor in Human Atherosclerosis: Using Genomic Technology to Identify a Potential Novel Pathway of Vascular Inflammation. Circulation 2000, 102, 2185–2189. [Google Scholar] [CrossRef] [Green Version]
- Boshuizen, M.C.S.; Hoeksema, M.A.; Neele, A.E.; van der Velden, S.; Hamers, A.A.J.; Van den Bossche, J.; Lutgens, E.; de Winther, M.P.J. Interferon-β Promotes Macrophage Foam Cell Formation by Altering Both Cholesterol Influx and Efflux Mechanisms. Cytokine 2016, 77, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Wang, L.; Li, F.; Yang, M.; Song, L.; Tian, F.; Yukht, A.; Shah, P.K.; Rothenberg, M.E.; Sharifi, B.G. Oxidized LDL Activated Eosinophil Polarize Macrophage Phenotype from M2 to M1 through Activation of CD36 Scavenger Receptor. Atherosclerosis 2017, 263, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Chatzigeorgiou, A.; Chavakis, T. Immune Cells and Metabolism. Handb. Exp. Pharmacol. 2016, 233, 221–249. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.Y.; Park, Y.J.; Ham, M.; Kim, J.B. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol. Cells 2014, 37, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab. 2019, 29, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbons, T.P.; Kogan, S.; Aouadi, M.; Hendricks, G.M.; Straubhaar, J.; Czech, M.P. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1425–H1437. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, T.; Nitta, T.; Maruno, K.; Yeh, Y.-S.; Kuwata, H.; Tomita, K.; Goto, T.; Takahashi, N.; Kawada, T. Macrophage Infiltration into Obese Adipose Tissues Suppresses the Induction of UCP1 Level in Mice. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E676–E687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts-Toler, C.; O’Neill, B.T.; Cypess, A.M. Diet-induced obesity causes insulin resistance in mouse brown adipose tissue. Obesity 2015, 23, 1765–1770. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.; Picard, F. Brown fat biology and thermogenesis. Front. Biosci. 2011, 16, 1233–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P.; Spiegelman, B.M. Brown and Beige Fat: Molecular Parts of a Thermogenic Machine. Diabetes 2015, 64, 2346–2351. [Google Scholar] [CrossRef] [Green Version]
- Neinast, M.D.; Frank, A.P.; Zechner, J.F.; Li, Q.; Vishvanath, L.; Palmer, B.F.; Aguirre, V.; Gupta, R.K.; Clegg, D.J. Activation of Natriuretic Peptides and the Sympathetic Nervous System Following Roux-En-Y Gastric Bypass Is Associated with Gonadal Adipose Tissues Browning. Mol. Metab. 2015, 4, 427–436. [Google Scholar] [CrossRef]
- den Hartigh, L.J.; Wang, S.; Goodspeed, L.; Wietecha, T.; Houston, B.; Omer, M.; Ogimoto, K.; Subramanian, S.; Gowda, G.A.N.; O’Brien, K.D.; et al. Metabolically Distinct Weight Loss by 10, 12 CLA and Caloric Restriction Highlight the Importance of Subcutaneous White Adipose Tissue for Glucose Homeostasis in Mice. PLoS ONE 2017, 12, e0172912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorbani, M.; Himms-Hagen, J. Appearance of brown adipocytes in white adipose tissue during CL 316,243-induced reversal of obesity and diabetes in Zucker fa/fa rats. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Sidossis, L.S.; Porter, C.; Saraf, M.K.; Børsheim, E.; Radhakrishnan, R.S.; Chao, T.; Ali, A.; Chondronikola, M.; Mlcak, R.; Finnerty, C.C.; et al. Browning of Subcutaneous White Adipose Tissue in Humans after Severe Adrenergic Stress. Cell Metab. 2015, 22, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Fukui, Y.; Masui, S.; Osada, S.; Umesono, K.; Motojima, K. A new thiazolidinedione, NC-2100, which is a weak PPAR-gamma activator, exhibits potent antidiabetic effects and induces uncoupling protein 1 in white adipose tissue of KKAy obese mice. Diabetes 2000, 49, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Villarroya, F.; Cereijo, R.; Gavaldà-Navarro, A.; Villarroya, J.; Giralt, M. Inflammation of brown/beige adipose tissues in obesity and metabolic disease. J. Intern. Med. 2018, 284, 492–504. [Google Scholar] [CrossRef] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Gaiz, A.; Mosawy, S.; Colson, N.; Singh, I. Thrombotic and cardiovascular risks in type two diabetes; Role of platelet hyperactivity. Biomed. Pharmacother. 2017, 94, 679–686. [Google Scholar] [CrossRef]
- Weber, C. Platelets and chemokines in atherosclerosis: Partners in crime. Circ. Res. 2005, 96, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Ferroni, P.; Basili, S.; Falco, A.; Davì, G. Platelet activation in type 2 diabetes mellitus. J. Thromb. Haemost. 2004, 2, 1282–1291. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.D.; Frenette, P.S. The vessel wall and its interactions. Blood 2008, 111, 5271–5281. [Google Scholar] [CrossRef]
- Nording, H.; Baron, L.; Langer, H.F. Platelets as therapeutic targets to prevent atherosclerosis. Atherosclerosis 2020, 307, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Kakouros, N.; Rade, J.J.; Kourliouros, A.; Resar, J.R. Platelet Function in Patients with Diabetes Mellitus: From a Theoretical to a Practical Perspective. Int. J. Endocrinol. 2011, 2011, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Abe, J.-i.; Takahashi, M.; Ishida, M.; Lee, J.D.; Berk, B.C. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. J. Biol. Chem. 1997, 272, 20389–20394. [Google Scholar] [CrossRef] [Green Version]
- Redondo, P.C.; Ben-Amor, N.; Salido, G.M.; Bartegi, A.; Pariente, J.A.; Rosado, J.A. Ca2+-independent activation of Bruton’s tyrosine kinase is required for store-mediated Ca2+ entry in human platelets. Cell. Signal. 2005, 17, 1011–1021. [Google Scholar] [CrossRef]
- Senis, Y.A.; Mazharian, A.; Mori, J. Src family kinases: At the forefront of platelet activation. Blood 2014, 124, 2013–2024. [Google Scholar] [CrossRef] [Green Version]
- Rawish, E.; Nording, H.; Münte, T.; Langer, H.F. Platelets as Mediators of Neuroinflammation and Thrombosis. Front. Immunol. 2020, 11, 548631. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Varghese, S.; Vitseva, O.; Tanriverdi, K.; Freedman, J.E. CD40 ligand influences platelet release of reactive oxygen intermediates. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2428–2434. [Google Scholar] [CrossRef] [Green Version]
- Vinik, A.I.; Erbas, T.; Park, T.S.; Nolan, R.; Pittenger, G.L. Platelet dysfunction in type 2 diabetes. Diabetes Care 2001, 24, 1476–1485. [Google Scholar] [CrossRef] [Green Version]
- Jennings, P.E.; McLaren, M.; Scott, N.A.; Saniabadi, A.R.; Belch, J.J. The relationship of oxidative stress to thrombotic tendency in type 1 diabetic patients with retinopathy. Diabet. Med. 1991, 8, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Jennings, P.E. From hemobiology to vascular disease: A review of the potential of gliclazide to influence the pathogenesis of diabetic vascular disease. J. Diabetes Complicat. 1994, 8, 226–230. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, C. Regulation of Microvascular Function by Adipose Tissue in Obesity and Type 2 Diabetes: Evidence of an Adipose-Vascular Loop. Am. J. Biomed. Sci. 2009, 1, 133. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Maenhaut, N.; Van de Voorde, J. Regulation of vascular tone by adipocytes. BMC Med. 2011, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Tozawa, K.; Ono-Uruga, Y.; Yazawa, M.; Mori, T.; Murata, M.; Okamoto, S.; Ikeda, Y.; Matsubara, Y. Megakaryocytes and Platelets from a Novel Human Adipose Tissue-Derived Mesenchymal Stem Cell Line. Blood 2019, 133, 633–643. [Google Scholar] [CrossRef]
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part I: Basic mechanisms and in vivo monitoring of ROS. Circulation 2003, 108, 1912–1916. [Google Scholar] [CrossRef] [Green Version]
- Carresi, C.; Mollace, R.; Macrì, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Coppoletta, A.R.; Guarnieri, L.; Ruga, S.; Zito, M.C.; et al. Oxidative Stress Triggers Defective Autophagy in Endothelial Cells: Role in Atherothrombosis Development. Antioxidants 2021, 10, 387. [Google Scholar] [CrossRef]
- McCann, S.K.; Dusting, G.J.; Roulston, C.L. Early increase of Nox4 NADPH oxidase and superoxide generation following endothelin-1-induced stroke in conscious rats. J. Neurosci. Res. 2008, 86, 2524–2534. [Google Scholar] [CrossRef]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrant, J.R.; Seals, D.R.; Connell, M.L.; Russell, M.J.; Lawson, B.R.; Folian, B.J.; Donato, A.J.; Lesniewski, L.A. Voluntary Wheel Running Restores Endothelial Function in Conduit Arteries of Old Mice: Direct Evidence for Reduced Oxidative Stress, Increased Superoxide Dismutase Activity and down-Regulation of NADPH Oxidase. J. Physiol. 2009, 587, 3271–3285. [Google Scholar] [CrossRef]
- Fang, J.; Seki, T.; Maeda, H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv. Drug Deliv. Rev. 2009, 61, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Khandrika, L.; Kumar, B.; Koul, S.; Maroni, P.; Koul, H.K. Oxidative stress in prostate cancer. Cancer Lett. 2009, 282, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklin, D.; Lambris, J.D. Complement in immune and inflammatory disorders: Pathophysiological mechanisms. J. Immunol. 2013, 190, 3831–3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, T.; Moldow, C.F.; Craddock, P.R.; Bowers, T.K.; Jacob, H.S. Oxygen radicals mediate endothelial cell damage by complement-stimulated granulocytes. An in vitro model of immune vascular damage. J. Clin. Investig. 1978, 61, 1161–1167. [Google Scholar] [CrossRef] [Green Version]
- Stahl, G.L.; Shernan, S.K.; Smith, P.K.; Levy, J.H. Complement activation and cardiac surgery: A novel target for improving outcomes. Anesth. Analg. 2012, 115, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Berger, H.J.; Taratuska, A.; Smith, T.W.; Halperin, J.A. Activated complement directly modifies the performance of isolated heart muscle cells from guinea pig and rat. Am. J. Physiol. 1993, 265 Pt 2, H267–H272. [Google Scholar] [CrossRef]
- Vakeva, A.P.; Agah, A.; Rollins, S.A.; Matis, L.A.; Li, L.; Stahl, G.L. Myocardial infarction and apoptosis after myocardial ischemia and reperfusion: Role of the terminal complement components and inhibition by anti-C5 therapy. Circulation 1998, 97, 2259–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, G.; Li, B.; Liu, X.; Zhang, M.; Gao, F.; Zhao, Y.; An, F.; Zhang, Y.; Zhang, C. Overexpression of Complement Component C5a Accelerates the Development of Atherosclerosis in ApoE-Knockout Mice. Oncotarget 2016, 7, 56060–56070. [Google Scholar] [CrossRef] [Green Version]
- Humbles, A.A.; Lu, B.; Nilsson, C.A.; Lilly, C.; Israel, E.; Fujiwara, Y.; Gerard, N.P.; Gerard, C. A Role for the C3a Anaphylatoxin Receptor in the Effector Phase of Asthma. Nature 2000, 406, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Shibata, K.; Akatsu, H.; Shimizu, N.; Sakata, N.; Katsuragi, T.; Okada, H. Contribution of Anaphylatoxin C5a to Late Airway Responses after Repeated Exposure of Antigen to Allergic Rats. J. Immunol. 2001, 167, 4651–4660. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, K.S.; Friedrichs, G.S.; Homeister, J.W.; Lucchesi, B.R. The complement system in myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 1994, 28, 437–444. [Google Scholar] [CrossRef]
- Braquet, P.; Paubert-Braquet, M.; Bourgain, R.H.; Bussolino, F.; Hosford, D. PAF/cytokine auto-generated feedback networks in microvascular immune injury: Consequences in shock, ischemia and graft rejection. J. Lipid Mediat. 1989, 1, 75–112. [Google Scholar]
- Suffritti, C.; Tobaldini, E.; Schiavon, R.; Strada, S.; Maggioni, L.; Mehta, S.; Sandrone, G.; Toschi-Dias, E.; Cicardi, M.; Montano, N. Complement and Contact System Activation in Acute Congestive Heart Failure Patients. Clin. Exp. Immunol. 2017, 190, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Muscari, A.; Antonelli, S.; Bianchi, G.; Cavrini, G.; Dapporto, S.; Ligabue, A.; Ludovico, C.; Magalotti, D.; Poggiopollini, G.; Zoli, M.; et al. Serum C3 Is a Stronger Inflammatory Marker of Insulin Resistance than C-Reactive Protein, Leukocyte Count, and Erythrocyte Sedimentation Rate: Comparison Study in an Elderly Population. Diabetes Care 2007, 30, 2362–2368. [Google Scholar] [CrossRef] [Green Version]
- Weyer, C.; Tataranni, P.A.; Pratley, R.E. Insulin action and insulinemia are closely related to the fasting complement C3, but not acylation stimulating protein concentration. Diabetes Care 2000, 23, 779–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertle, E.; van Greevenbroek, M.M.; Arts, I.C.; van der Kallen, C.J.; Geijselaers, S.L.; Feskens, E.J.; Jansen, E.H.; Schalkwijk, C.G.; Stehouwer, C.D. Distinct Associations of Complement C3a and Its Precursor C3 with Atherosclerosis and Cardiovascular Disease. The CODAM Study. Thromb. Haemost. 2014, 111, 1102–1111. [Google Scholar] [CrossRef]
- Shim, K.; Begum, R.; Yang, C.; Wang, H. Complement activation in obesity, insulin resistance, and type 2 diabetes mellitus. World J. Diabetes 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Onat, A.; Uzunlar, B.; Hergenç, G.; Yazici, M.; Sari, I.; Uyarel, H.; Can, G.; Sansoy, V. Cross-Sectional Study of Complement C3 as a Coronary Risk Factor among Men and Women. Clin. Sci. 2005, 108, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Rawish, E.; Nickel, L.; Schuster, F.; Stölting, I.; Frydrychowicz, A.; Saar, K.; Hübner, N.; Othman, A.; Kuerschner, L.; Raasch, W. Telmisartan Prevents Development of Obesity and Normalizes Hypothalamic Lipid Droplets. J. Endocrinol. 2020, 244, 95–110. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Nilsson, C.; Nielsen, M.O.; Grove, K.; Raun, K. Hypothalamic oxidative stress and inflammation, and peripheral glucose homeostasis in Sprague-Dawley rat offspring exposed to maternal and postnatal chocolate and soft drink. Nutr. Diabetes 2018, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, G.; Viggiano, E.; Trinchese, G.; De Filippo, C.; Messina, A.; Monda, V.; Valenzano, A.; Cincione, R.I.; Zammit, C.; Cimmino, F.; et al. Long Feeding High-Fat Diet Induces Hypothalamic Oxidative Stress and Inflammation, and Prolonged Hypothalamic AMPK Activation in Rat Animal Model. Front. Physiol. 2018, 9, 818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Git, K.C.G.; Adan, R.A.H. Leptin resistance in diet-induced obesity: The role of hypothalamic inflammation. Obes. Rev. 2015, 16, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Kuo, D.-Y.; Chen, P.-N.; Hsieh, Y.-S. Targeting oxidative stress in the hypothalamus: The effect of transcription factor STAT3 knockdown on endogenous antioxidants-mediated appetite control. Arch. Toxicol. 2015, 89, 87–100. [Google Scholar] [CrossRef]
- Samodien, E.; Johnson, R.; Pheiffer, C.; Mabasa, L.; Erasmus, M.; Louw, J.; Chellan, N. Diet-Induced Hypothalamic Dysfunction and Metabolic Disease, and the Therapeutic Potential of Polyphenols. Mol. Metab. 2019, 27, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.J.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Mercer, J.R.; Yu, E.; Figg, N.; Cheng, K.-K.; Prime, T.A.; Griffin, J.L.; Masoodi, M.; Vidal-Puig, A.; Murphy, M.P.; Bennett, M.R. The Mitochondria-Targeted Antioxidant MitoQ Decreases Features of the Metabolic Syndrome in ATM+/−/ApoE−/− Mice. Free Radic. Biol. Med. 2012, 52, 841–849. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Feng, S.; Yang, Q.; Liu, M.; Li, W.; Yuan, W.; Zhang, S.; Wu, B.; Li, J. Edaravone for Acute Ischaemic Stroke. Cochrane Database Syst. Rev. 2011, CD007230. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.M.; Wingler, K.; Schmidt, H.H.H.W. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, A.M.; Russell, J.W.; Sullivan, K.A.; Backus, C.; Hayes, J.M.; McLean, L.L.; Feldman, E.L. SOD2 Protects Neurons from Injury in Cell Culture and Animal Models of Diabetic Neuropathy. Exp. Neurol. 2007, 208, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Otero, P.; Bonet, B.; Herrera, E.; Rabano, A. Development of atherosclerosis in the diabetic BALB/c mice. Prevention with Vitamin E administration. Atherosclerosis 2005, 182, 259–265. [Google Scholar] [CrossRef]
- Zhang, Y.; Wada, J.; Hashimoto, I.; Eguchi, J.; Yasuhara, A.; Kanwar, Y.S.; Shikata, K.; Makino, H. Therapeutic Approach for Diabetic Nephropathy Using Gene Delivery of Translocase of Inner Mitochondrial Membrane 44 by Reducing Mitochondrial Superoxide Production. J. Am. Soc. Nephrol. 2006, 17, 1090–1101. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, V.; Xiong, Y.; Ho, Y.-S. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic. Biol. Med. 2006, 41, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- DeRubertis, F.R.; Craven, P.A.; Melhem, M.F. Acceleration of diabetic renal injury in the superoxide dismutase knockout mouse: Effects of tempol. Metabolism 2007, 56, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
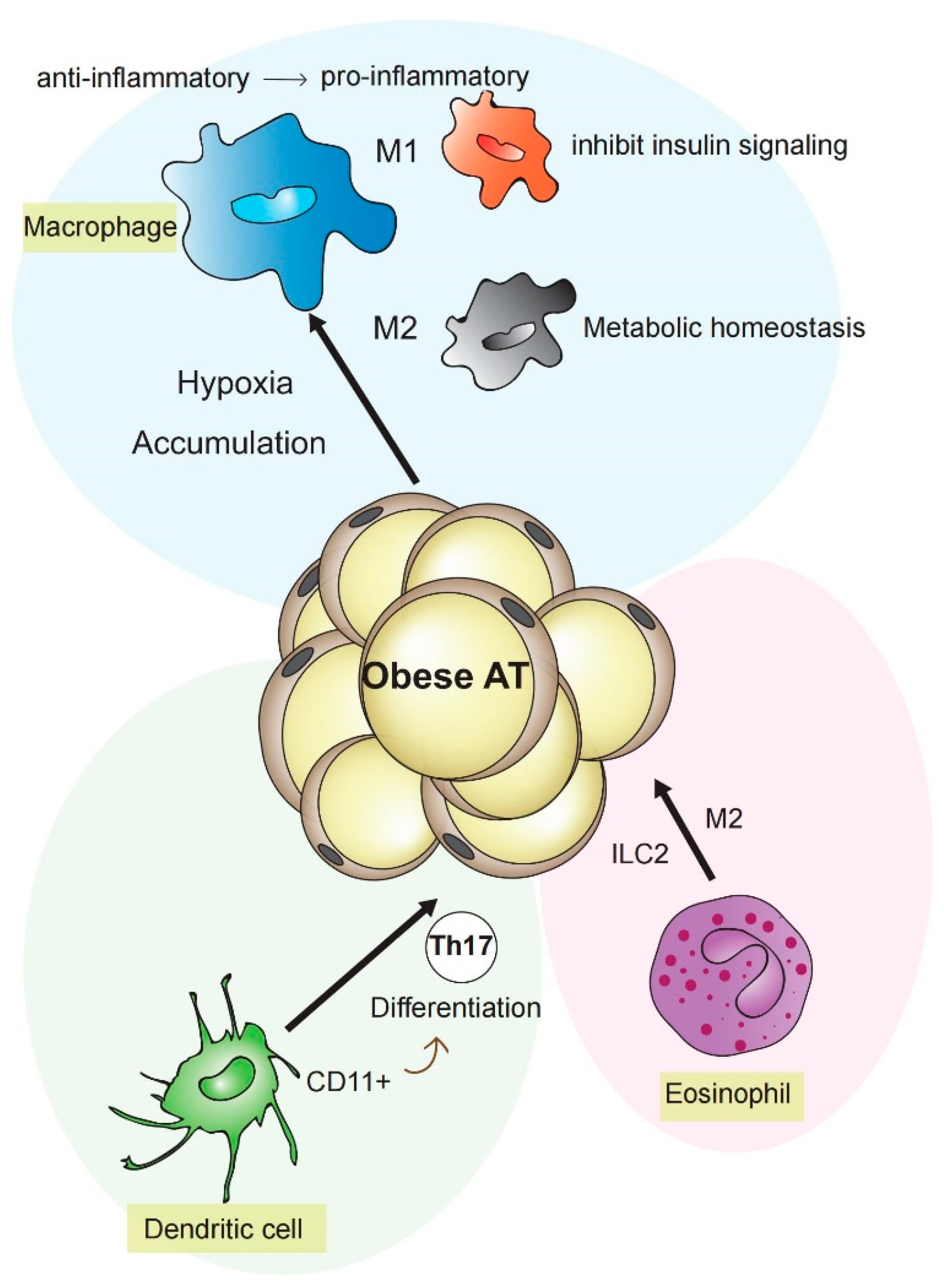
Figure 1.
Contribution of different cell types to inflammation in adipose tissue. Mature CD11+DCs induce differentiation of Th17 under obesity-related insulin resistance and contribute to adipose tissue inflammation. Eosinophils, M2 macrophages, and type 2 innate lymphoid cells regulate metabolic homeostasis in adipose tissue by cytokine expression or dependence upon the γc cytokine chain. Under obesity conditions, M1-like macrophages lead to insulin resistance. Following the adipose increase, macrophage phenotype changed from anti-inflammatory M2 to pro-inflammatory M1 type.
Figure 1.
Contribution of different cell types to inflammation in adipose tissue. Mature CD11+DCs induce differentiation of Th17 under obesity-related insulin resistance and contribute to adipose tissue inflammation. Eosinophils, M2 macrophages, and type 2 innate lymphoid cells regulate metabolic homeostasis in adipose tissue by cytokine expression or dependence upon the γc cytokine chain. Under obesity conditions, M1-like macrophages lead to insulin resistance. Following the adipose increase, macrophage phenotype changed from anti-inflammatory M2 to pro-inflammatory M1 type.

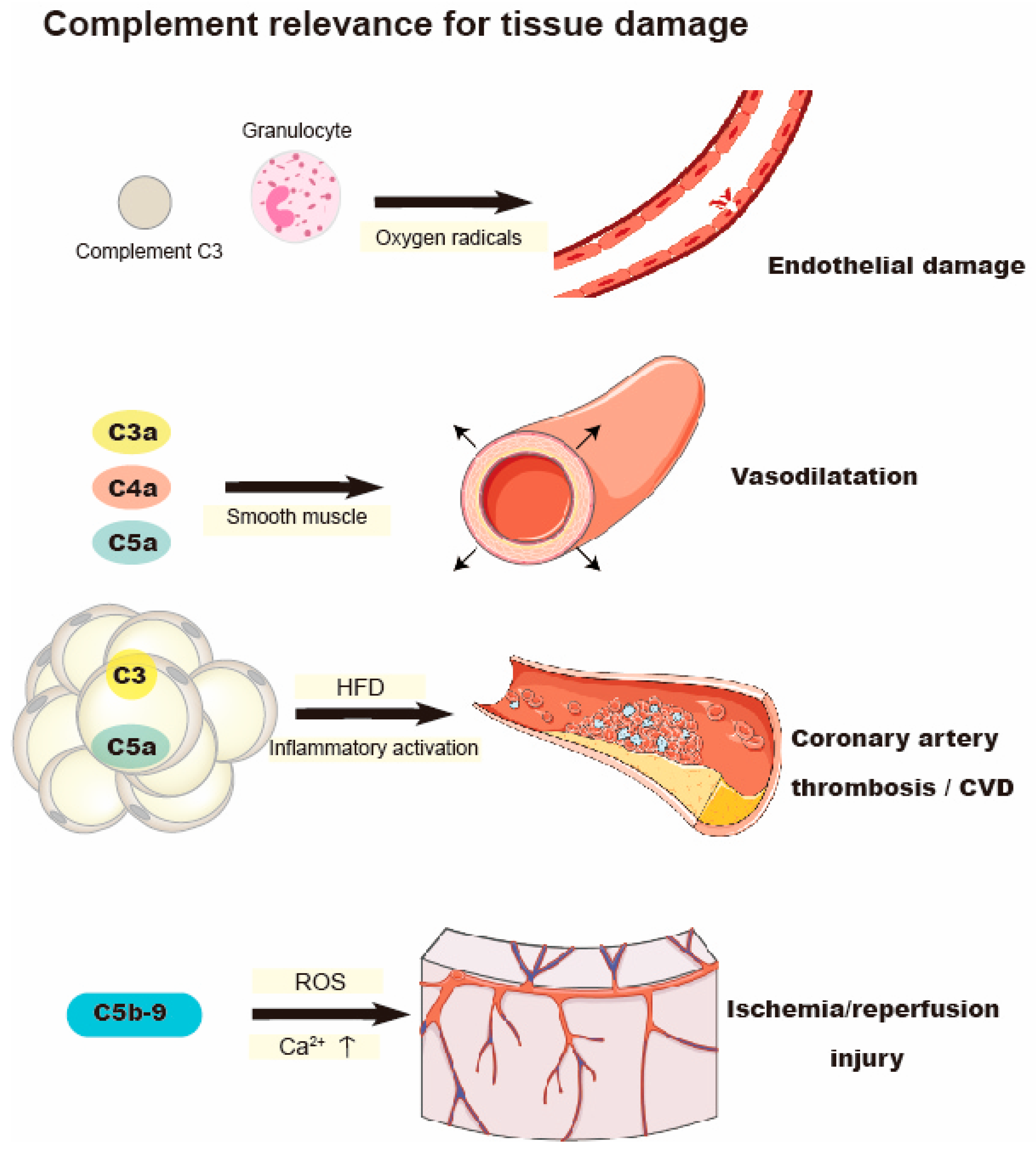
Figure 2.
Complement-mediated oxidative stress and its relevance to tissue damage. After exposure to complement C3, oxygen radicals produced by granulocytes contribute to the endothelial damage. C3, C3a, C4a, and C5a are involved in vessel damage in CVD. Activated C5b-9 aggravates ischemia/reperfusion injury.
Figure 2.
Complement-mediated oxidative stress and its relevance to tissue damage. After exposure to complement C3, oxygen radicals produced by granulocytes contribute to the endothelial damage. C3, C3a, C4a, and C5a are involved in vessel damage in CVD. Activated C5b-9 aggravates ischemia/reperfusion injury.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sun, Y.; Rawish, E.; Nording, H.M.; Langer, H.F. Inflammation in Metabolic and Cardiovascular Disorders—Role of Oxidative Stress. Life 2021, 11, 672. https://0-doi-org.brum.beds.ac.uk/10.3390/life11070672
AMA Style
Sun Y, Rawish E, Nording HM, Langer HF. Inflammation in Metabolic and Cardiovascular Disorders—Role of Oxidative Stress. Life. 2021; 11(7):672. https://0-doi-org.brum.beds.ac.uk/10.3390/life11070672
Chicago/Turabian StyleSun, Ying, Elias Rawish, Henry M. Nording, and Harald F. Langer. 2021. "Inflammation in Metabolic and Cardiovascular Disorders—Role of Oxidative Stress" Life 11, no. 7: 672. https://0-doi-org.brum.beds.ac.uk/10.3390/life11070672
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.