Characterization of Structural and Energetic Differences between Conformations of the SARS-CoV-2 Spike Protein

 ,
,  ,
,  , and
, and 
Abstract
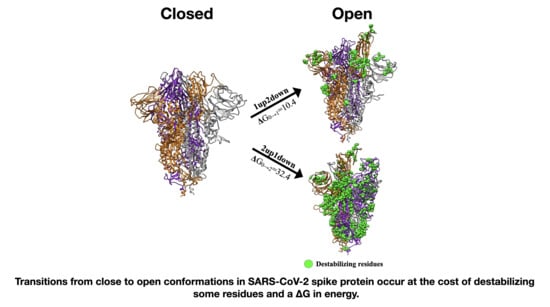
:
1. Introduction
2. Materials and Methods
2.1. Modeling of the Full SARS-CoV-2 Spike Structures
2.2. All-Atom MD of the SARS-CoV-2 Spike and Its Conformations
2.3. Differential Contact Map (dCM) Analysis
2.4. Poisson Boltzmann Calculations for the Spike Protein Energetic Characterization
- Coronavirus spike protein structures (Ω1).
- External (Ω2), representing the solver.
(∇2 − κ22) ϕ2 = 0
ϕ1 = ϕ2
∈1(∂ϕ1/∂n) = ∈2(∂ϕ2/∂n)
3. Results and Discussion
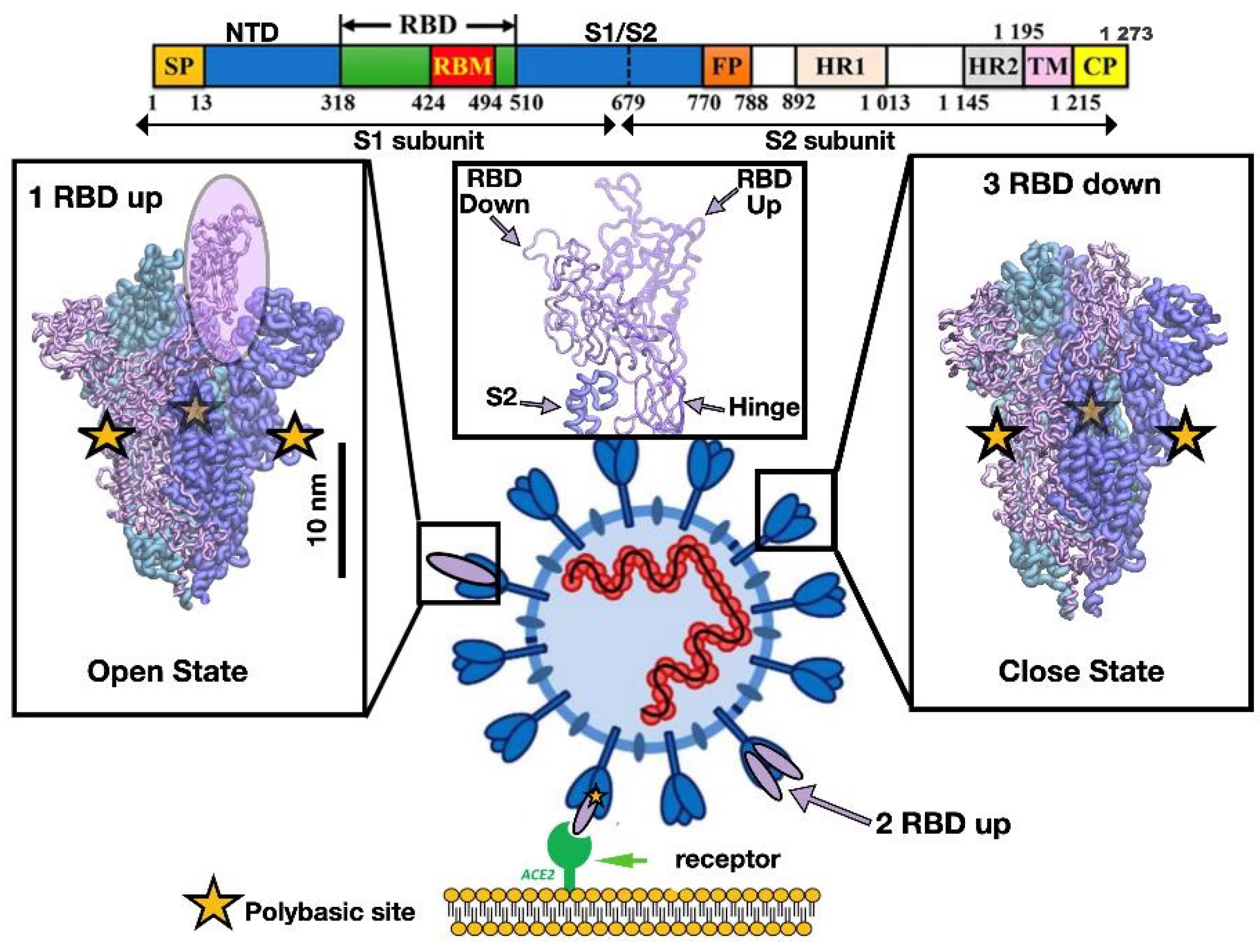
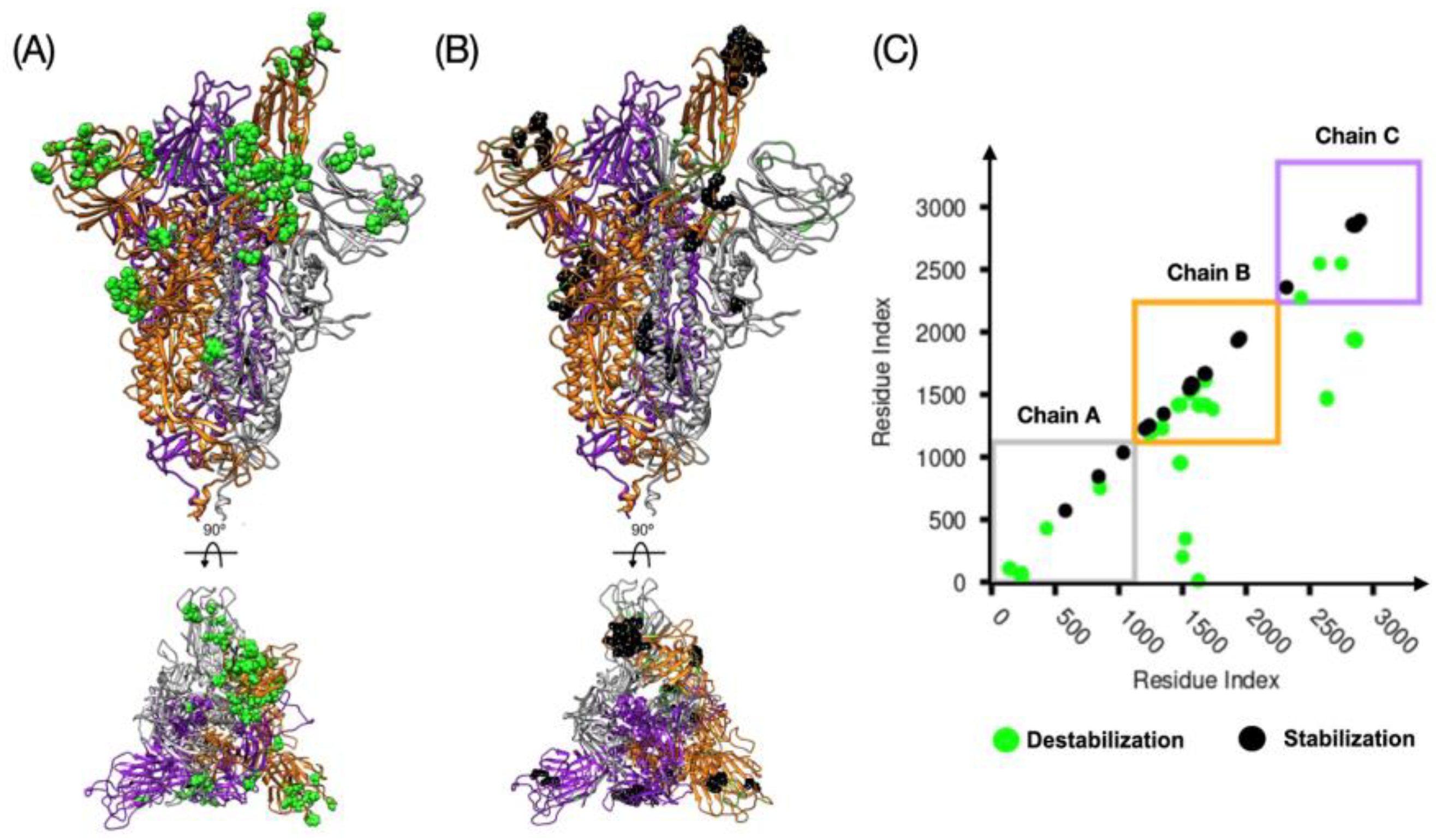
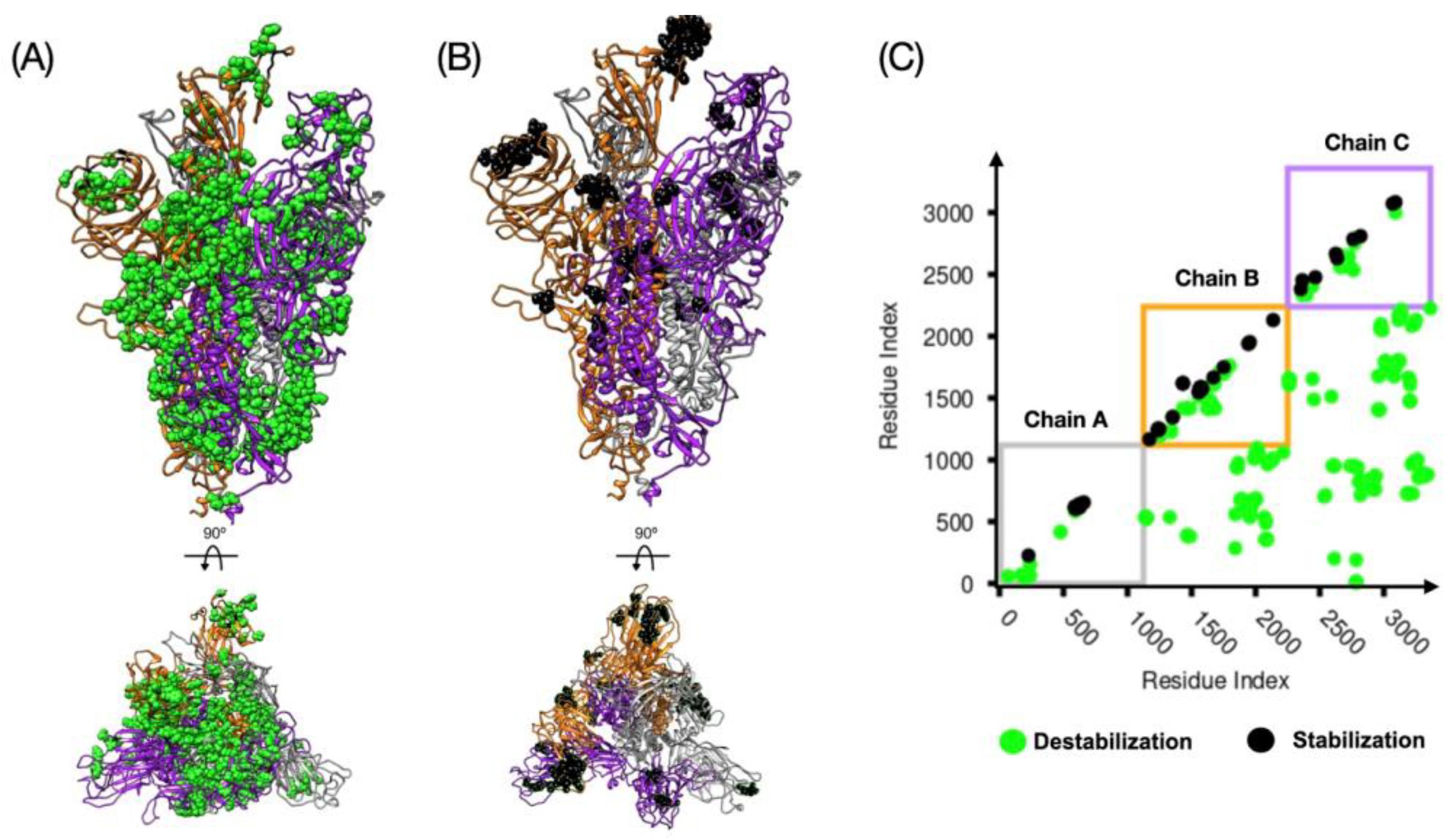
3.1. Relative Stability Analysis of the SARS-CoV-2 Spike Protein Conformation
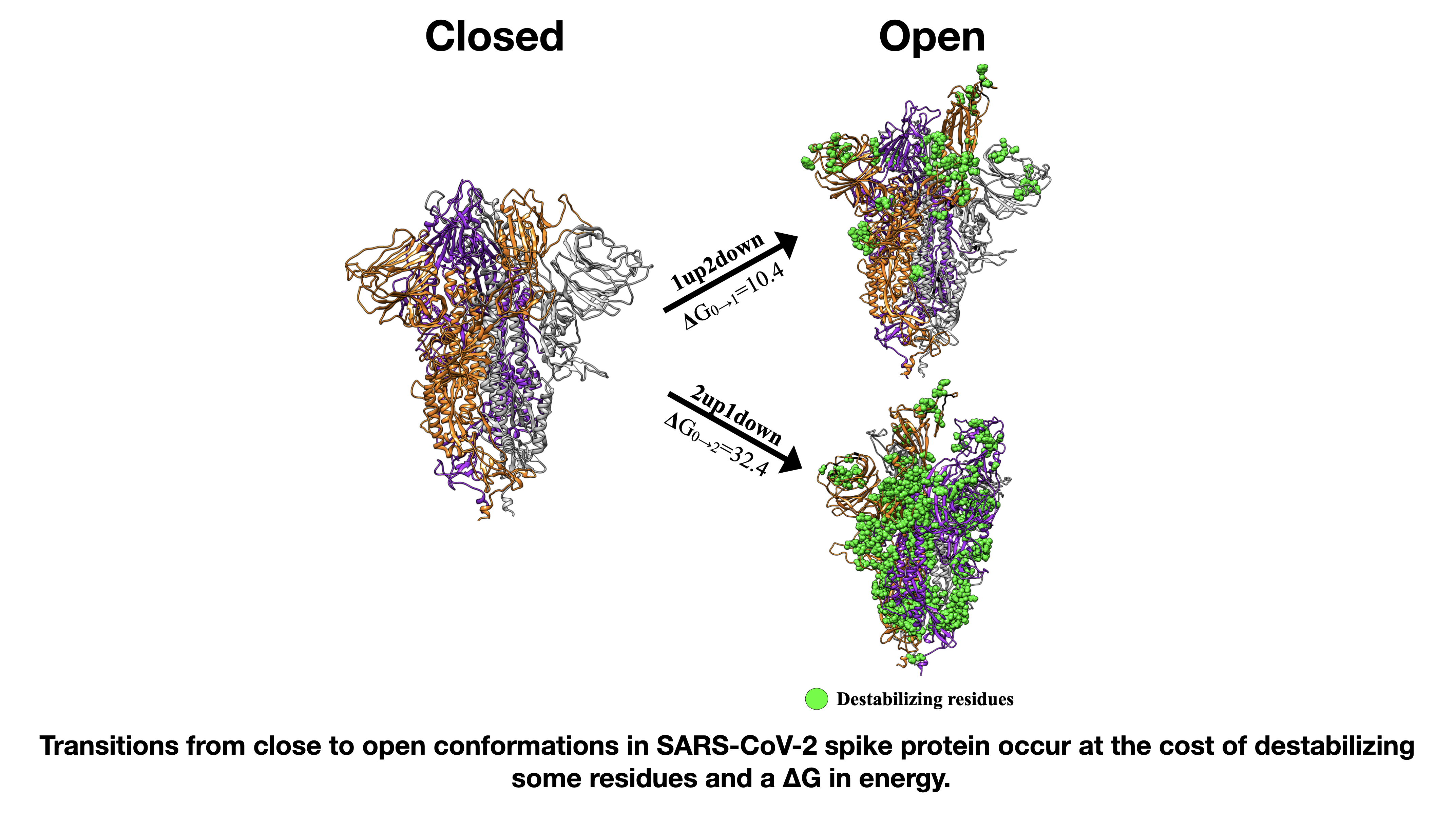
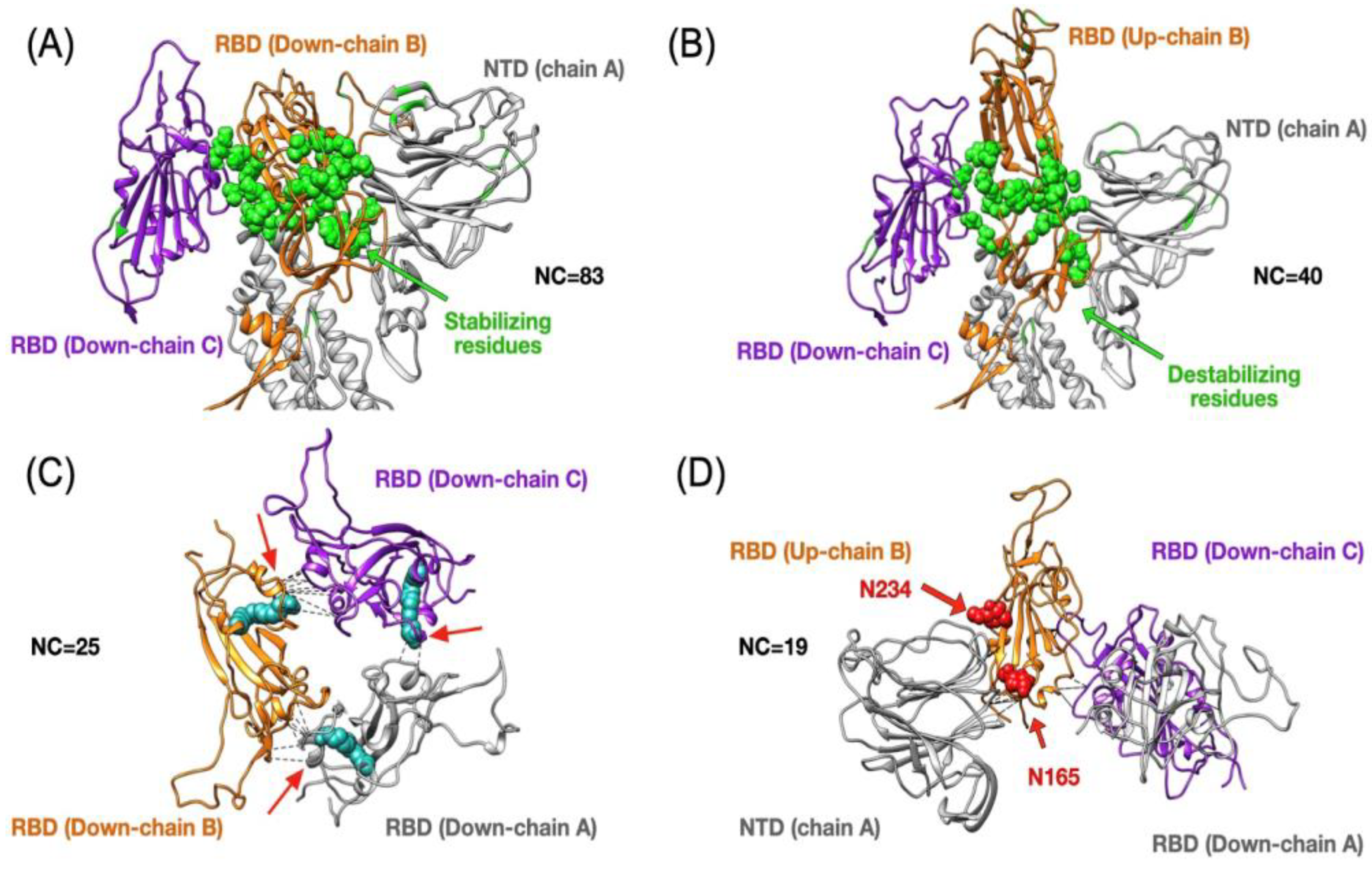
3.2. Insight into the RBD SARS-CoV-2 Conformations
3.3. Energetic Calculations of the Spike Protein Conformations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cherry, J.D. The chronology of the 2002–2003 SARS mini pandemic. Paediatr. Respir. Rev. 2004, 5, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Standl, F.; Jöckel, K.-H.; Brune, B.; Schmidt, B.; Stang, A. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Baldwin, R.; Weder di Mauro, B. Economics in the Time of COVID-19; VOX, CEPR Policy Portal: London, UK, 2020; ISBN 9781912179282. [Google Scholar]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.-C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, A.S.F.; Ibarra, A.A.; Bermudez, I.; Casalino, L.; Gaieb, Z.; Shoemark, D.K.; Gallagher, T.; Sessions, R.B.; Amaro, R.E.; Mulholland, A.J. Simulations support the interaction of the SARS-CoV-2 spike protein with nicotinic acetylcholine receptors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hou, Y.; Zhao, J.; Martin, W.; Kallianpur, A.; Chung, M.K.; Jehi, L.; Sharifi, N.; Erzurum, S.; Eng, C.; Cheng, F. New insights into genetic susceptibility of COVID-19: An ACE2 and TMPRSS2 polymorphism analysis. BMC Med. 2020, 18, 216. [Google Scholar] [CrossRef]
- Corrêa Giron, C.; Laaksonen, A.; Barroso da Silva, F.L. On the interactions of the receptor-binding domain of SARS-CoV-1 and SARS-CoV-2 spike proteins with monoclonal antibodies and the receptor ACE2. Virus Res. 2020, 285, 198021. [Google Scholar] [CrossRef]
- Xia, S.; Lan, Q.; Su, S.; Wang, X.; Xu, W.; Liu, Z.; Zhu, Y.; Wang, Q.; Lu, L.; Jiang, S. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct. Target. Ther. 2020, 5, 92. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Brielle, E.S.; Schneidman-Duhovny, D.; Linial, M. The SARS-CoV-2 Exerts a Distinctive Strategy for Interacting with the ACE2 Human Receptor. Viruses 2020, 12, 497. [Google Scholar] [CrossRef] [PubMed]
- Moreira, R.A.; Chwastyk, M.; Baker, J.L.; Guzman, H.V.; Poma, A.B. Quantitative determination of mechanical stability in the novel coronavirus spike protein. Nanoscale 2020, 12, 16409–16413. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.S.; Gruber, S.; Milles, L.F.; Nicolaus, T.; Schendel, L.C.; Gaub, H.E.; Lipfert, J. A Tethered Ligand Assay to Probe the SARS-CoV-2 ACE2 Interaction under Constant Force. bioRxiv 2020. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020. [Google Scholar] [CrossRef]
- Qiao, B.; Olvera de la Cruz, M. Enhanced Binding of SARS-CoV-2 Spike Protein to Receptor by Distal Polybasic Cleavage Sites. ACS Nano 2020, 14, 10616–10623. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Rangarajan, E.S.; Izard, T.; Farzan, M.; Choe, H. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Turoňová, B.; Sikora, M.; Schürmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals lexibility mediated by three hinges. Science 2020. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Kavanagh Williamson, M.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020. [Google Scholar] [CrossRef]
- Watanabe, Y.; Bowden, T.A.; Wilson, I.A.; Crispin, M. Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1480–1497. [Google Scholar] [CrossRef]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Amanat, F.; Krammer, F. SARS-CoV-2 Vaccines: Status Report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, W.; Drabek, D.; Okba, N.M.A.; van Haperen, R.; Osterhaus, A.D.M.E.; van Kuppeveld, F.J.M.; Haagmans, B.L.; Grosveld, F.; Bosch, B.-J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 2020, 11, 2251. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef]
- Chikhale, R.V.; Gurav, S.S.; Patil, R.B.; Sinha, S.K.; Prasad, S.K.; Shakya, A.; Shrivastava, S.K.; Gurav, N.S.; Prasad, R.S. Sars-cov-2 host entry and replication inhibitors from Indian ginseng: An approach. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Sarah, R.; Tabassum, B.; Idrees, N.; Hussain, M.K. Bio-active Compounds Isolated from Neem Tree and Their Applications. In Natural Bio-Active Compounds; Akhtar, M.S., Swamy, M.K., Sinniah, U.R., Eds.; Springer: Singapore, 2019; Volume 46, pp. 509–528. ISBN 9789811371530. [Google Scholar]
- Sasidharan, S.; Selvaraj, C.; Singh, S.K.; Dubey, V.K.; Kumar, S.; Fialho, A.M.; Saudagar, P. Bacterial protein azurin and derived peptides as potential anti-SARS-CoV-2 agents: Insights from molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Chowdhury, P. In silico investigation of phytoconstituents from Indian medicinal herb “(giloy)” against SARS-CoV-2 (COVID-19) by molecular dynamics approach. J. Biomol. Struct. Dyn. 2020, 1–18. [Google Scholar] [CrossRef]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-time conformational dynamics of SARS-CoV-2 spikes on virus particles. bioRxiv 2020. [Google Scholar] [CrossRef]
- Roy, S.; Jaiswar, A.; Sarkar, R. Dynamic Asymmetry Exposes 2019-nCoV Prefusion Spike. J. Phys. Chem. Lett. 2020, 11, 7021–7027. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef]
- Vay, J.L.; Fawley, W. AMBER User’s Manual; Technical Report; U.S. Department of Energy: Columbia, DC, USA, 2000. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef]
- Moreira, R.A.; Guzman, H.V.; Boopathi, S.; Baker, J.L.; Poma, A.B. Characterization of structural and energetic differences between conformations of the SARS-CoV-2 spike protein. Snapshots, frequency contact maps analysis, Poisson Boltzmann calculations, and data scripts. bioRxiv 2020. [Google Scholar] [CrossRef]
- Poma, A.B.; Li, M.S.; Theodorakis, P.E. Generalization of the elastic network model for the study of large conformational changes in biomolecules. Phys. Chem. Chem. Phys. 2018, 20, 17020–17028. [Google Scholar] [CrossRef] [Green Version]
- Senapati, S.; Poma, A.B.; Cieplak, M.; Filipek, S.; Park, P.S.H. Differentiating between Inactive and Active States of Rhodopsin by Atomic Force Microscopy in Native Membranes. Anal. Chem. 2019, 1, 7226–7235. [Google Scholar] [CrossRef]
- Chwastyk, M.; Bernaola, A.P.; Cieplak, M. Statistical radii associated with amino acids to determine the contact map: Fixing the structure of a type I cohesin domain in the Clostridium thermocellum cellulosome. Phys. Biol. 2015, 12, 046002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poma, A.B.; Guzman, H.V.; Li, M.S.; Theodorakis, P.E. Mechanical and thermodynamic properties of Aβ, Aβ, and α-synuclein fibrils: A coarse-grained method to complement experimental studies. Beilstein J. Nanotechnol. 2019, 10, 500–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wołek, K.; Gómez-Sicilia, À.; Cieplak, M. Determination of contact maps in proteins: A combination of structural and chemical approaches. J. Chem. Phys. 2015, 143, 243105. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Gelain, F. Probing mechanical properties and failure mechanisms of fibrils of self-assembling peptides. Nanoscale Adv. 2020, 2, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Liguori, N.; Croce, R.; Marrink, S.J.; Thallmair, S. Molecular dynamics simulations in photoynthesis. Photosynth. Res. 2020, 144, 273–295. [Google Scholar] [CrossRef] [Green Version]
- Makshakova, O.; Breton, C.; Perez, S. Unraveling the complex enzymatic machinery making a key galactolipid in chloroplast membrane: A multiscale computer simulation. Sci. Rep. 2020, 10, 13514. [Google Scholar] [CrossRef]
- Götz, A.; Mylonas, N.; Högel, P.; Silber, M.; Heinel, H.; Menig, S.; Vogel, A.; Feyrer, H.; Huster, D.; Luy, B.; et al. Modulating Hinge Flexibility in the APP Transmembrane Domain Alters γ-Secretase Cleavage. Biophys. J. 2019, 116, 2103–2120. [Google Scholar] [CrossRef]
- Gur, M.; Taka, E.; Yilmaz, S.Z.; Kilinc, C.; Aktas, U.; Golcuk, M. Conformational transition of SARS-CoV-2 spike glycoprotein between its closed and open states. J. Chem. Phys. 2020, 153, 075101. [Google Scholar] [CrossRef]
- Jarzynski, C. Nonequilibrium Equality for Free Energy Differences. Phys. Rev. Lett. 1997, 78, 2690–2693. [Google Scholar] [CrossRef] [Green Version]
- Martínez, M.; Cooper, C.D.; Poma, A.B.; Guzman, H.V. Free Energies of the Disassembly of Viral Capsids from a Multiscale Molecular Simulation Approach. J. Chem. Inf. Model. 2020, 60, 974–981. [Google Scholar] [CrossRef] [Green Version]
- Armacost, K.A.; Riniker, S.; Cournia, Z. Novel Directions in Free Energy Methods and Applications. J. Chem. Inf. Model. 2020, 60, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.B.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry and Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spike | dCM | Chain A | Chain B | Chain C | |||
|---|---|---|---|---|---|---|---|
| 1up2down | 3down | 24 (9) | 6(3) | 54 (26) | 32 (24) | 14 (3) | 14 (11) |
| 2up1down | 3down | 156 (18) | 18(9) | 196 (61) | 47 (32) | 150 (10) | 17 (13) |
| 2up1down | 1up2down | 122 (15) | 12(6) | 111 (18) | 6 (3) | 110 (2) | 4 (2) |
| Spike Conformation “C” | Spike Conformation Reference “R” | Δ(C-R) PB Energies (kcal/mol) |
|---|---|---|
| 1up2down | 3down | 10.4 |
| 2up1down | 3down | 32.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, R.A.; Guzman, H.V.; Boopathi, S.; Baker, J.L.; Poma, A.B. Characterization of Structural and Energetic Differences between Conformations of the SARS-CoV-2 Spike Protein. Materials 2020, 13, 5362. https://0-doi-org.brum.beds.ac.uk/10.3390/ma13235362
Moreira RA, Guzman HV, Boopathi S, Baker JL, Poma AB. Characterization of Structural and Energetic Differences between Conformations of the SARS-CoV-2 Spike Protein. Materials. 2020; 13(23):5362. https://0-doi-org.brum.beds.ac.uk/10.3390/ma13235362
Chicago/Turabian StyleMoreira, Rodrigo A., Horacio V. Guzman, Subramanian Boopathi, Joseph L. Baker, and Adolfo B. Poma. 2020. "Characterization of Structural and Energetic Differences between Conformations of the SARS-CoV-2 Spike Protein" Materials 13, no. 23: 5362. https://0-doi-org.brum.beds.ac.uk/10.3390/ma13235362