
Genomic and Ancestral Variation Underlies the Severity of COVID-19 Clinical Manifestation in Individuals of European Descent


Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Generation of Genomic Datasets
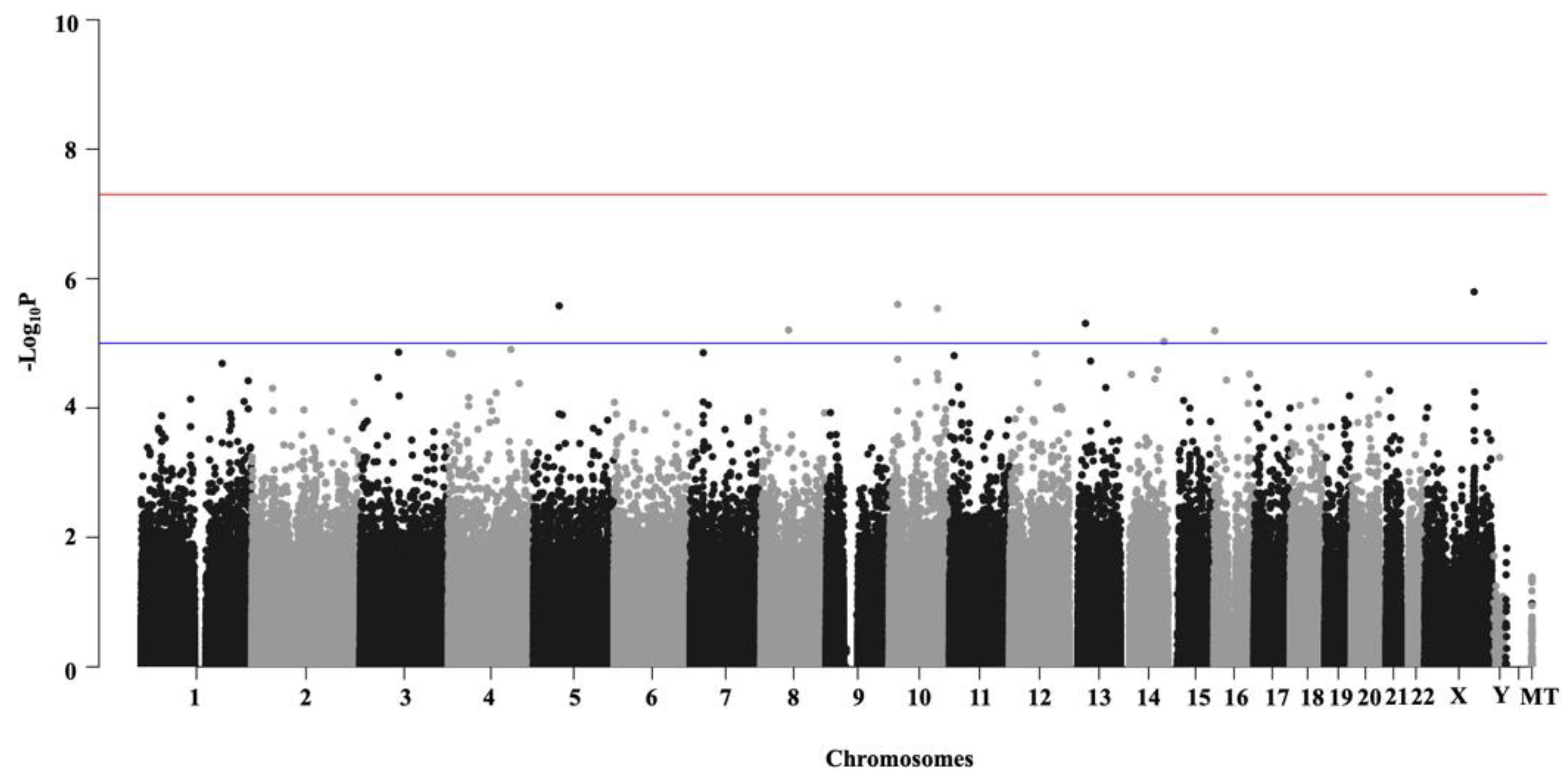
2.3. Genome-Wide Association Analyses (GWAS)
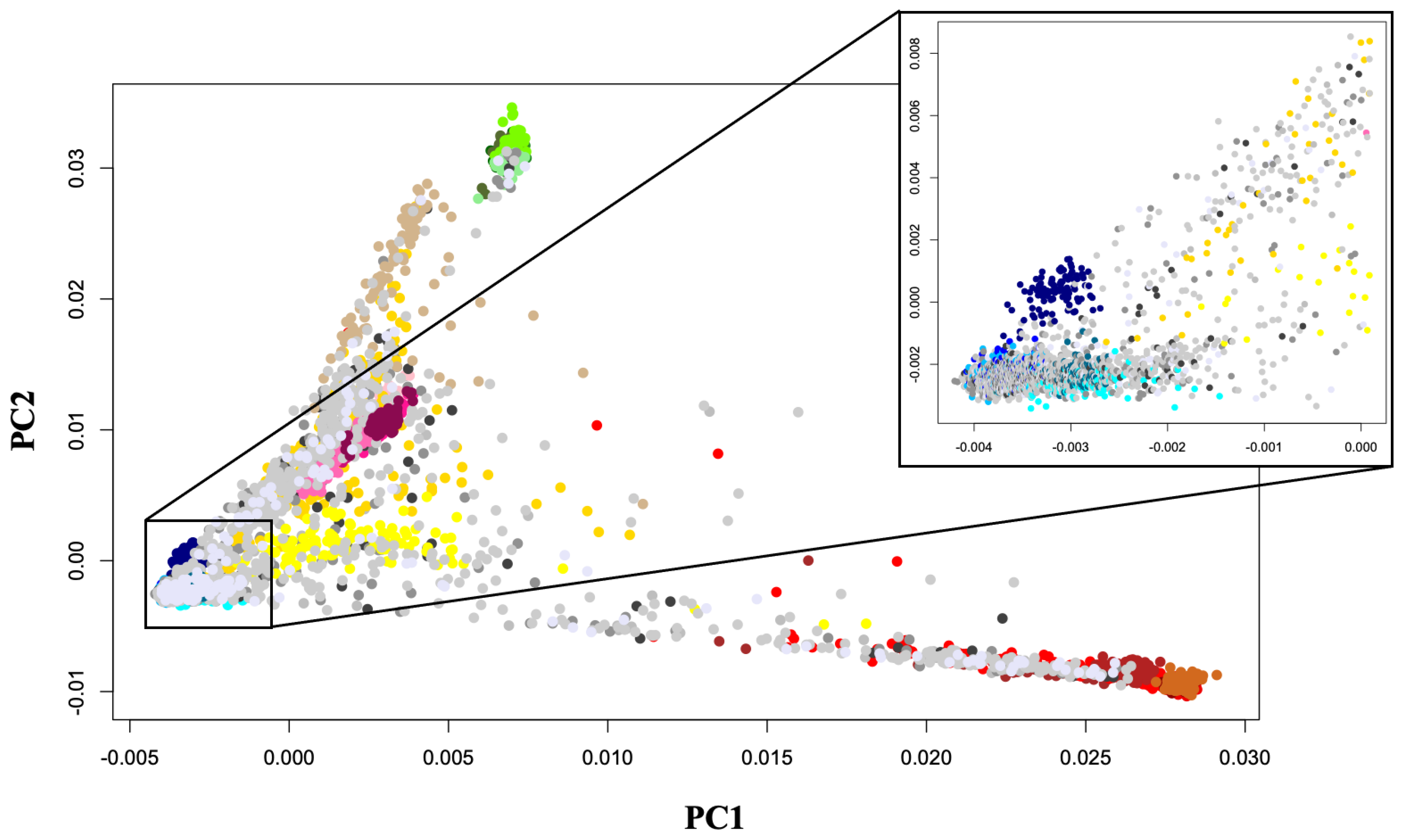
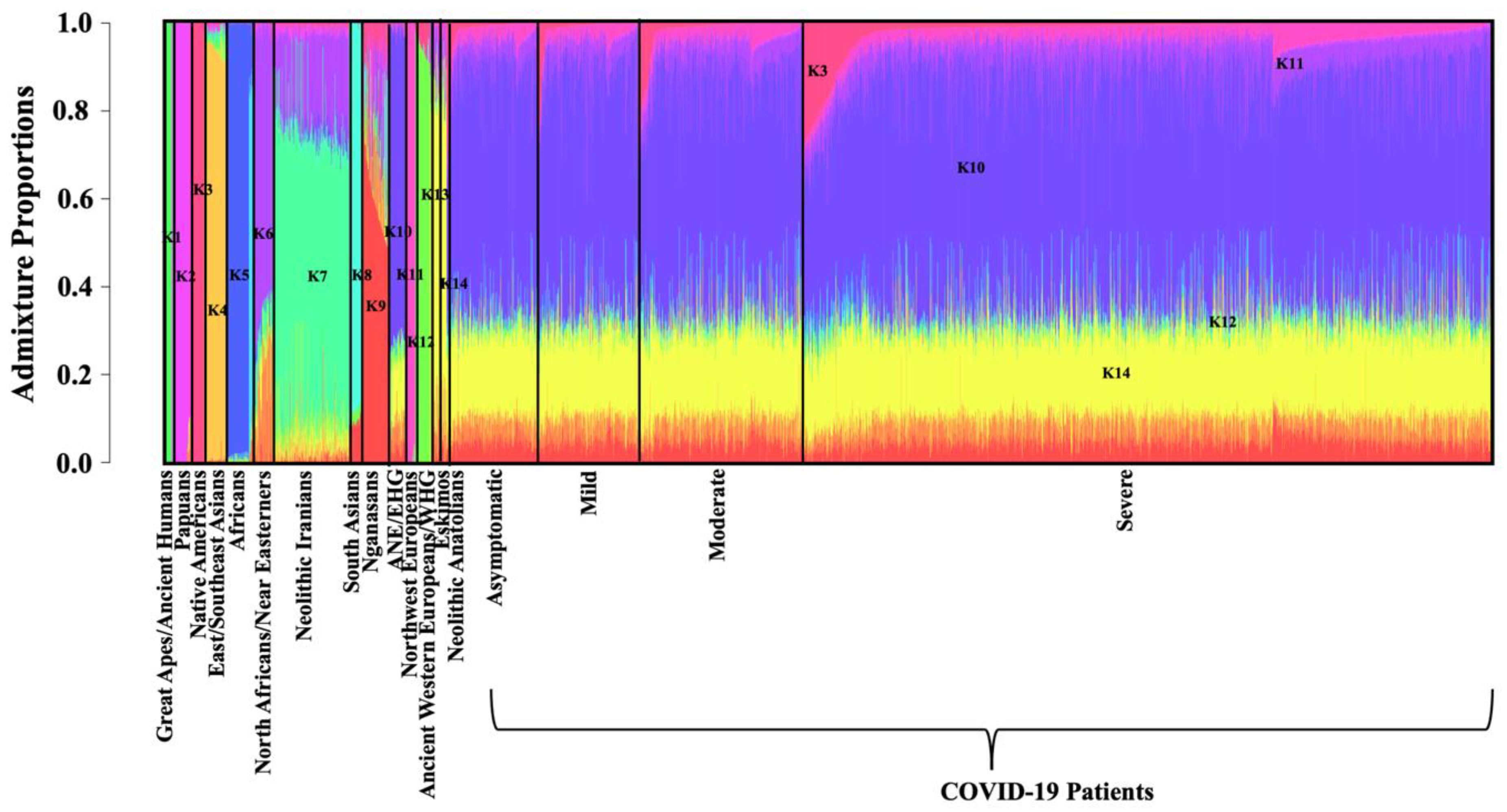
2.4. Population Clustering and Ancestry Determination
2.5. Ancestry Proportions among COVID-19 Patients of European Ancestry
3. Results
3.1. Genome-Wide Association Analyses (GWAS)
3.2. ADMIXTURE Analysis
3.3. Multiple Regression
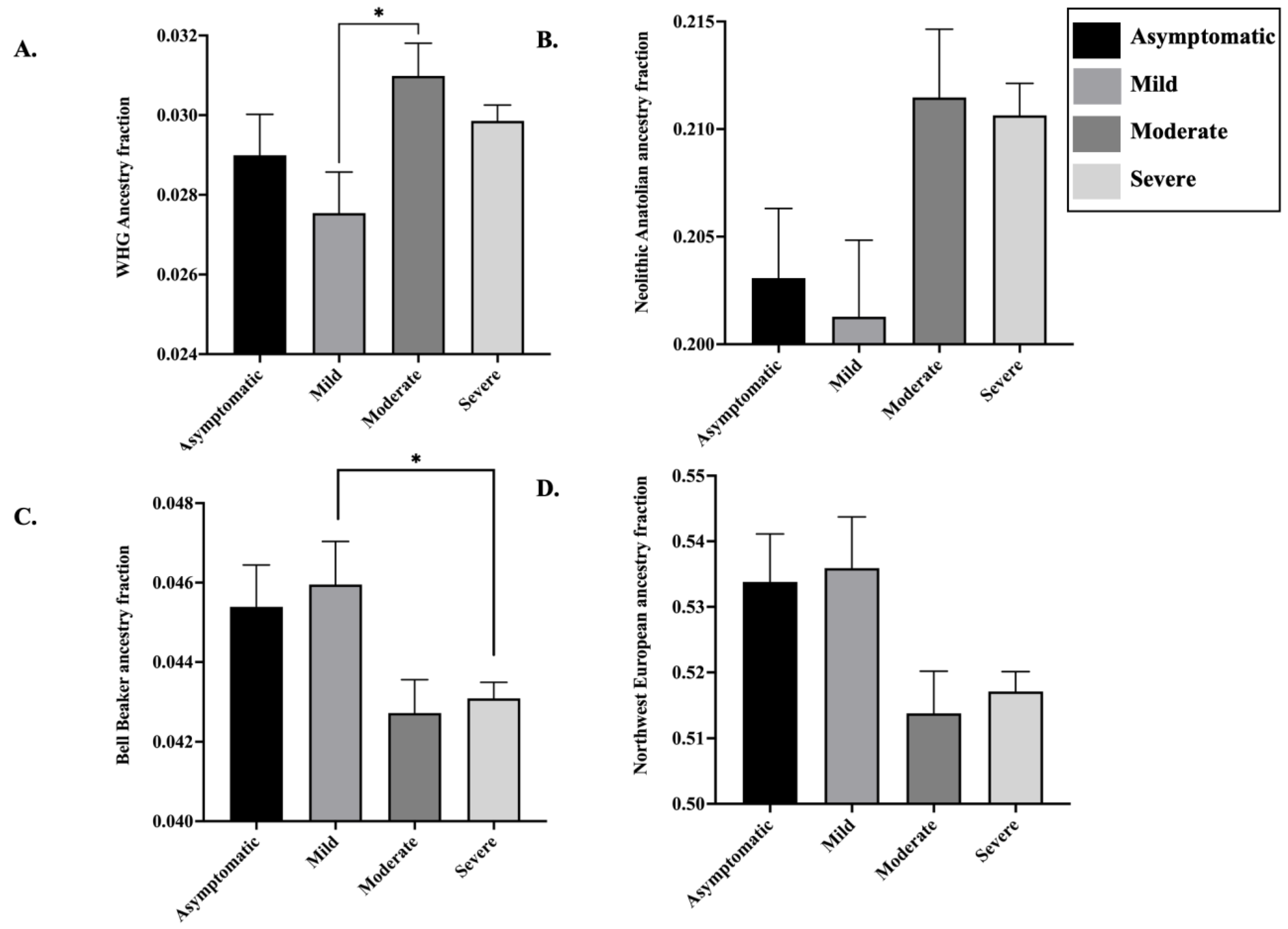
3.4. Ancestry Proportions in the European Genomes Present in COVID-19 Patient Dataset
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Xu, Y.; Sun, C.; Wang, X.; Guo, Y.; Qiu, S.; Ma, K. A systematic review of asymptomatic infections with COVID-19. J. Microbiol. Immunol. Infect. 2021, 54, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Niazkar, H.R.; Zibaee, B.; Nasimi, A.; Bahri, N. The neurological manifestations of COVID-19: A review article. Neurol Sci. 2020, 41, 1667–1671. [Google Scholar] [CrossRef] [PubMed]
- Machhi, J.; Herskovitz, J.; Senan, A.M.; Dutta, D.; Nath, B.; Oleynikov, M.D.; Blomberg, W.R.; Meigs, D.D.; Hasan, M.; Patel, M.; et al. The Natural History, Pathobiology, and Clinical Manifestations of SARS-CoV-2 Infections. J. Neuroimmune Pharmacol. 2020, 15, 359–386. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Lu, Y.; Li, X.; Geng, D.; Mei, N.; Wu, P.Y.; Huang, C.C.; Jia, T.; Zhao, Y.; Wang, D.; Xiao, A.; et al. Cerebral Micro-Structural Changes in COVID-19 Patients—An MRI-based 3-month Follow-up Study. EClinicalMedicine 2020, 25, 100484. [Google Scholar] [CrossRef] [PubMed]
- Docherty, A.B.; Harrison, E.M.; Green, C.A.; Hardwick, H.E.; Pius, R.; Norman, L.; Holden, K.A.; Read, J.M.; Dondelinger, F.; Carson, G.; et al. Features of 20 133 UK patients in hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: Prospective observational cohort study. BMJ 2020, 369, m1985. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Morales, A.J.; Cardona-Ospina, J.A.; Gutiérrez-Ocampo, E.; Villamizar-Peña, R.; Holguin-Rivera, Y.; Escalera-Antezana, J.P.; Alvarado-Arnez, L.E.; Bonilla-Aldana, D.K.; Franco-Paredes, C.; Henao-Martinez, A.F.; et al. Clinical, laboratory and imaging features of COVID-19: A systematic review and meta-analysis. Travel Med. Infect. Dis. 2020, 34, 101623. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; Cohen, S.L.; et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- Liu, W.; Tao, Z.W.; Wang, L.; Yuan, M.L.; Liu, K.; Zhou, L.; Wei, S.; Deng, Y.; Liu, J.; Liu, H.G.; et al. Analysis of factors associated with disease outcomes in hospitalized patients with 2019 novel coronavirus disease. Chin. Med. J. 2020, 133, 1032–1038. [Google Scholar] [CrossRef]
- Wang, P.; Sha, J.; Meng, M.; Wang, C.; Yao, Q.; Zhang, Z.; Sun, W.; Wang, X.; Qie, G.; Bai, X.; et al. Risk factors for severe COVID-19 in middle-aged patients without comorbidities: A multicentre retrospective study. J. Transl. Med. 2020, 18, 461. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; Asselta, R.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Cao, Y.; Li, L.; Feng, Z.; Wan, S.; Huang, P.; Sun, X.; Wen, F.; Huang, X.; Ning, G.; Wang, W. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Benetti, E.; Tita, R.; Spiga, O.; Ciolfi, A.; Birolo, G.; Bruselles, A.; Doddato, G.; Giliberti, A.; Marconi, C.; Musacchia, F.; et al. ACE2 gene variants may underlie interindividual variability and susceptibility to COVID-19 in the Italian population. Eur. J. Hum. Genet. 2020, 28, 1602–1614. [Google Scholar] [CrossRef]
- Srivastava, A.; Pandey, R.K.; Singh, P.P.; Kumar, P.; Rasalkar, A.A.; Tamang, R.; van Driem, G.; Shrivastava, P.; Chaubey, G. Most frequent South Asian haplotypes of ACE2 share identity by descent with East Eurasian populations. PLoS ONE 2020, 15, e0238255. [Google Scholar] [CrossRef] [PubMed]
- Zeberg, H.; Pääbo, S. A genomic region associated with protection against severe COVID-19 is inherited from Neandertals. Proc. Natl. Acad. Sci. USA 2021, 118, e2026309118. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.F.; Shastri, A.J.; Ye, C.; Weldon, C.H.; Filshtein-Sonmez, T.; Coker, D.; Symons, A.; Esparza-Gordillo, J.; The 23andMe COVID-19 Team; Aslibekyan, S.; et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 2021, 53, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Initiative, C.-H.G. Mapping the human genetic architecture of COVID-19. Nature 2021. [Google Scholar] [CrossRef]
- Roberts, G.H.L.; Park, D.S.; Coignet, M.V.; McCurdy, S.R.; Knight, S.C.; Partha, R.; Rhead, B.; Zhang, M.; Berkowitz, N.; Haug Baltzell, A.K.; et al. AncestryDNA COVID-19 Host Genetic Study Identifies Three Novel Loci. medRxiv 2020. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, S. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Source Softw. 2018. [Google Scholar] [CrossRef] [Green Version]
- Dayem Ullah, A.Z.; Oscanoa, J.; Wang, J.; Nagano, A.; Lemoine, N.R.; Chelala, C. SNVnexus: Assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Res. 2018, 46, W109–W113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haak, W.; Lazaridis, I.; Patterson, N.; Rohland, N.; Mallick, S.; Llamas, B.; Brandt, G.; Nordenfelt, S.; Harney, E.; Stewardson, K.; et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 2015, 522, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, N.; Moorjani, P.; Luo, Y.; Mallick, S.; Rohland, N.; Zhan, Y.; Genschoreck, T.; Webster, T.; Reich, D. Ancient admixture in human history. Genetics 2012, 192, 1065–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazaridis, I.; Nadel, D.; Rollefson, G.; Merrett, D.C.; Rohland, N.; Mallick, S.; Fernandes, D.; Novak, M.; Gamarra, B.; Sirak, K.; et al. Genomic insights into the origin of farming in the ancient Near East. Nature 2016, 536, 419–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourier, T.; Sadykov, M.; Carr, M.J.; Gonzalez, G.; Hall, W.W.; Pain, A. Host-directed editing of the SARS-CoV-2 genome. Biochem. Biophys. Res. Commun. 2021, 538, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Nevill, A.M.; Metsios, G.S. The need to redefine age- and gender-specific overweight and obese body mass index cutoff points. Nutr. Diabetes 2015, 5, e186. [Google Scholar] [CrossRef]
- Lewis, D.D.; Cropp, C.D. The Impact of African Ancestry on Prostate Cancer Disparities in the Era of Precision Medicine. Genes 2020, 11, 1471. [Google Scholar] [CrossRef]
- Kathiresan, S.; Srivastava, D. Genetics of human cardiovascular disease. Cell 2012, 148, 1242–1257. [Google Scholar] [CrossRef] [Green Version]
- Zekavat, S.M.; Ruotsalainen, S.; Handsaker, R.E.; Alver, M.; Bloom, J.; Poterba, T.; Seed, C.; Ernst, J.; Chaffin, M.; Engreitz, J.; et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat. Commun. 2018, 9, 2606. [Google Scholar] [CrossRef] [Green Version]
- Abdel Hameid, R.; Cormet-Boyaka, E.; Kuebler, W.M.; Uddin, M.; Berdiev, B.K. SARS-CoV-2 may hijack GPCR signaling pathways to dysregulate lung ion and fluid transport. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 320, L430–L435. [Google Scholar] [CrossRef]
- Birch, C.A.; Molinar-Inglis, O.; Trejo, J. Subcellular hot spots of GPCR signaling promote vascular inflammation. Curr. Opin. Endocr. Metab. Res. 2021, 16, 37–42. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Choi, E.Y.; Park, H.H.; Kim, H.; Kim, H.N.; Kim, I.; Jeon, S.; Kim, W.; Bae, J.S.; Lee, W. Wnt5a and Wnt11 as acute respiratory distress syndrome biomarkers for severe acute respiratory syndrome coronavirus 2 patients. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef]
- Ramasamy, S.; Subbian, S. Critical Determinants of Cytokine Storm and Type I Interferon Response in COVID-19 Pathogenesis. Clin. Microbiol. Rev. 2021, 34. [Google Scholar] [CrossRef] [PubMed]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodriguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- Wei, C.; Wan, L.; Yan, Q.; Wang, X.; Zhang, J.; Yang, X.; Zhang, Y.; Fan, C.; Li, D.; Deng, Y.; et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. 2020, 2, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.; Aziz, R.; Al Mahri, S.; Malik, S.S.; Haji, E.; Khan, A.H.; Khatlani, T.S.; Bouchama, A. Obesity and COVID-19: What makes obese host so vulnerable? Immun. Ageing 2021, 18. [Google Scholar] [CrossRef]
- Olalde, I.; Brace, S.; Allentoft, M.E.; Armit, I.; Kristiansen, K.; Booth, T.; Rohland, N.; Mallick, S.; Szécsényi-Nagy, A.; Mittnik, A.; et al. The Beaker phenomenon and the genomic transformation of northwest Europe. Nature 2018, 555, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Ivanisenko, V.A.; Anashkina, A.A.; Upadhyai, P. The story of the lost twins: Decoding the genetic identities of the Kumhar and Kurcha populations from the Indian subcontinent. BMC Genet. 2020, 21, 117. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
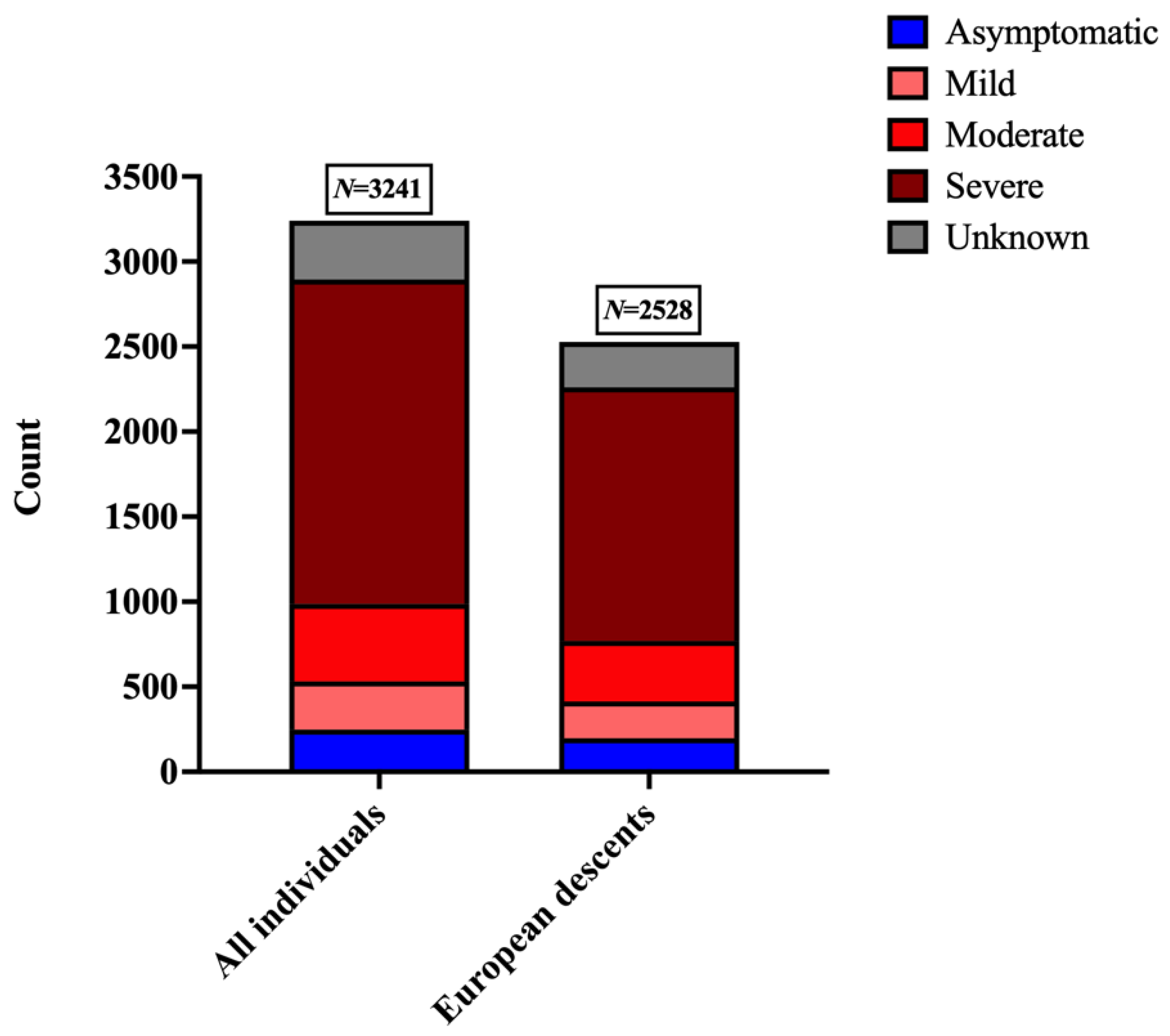
| Category | Criteria |
|---|---|
| Asymptomatic | individuals reported experiencing no perceptible symptoms |
| Mild | (a) All symptoms were reported as mild (b) One of the symptoms was reported as moderate, and the rest were mild |
| Moderate | (a) Two or more symptoms were reported as moderate (b) One of the symptoms was reported as severe,* and the rest are listed as moderate |
| Severe | (a) Two or more severe symptoms reported (b) Pneumonia |
| Unknown | symptom responses are unavailable |
| Patient Category | WHG Ancestry | ANE Ancestry | Neolithic Iranian Ancestry |
|---|---|---|---|
| Asymptomatic | 22.9% | 28.9% | 48.2% |
| Mild | 22.5% | 28.7% | 48.7% |
| Moderate | 23.5% | 27.9% | 48.6% |
| Severe | 24.0% | 27.1% | 48.9% |
| Patient Category | WHG Ancestry | EHG Ancestry | Neolithic Iranian Ancestry |
|---|---|---|---|
| Asymptomatic | 20.4% | 22.2% | 57.4% |
| Mild | 19.6% | 22.2% | 58.3% |
| Moderate | 20.9% | 21.5% | 57.7% |
| Severe | 21.5% | 21.0% | 57.5% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Upadhyai, P.; Suresh, G.; Parit, R.; Das, R. Genomic and Ancestral Variation Underlies the Severity of COVID-19 Clinical Manifestation in Individuals of European Descent. Life 2021, 11, 921. https://0-doi-org.brum.beds.ac.uk/10.3390/life11090921
Upadhyai P, Suresh G, Parit R, Das R. Genomic and Ancestral Variation Underlies the Severity of COVID-19 Clinical Manifestation in Individuals of European Descent. Life. 2021; 11(9):921. https://0-doi-org.brum.beds.ac.uk/10.3390/life11090921
Chicago/Turabian StyleUpadhyai, Priyanka, Gokul Suresh, Rahul Parit, and Ranajit Das. 2021. "Genomic and Ancestral Variation Underlies the Severity of COVID-19 Clinical Manifestation in Individuals of European Descent" Life 11, no. 9: 921. https://0-doi-org.brum.beds.ac.uk/10.3390/life11090921