
Low Temperature Synthesis and Characterization of AlScMo3O12


Abstract
:
1. Introduction

2. Experimental Section
3. Results and Discussion
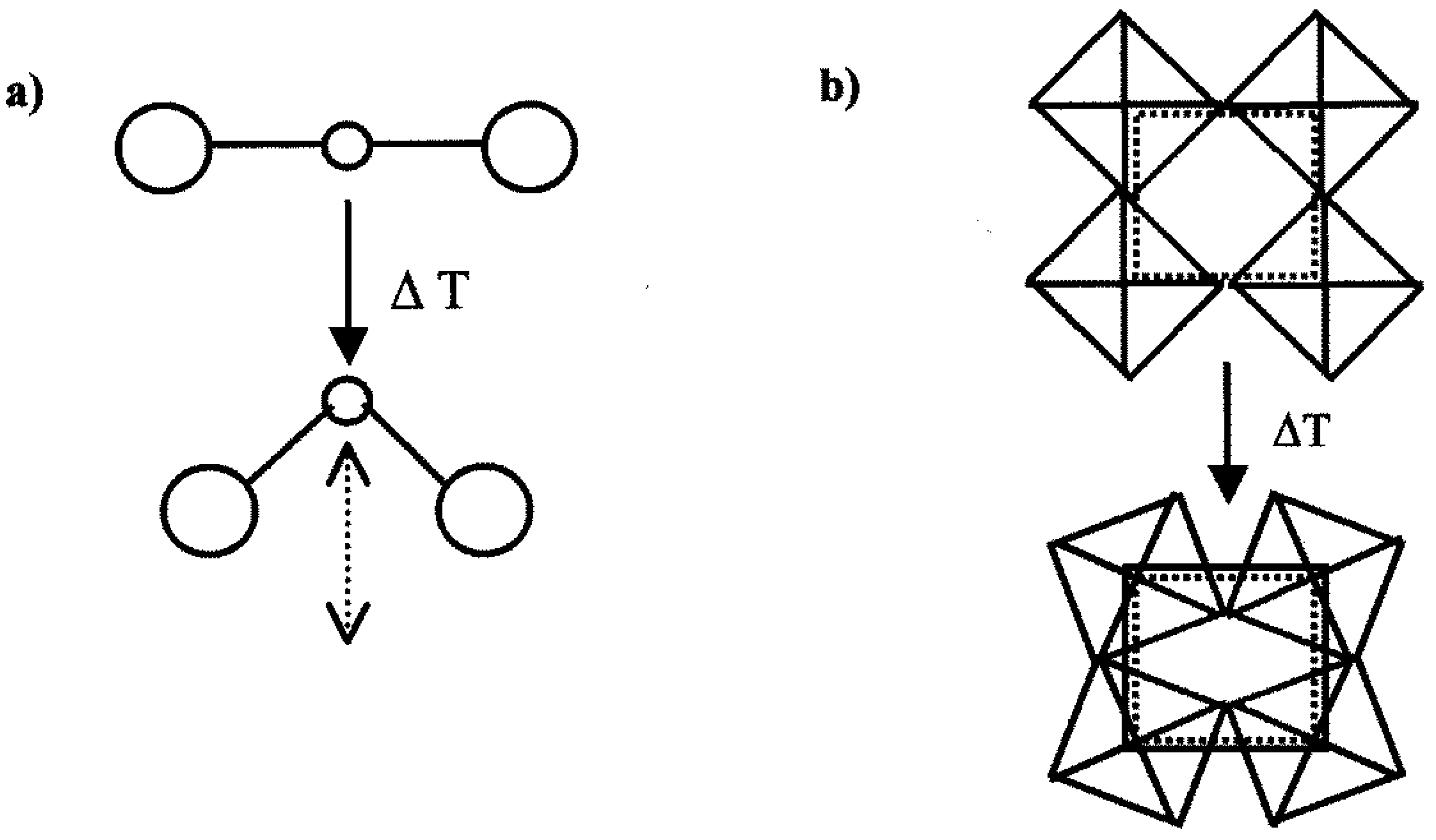
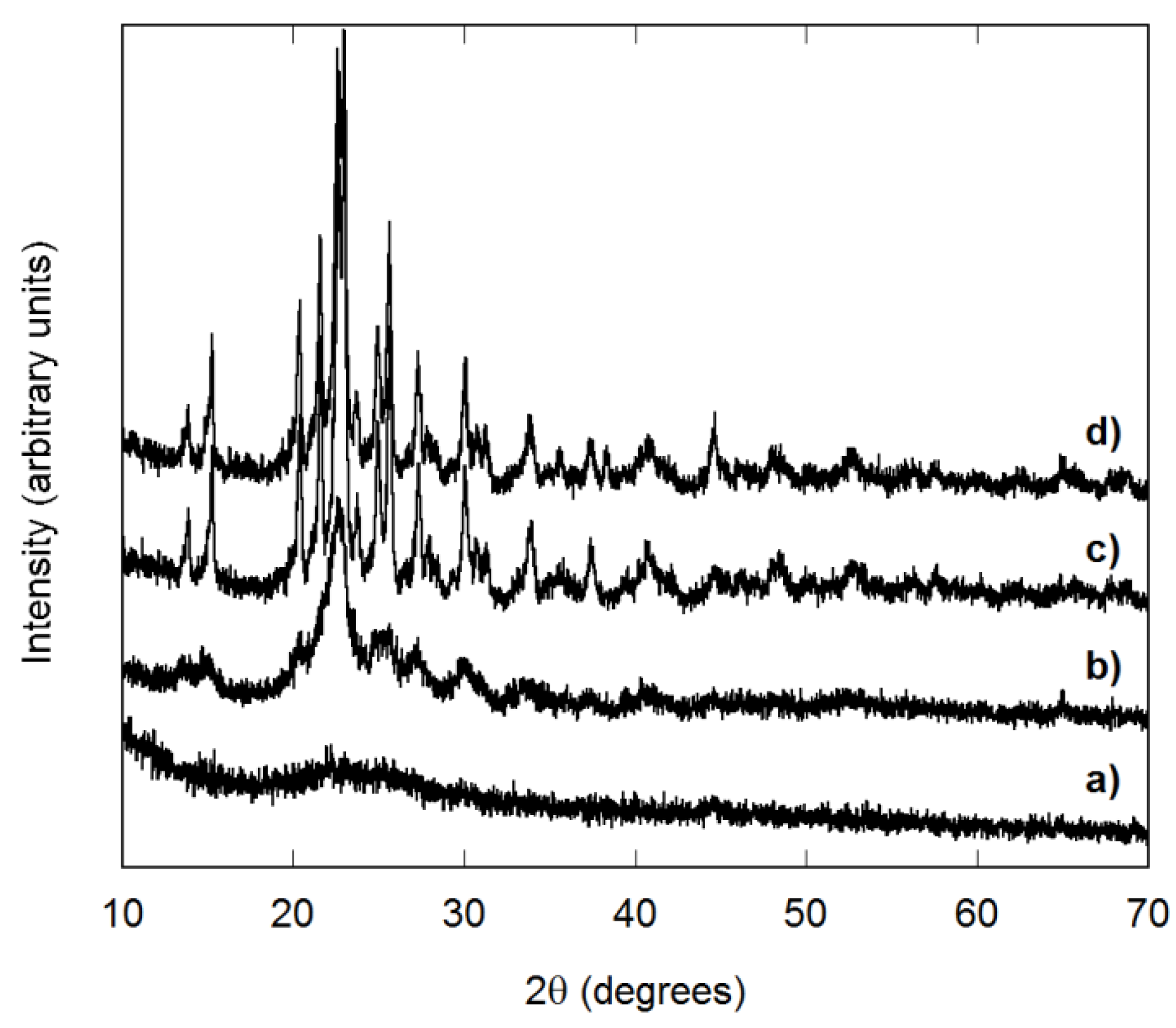
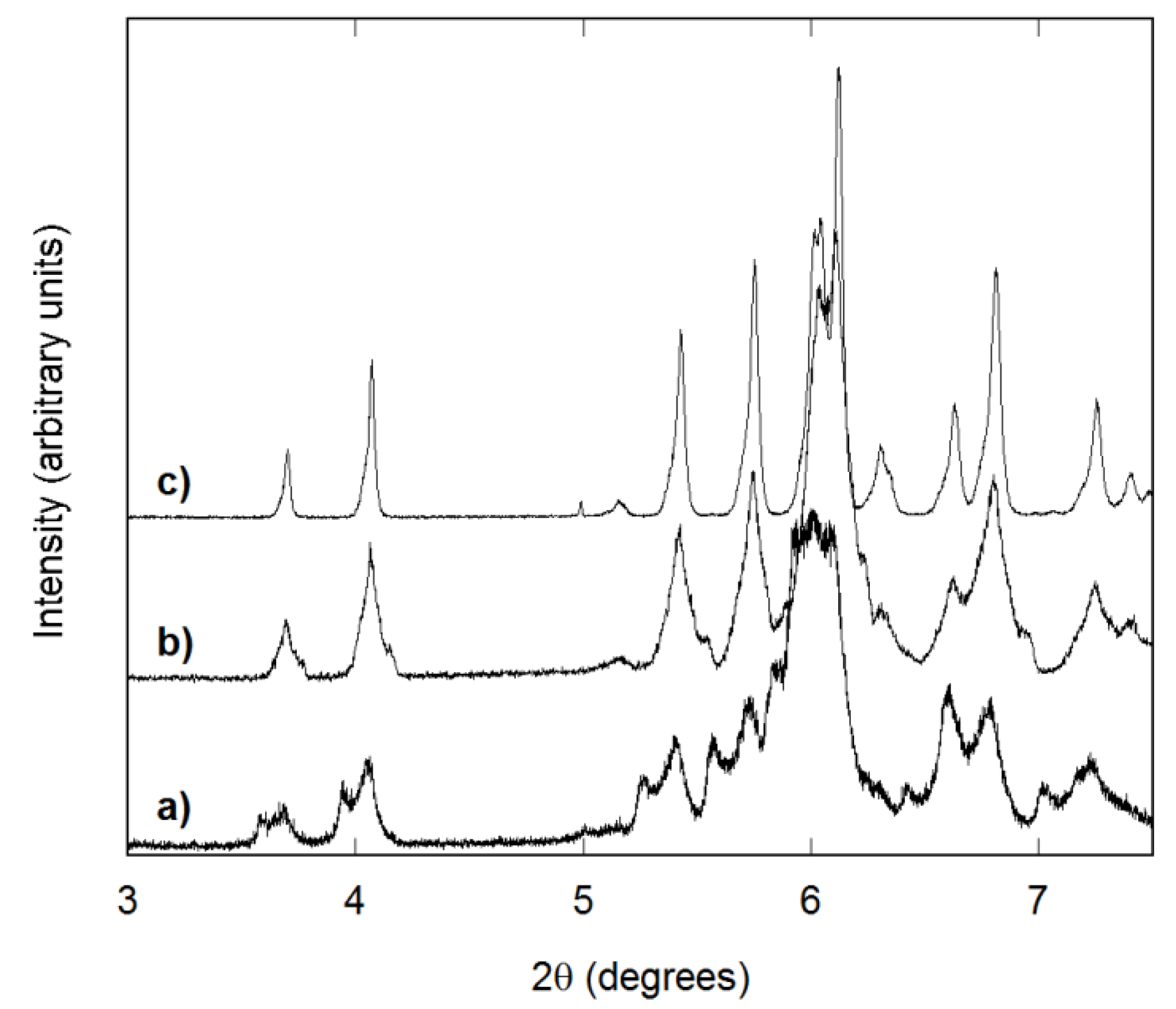
3.1. Reaction Completeness and Crystallization Behavior

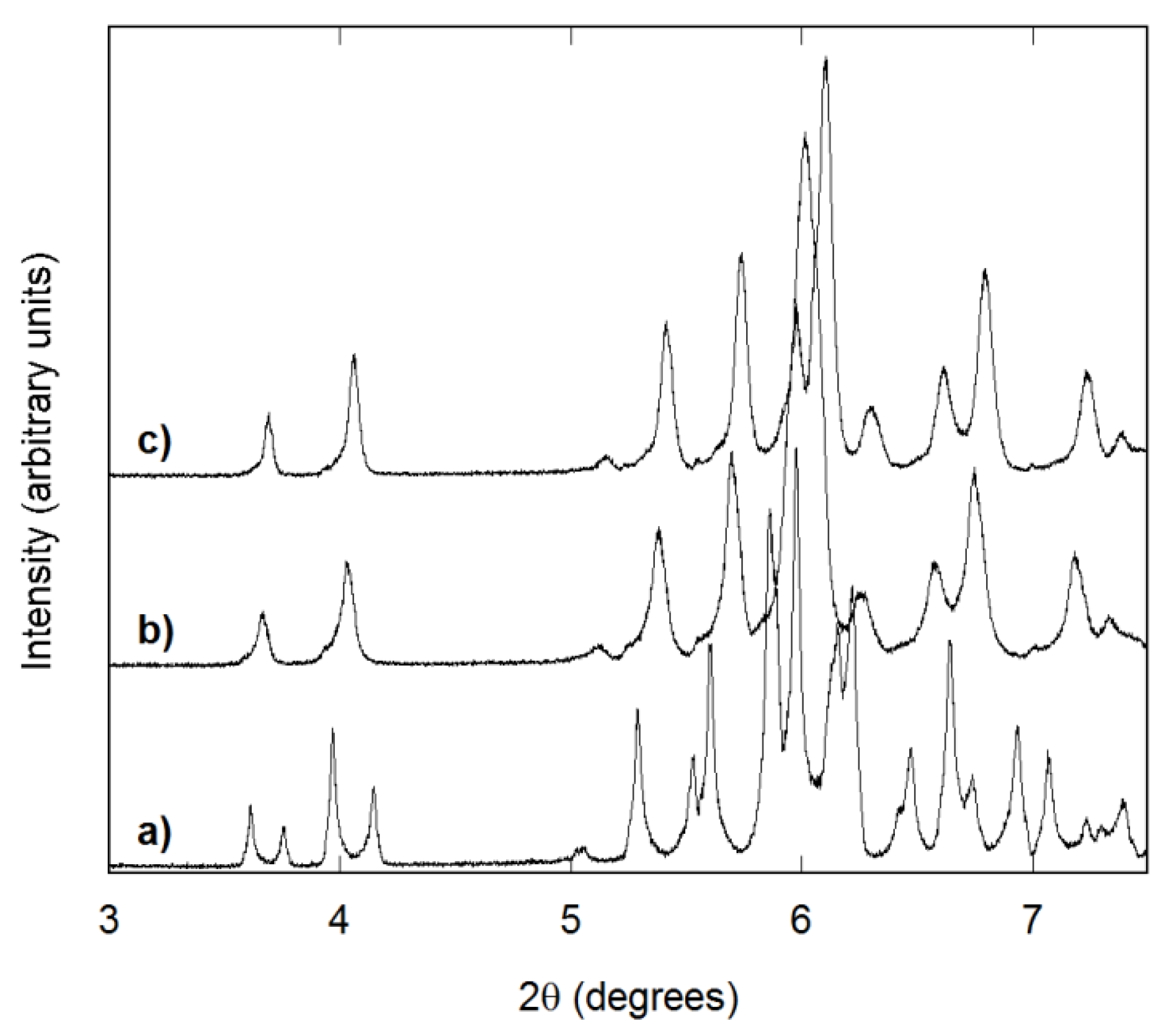
3.2. Structure and Homogeneity of Samples



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tsynth (°C) | Solvent | Vsolv (mL) | tsynth (days) | # of phases | Space group | Phase fraction | V (Å3) | %Sc Vegard |
|---|---|---|---|---|---|---|---|---|
| 130 | CH3CN | 10 | 7 | 4+ | Pnca * | 11% | 1022.11 | 4 |
| 31% | 1088.09 | 36 | ||||||
| 27% | 1123.28 | 54 | ||||||
| 31% | 1153.42 | 68 | ||||||
| 130 | CH3CN | 10 | 17 | 2 | Pnca | 89% | 1124.95 | 54 |
| Pnca | 11% | 1184.49 | 84 | |||||
| 130 | CH3CN | 10 | 27 | 2 | Pnca | 86% | 1107.58 | 46 |
| Pnca | 14% | 1154.62 | 69 | |||||
| 150 | CH3CN | 9 | 14 | 2 | Pnca | 74% | 1125.15 | 54 |
| Pnca | 26% | 1173.71 | 78 | |||||
| 170 | CH3CN | 9 | 7 | 1 | Pnca | 100% | 1115.99 | 50 |
| 170 | CH3CN | 9 | 7 | 2 | Pnca | 56% | 1075.86 | 30 |
| Pnca | 44% | 1131.80 | 58 | |||||
| 110 | CHCl3 | 5 | 3 | 2 | Pnca | 71% | 1112.73 | 48 |
| Pnca | 29% | 1200.11 | 91 | |||||
| 110 | CHCl3 | 10 | 7 | 1 | Pnca | 100% | 1100.98 | 43 |
| 110 | CHCl3 | 15 | 12 | 1 | Pnca | 100% | 1102.11 | 43 |
| 130 | CHCl3 | 15 | 7 | 2 | P 21/c | 6% | 2060.65 | 8 |
| Pnca | 94% | 1121.07 | 52 |

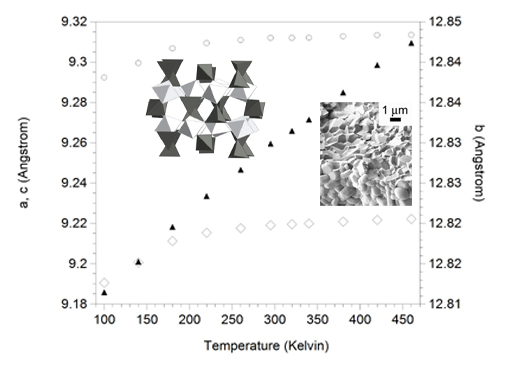
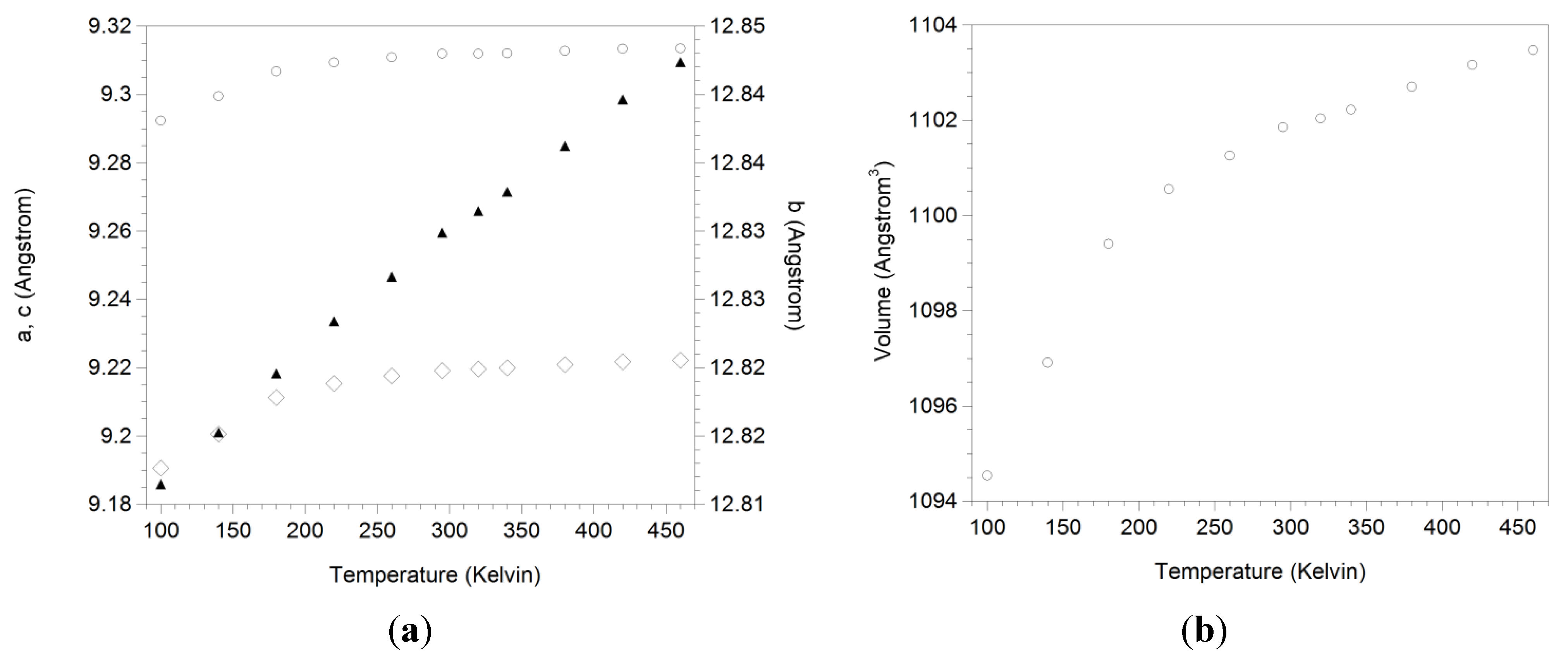
3.3. Expansion Behavior of AlScMo3O12
| T (K) | Rp (%) | a (Å) | b (Å) | c (Å) | β (°) | V (Å3) | %Sc |
|---|---|---|---|---|---|---|---|
| 100 | 11.7 | 15.771 | 9.291 | 18.382 | 125.64 | 2189.04 | 46.4 |
| 100 | 9.2 | 9.292 | 12.816 | 9.191 | 90.0 | 1094.54 | 51.6 |
| 140 | 9.1 | 9.299 | 12.820 | 9.201 | 90.0 | 1096.91 | 50.9 |
| 180 | 8.7 | 9.307 | 12.825 | 9.211 | 90.0 | 1099.40 | 50.7 |
| 220 | 8.4 | 9.309 | 12.828 | 9.215 | 90.0 | 1100.55 | 50.6 |
| 260 | 8.6 | 9.311 | 12.832 | 9.218 | 90.0 | 1101.26 | 50.5 |
| 295 | 8.5 | 9.312 | 12.835 | 9.219 | 90.0 | 1101.85 | 50.4 |
| 320 | 8.7 | 9.312 | 12.836 | 9.220 | 90.0 | 1102.04 | 50.6 |
| 340 | 8.8 | 9.312 | 12.838 | 9.220 | 90.0 | 1102.22 | 50.7 |
| 380 | 9.0 | 9.313 | 12.841 | 9.221 | 90.0 | 1102.70 | 50.5 |
| 420 | 9.0 | 9.313 | 12.845 | 9.222 | 90.0 | 1103.16 | 52.0 |
| 460 | 9.1 | 9.313 | 12.847 | 9.222 | 90.0 | 1103.47 | 50.4 |

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Takenaka, K. Negative thermal expansion materials: Technological key for control of thermal expansion. Sci. Technol. Adv. Mater. 2012, 13, 013001:1–013001:11. [Google Scholar] [CrossRef]
- Roy, R.; Agrawal, D.K.; McKinstry, H.A. Very low thermal expansion coefficient materials. Annu. Rev. Mater. Sci. 1989, 19, 59–81. [Google Scholar] [CrossRef]
- Lommens, P.; de Meyer, C.; Bruneel, E.; de Buysser, K.; van Driessche, I.; Hoste, S. Synthesis and thermal expansion of ZrO2/ZrW2O8 composites. J. Eur. Ceram. Soc. 2005, 25, 3605–3610. [Google Scholar] [CrossRef]
- Tani, J.-I.; Takahashi, M.; Kido, H. Fabrication and thermal expansion properties of ZrW2O8/Zr2WP2O12 composites. J. Eur. Ceram. Soc. 2010, 30, 1483–1488. [Google Scholar] [CrossRef]
- Phillips, A.E.; Goodwin, A.L.; Halder, G.J.; Southon, P.D.; Kepert, C.J. Nanoporosity and exceptional negative thermal expansion in single-network cadmium cyanide. Angew. Chem. 2008, 47, 1396–1399. [Google Scholar] [CrossRef] [Green Version]
- Sleight, A.W. Isotropic negative thermal expansion. Annu. Rev. Mater. Sci. 1998, 28, 29–43. [Google Scholar] [CrossRef]
- Attfield, M.P.; Sleight, A.W. Strong negative thermal expansion in siliceous faujasite. Chem. Commun. 1998, 5, 601–602. [Google Scholar] [CrossRef]
- Reisner, B.A.; Lee, Y.; Hanson, J.C.; Jones, G.A.; Parise, J.B.; Corbin, D.R.; Toby, B.H.; Freitag, A.; Larese, J.Z.; Kahlenberg, V. Understanding negative thermal expansion and “trap door” cation relocations in zeolite rho. Chem. Commun. 2000, 22, 2221–2222. [Google Scholar] [CrossRef]
- Attfield, M.P.; Sleight, A.W. Exceptional negative thermal expansion in AlPO4-17. Chem. Mater. 1998, 10, 2013–2019. [Google Scholar] [CrossRef]
- Chapman, K.W.; Chupas, P.J. Anomalous thermal expansion of cuprites: A combined high resolution pair distribution function and geometric analysis. Chem. Mater. 2009, 21, 425–431. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Mary, T.A.; Vogt, T.; Subramanian, M.A.; Sleight, A.W. Negative thermal expansion in ZrW2O8 and HfW2O8. Chem. Mater. 1996, 8, 2809–2823. [Google Scholar] [CrossRef]
- Korthuis, V.; Khosrovani, N.; Sleight, A.W.; Roberts, N.; Dupree, R.; Warren, W.W. Negative thermal expansion and phase transitions in the ZrV2−xPxO7 series. Chem. Mater. 1995, 7, 412–417. [Google Scholar] [CrossRef]
- Li, J.; Yokochi, A.; Amos, T.G.; Sleight, A.W. Strong negative thermal expansion along the O-Cu-O linkage in CuScO2. Chem. Mater. 2002, 14, 2602–2606. [Google Scholar] [CrossRef]
- Lind, C.; Wilkinson, A.P.; Hu, Z.B.; Short, S.; Jorgensen, J.D. Synthesis and properties of the negative thermal expansion material cubic ZrMo2O8. Chem. Mater. 1998, 10, 2335–2337. [Google Scholar] [CrossRef]
- Kozy, L.C.; Tahir, M.N.; Lind, C.; Tremel, W. Particle size and morphology control of the negative thermal expansion material cubic zirconium tungstate. J. Mater. Chem. 2009, 19, 2760–2765. [Google Scholar] [CrossRef]
- Lightfoot, P.; Woodcock, D.A.; Maple, M.J.; Villaescusa, L.A.; Wright, P.A. The widespread occurrence of negative thermal expansion in zeolites. J. Mater. Chem. 2001, 11, 212–216. [Google Scholar] [CrossRef]
- Chapman, K.W.; Chupas, P.J.; Kepert, C.J. Direct observation of a transverse vibrational mechanism for negative thermal expansion in Zn(CN)2: An atomic pair distribution function analysis. J. Am. Chem. Soc. 2005, 127, 15630–15636. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.S.O.; Hanson, P.A.; Ibberson, R.M.; Duan, N.; Kameswari, U.; Sleight, A.W. Low-temperature oxygen migration and negative thermal expansion in ZrW2−xMoxO8. J. Am. Chem. Soc. 2000, 122, 8694–8699. [Google Scholar] [CrossRef]
- Korcok, J.L.; Katz, M.J.; Leznoff, D.B. Impact of metallophilicity on “colossal” positive and negative thermal expansion in a series of isostructural dicyanometallate coordination polymers. J. Am. Chem. Soc. 2009, 131, 4866–4871. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.S.O. Negative thermal expansion materials. J. Chem. Soc. Dalton Trans. 1999, 19, 3317–3326. [Google Scholar] [CrossRef]
- Lind, C. Two decades of negative thermal expansion research: Where do we stand? Materials 2012, 5, 1125–1154. [Google Scholar] [CrossRef]
- Ernst, G.; Broholm, C.; Kowach, G.R.; Ramirez, A.P. Phonon density of states and negative thermal expansion in ZrW2O8. Nature 1998, 396, 147–149. [Google Scholar] [CrossRef]
- Azuma, M.; Chen, W.-T.; Seki, H.; Czapski, M.; Olga, S.; Oka, K.; Mizumaki, M.; Watanuki, T.; Ishimatsu, N.; Kawamura, N.; et al. Colossal negative thermal expansion in BiNiO3 induced by intermetallic charge transfer. Nat. Commun. 2011, 2, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.P.; Kieffer, J. Structural origin of negative thermal expansion in high-temperature silica polymorphs. Phys. Rev. Lett. 2005, 95, 215901. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Chaplot, S.L.; Schober, H.; Mary, T.A. Origin of negative thermal expansion in cubic ZrW2O8 revealed by high pressure inelastic neutron scattering. Phys. Rev. Lett. 2001, 86, 4692–4695. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.P.; Kowach, G.R. Large low temperature specific heat in the negative thermal expansion compound ZrW2O8. Phys. Rev. Lett. 1998, 80, 4903–4906. [Google Scholar] [CrossRef]
- Tucker, M.G.; Goodwin, A.L.; Dove, M.T.; Keen, D.A.; Wells, S.A.; Evans, J.S.O. Negative thermal expansion in ZrW2O8: Mechanisms, rigid unit modes, and neutron total scattering. Phys. Rev. Lett. 2005, 95, 255501:1–255501:4. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Hu, Z.; Jorgensen, J.D.; Argyriou, D.N.; Short, S.; Sleight, A.W. Compressibility, phase transitions, and oxygen migration in zirconium tungstate, ZrW2O8. Science 1997, 275, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, A.L.; Calleja, M.; Conterio, M.J.; Dove, M.T.; Evans, J.S.O.; Keen, D.A.; Peters, L.; Tucker, M.G. Colossal positive and negative thermal expansion in the framework material Ag3[Co(CN)6]. Science 2008, 319, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Mary, T.A.; Evans, J.S.O.; Vogt, T.; Sleight, A.W. Negative thermal expansion from 0.3 to 1050 Kelvin in ZrW2O8. Science 1996, 272, 90–92. [Google Scholar] [CrossRef]
- Perottoni, C.A.; da Jornada, J.A.H. Pressure-induced amorphization and negative thermal expansion in ZrW2O8. Science 1998, 280, 886–889. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.S.O.; Mary, T.A.; Sleight, A.W. Negative thermal expansion in a large molybdate and tungstate family. J. Solid State Chem. 1997, 133, 580–583. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Mary, T.A.; Sleight, A.W. Negative thermal expansion materials. Phys. B 1997, 241, 311–316. [Google Scholar] [CrossRef]
- Khosrovani, N.; Sleight, A.W. Strong anisotropic thermal expansion in oxides. Int. J. Inorg. Mater. 1999, 1, 3–10. [Google Scholar] [CrossRef]
- Mary, T.A.; Sleight, A.W. Bulk thermal expansion for tungstate and molybdates of the type A2M3O12. J. Mater. Res. 1999, 14, 912–915. [Google Scholar] [CrossRef]
- Sleight, A. Materials science: Zero-expansion plan. Nature 2003, 425, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Sleight, A.W. Thermal contraction. Endeavour 1995, 19, 64–68. [Google Scholar] [CrossRef]
- Sleight, A.W. Compounds that contract on heating. Inorg. Chem. 1998, 37, 2854–2860. [Google Scholar] [CrossRef]
- David, W.I.F.; Evans, J.S.O.; Sleight, A.W. Direct evidence for a low-frequency phonon mode mechanism in the negative thermal expansion compound ZrW2O8. Europhys. Lett. 1999, 46, 661–666. [Google Scholar] [CrossRef]
- Pryde, A.K.A.; Hammonds, K.D.; Dove, M.T.; Heine, V.; Gale, J.D.; Warren, M.C. Rigid unit modes and the negative thermal expansion in ZrW2O8. Phase Transit. 1997, 61, 141–153. [Google Scholar] [CrossRef]
- Tao, J.Z.; Sleight, A.W. The role of rigid unit modes in negative thermal expansion. J. Solid State Chem. 2003, 173, 442–448. [Google Scholar] [CrossRef]
- Badrinarayanan, P.; Mac Murray, B.; Kessler, M.R. Zirconium tungstate reinforced cyanate ester composites with enhanced dimensional stability. J. Mater. Res. 2009, 24, 2235–2242. [Google Scholar] [CrossRef]
- Balch, D.K.; Dunand, D.C. Copper-zirconium tungstate composites exhibiting low and negative thermal expansion influenced by reinforcement phase transformations. Metall. Mater. Trans. A 2004, 35A, 1159–1165. [Google Scholar] [CrossRef]
- Chu, X.; Huang, R.; Yang, H.; Wu, Z.; Lu, J.; Zhou, Y.; Li, L. The cryogenic thermal expansion and mechanical properties of plasma modified ZrW2O8 reinforced epoxy. Mater. Sci. Eng. A 2011, 528, 3367–3374. [Google Scholar] [CrossRef]
- De Buysser, K.; Lommens, P.; de Meyer, C.; Bruneel, E.; Hoste, S.; van Driessche, I. ZrO2-ZrW2O8 composites with tailor-made thermal expansion. Ceramics-Silikaty 2004, 48, 139–144. [Google Scholar]
- Haman, K.; Badrinarayanan, P.; Kessler, M.R. Effect of a zirconium tungstate filler on the cure behavior of a cyanate ester resin. ACS Appl. Mater. Interfaces 2009, 1, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Lind, C.; Coleman, M.R.; Kozy, L.C.; Sharma, G.R. Zirconium tungstate/polymer nanocomposites: Challenges and opportunities. Phys. Status Solidi B 2011, 248, 123–129. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, X.; Yan, X.; Yang, J.; Fu, T.; Qiu, J. Synthesis of ZrO2/ZrW2O8 composites with low thermal expansion. Compos. Sci. Technol. 2007, 67, 1167–1171. [Google Scholar] [CrossRef]
- Suzuki, T.; Omote, A. Negative thermal expansion in (HfMg)(WO4)3. J. Am. Ceram. Soc. 2004, 87, 1365–1367. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Mary, T.A.; Sleight, A.W. Structure of Zr2(WO4)(PO4)2 from powder X-ray data—Cation ordering with no superstructure. J. Solid State Chem. 1995, 120, 101–104. [Google Scholar] [CrossRef]
- Sleight, A.W.; Brixner, L.H. A new ferroelastic transition in some A2(MO4)3 molybdates and tungstates. J. Solid State Chem. 1973, 7, 172–174. [Google Scholar] [CrossRef]
- Forster, P.M.; Yokochi, A.; Sleight, A.W. Enhanced negative thermal expansion in Lu2W3O12. J. Solid State Chem. 1998, 140, 157–158. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, J.; Cheng, X. Synthesis and tunable thermal expansion property of Al2−dScdW3O12. Solid State Sci. 2012, 14, 187–190. [Google Scholar] [CrossRef]
- Wu, M.M.; Liu, X.Z.; Chen, D.F.; Huang, Q.Z.; Wu, H.; Liu, Y.T. Structure, phase transition, and controllable thermal expansion behaviors of Sc2−xFexMo3O12. Inorg. Chem. 2014, 53, 9206–9212. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Peng, J.; Cheng, Y.Z.; Xiao, X.L.; Chen, D.F.; Hu, Z.B. Structural and controllable thermal expansion properties of Sc2−xAlxMo3O12. J. Alloy. Compd. 2013, 577, 295–298. [Google Scholar] [CrossRef]
- Wu, M.M.; Peng, J.; Han, S.B.; Hu, Z.B.; Liu, Y.T.; Chen, D.F. Phase transition and negative thermal expansion properties of Sc2−xCrxMo3O12. Ceram. Int. 2012, 38, 6525–6529. [Google Scholar] [CrossRef]
- Wu, M.M.; Peng, J.; Zu, Y.; Liu, R.D.; Hu, Z.B.; Liu, Y.T.; Chen, D.F. Thermal expansion properties of Lu2−xFexMo3O12. Chin. Phys. B 2012, 11, 1–6. [Google Scholar]
- Ari, M.; Jardim, P.M.; Marinkovic, B.A.; Rizzo, F.; Ferreira, F.F. Thermal expansion of Cr2xFe2−2xMo3O12, Al2xFe2−2xMo3O12 and Al2xCr2−2xMo3O12 solid solutions. J. Solid State Chem. 2008, 181, 1472–1479. [Google Scholar] [CrossRef]
- Baiz, T.I.; Heinrich, C.P.; Banek, N.A.; Vivekens, B.L.; Lind, C. In-situ non-ambient X-ray diffraction studies of indium tungstate. J. Solid State Chem. 2012, 187, 195–199. [Google Scholar] [CrossRef]
- Arnal, P.; Corriu, R.J.P.; Leclercq, D.; Mutin, P.H.; Vioux, A. Preparation of Transition metal oxides by a non-hydrolytic sol-gel process. Mater. Res. Soc. Symp. Proc. 1994, 346, 339–344. [Google Scholar] [CrossRef]
- Hay, J.N.; Raval, H.M. Preparation of inorganic oxides via a non-hydrolytic sol-gel route. J. Sol-Gel Sci. Technol. 1998, 13, 109–112. [Google Scholar] [CrossRef]
- Lafond, V.; Mutin, P.H.; Vioux, A. Non-hydrolytic sol-gel routes based on alkyl halide elimination: Toward better mixed oxide catalysts and new supports—Application to the preparation of a SiO2-TiO2 epoxidation catalyst. J. Mol. Catal. A 2002, 182, 81–88. [Google Scholar] [CrossRef]
- Lind, C.; Gates, S.D.; Pedoussaut, N.M.; Baiz, T.I. Novel materials through non-hydrolytic sol-gel processing: Negative thermal expansion oxides and beyond. Materials 2010, 3, 2567–2587. [Google Scholar] [CrossRef]
- Vioux, A.; Leclercq, D. Non-aqueous routes to sol-gel. Heterog. Chem. Rev. 1996, 3, 65–73. [Google Scholar] [CrossRef]
- Gates, S.D.; Colin, J.A.; Lind, C. Non-hydrolytic sol-gel synthesis, properties, and high-pressure behavior of gallium molybdate. J. Mater. Chem. 2006, 16, 4214–4219. [Google Scholar] [CrossRef]
- Gates, S.D.; Lind, C. Polymorphism in yttrium molybdate Y2Mo3O12. J. Solid State Chem. 2007, 180, 3510–3514. [Google Scholar] [CrossRef]
- Baiz, T.I.; Gindhart, A.M.; Kraemer, S.K.; Lind, C. Synthesis of MgHf(WO4)3 and MgZr(WO4)3 using a non-hydrolytic sol-gel method. J. Sol-Gel Sci. Technol. 2008, 47, 128–130. [Google Scholar] [CrossRef]
- Gates, S.D. Cation Influence on Negative Thermal Expansion in the A2M3O12 Family. Ph.D. Thesis, the University of Toledo, Toledo, OH, USA, 2008. [Google Scholar]
- Shannon, R.D. Revised effective ionic-radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Tyagi, A.K.; Achary, S.N.; Mathews, M.D. Phase transition and negative thermal expansion in A(2)(MoO4)(3) system (A = Fe3+, Cr3+ and Al3+). J. Alloy. Compd. 2002, 339, 207–210. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Mary, T.A. Structural phase transitions and negative thermal expansion in Sc2(MoO4)3. Int. J. Inorg. Mater. 2000, 2, 143–151. [Google Scholar] [CrossRef]
- Li, Z.Y.; Song, W.B.; Liang, E.J. Structures, phase transition, and crystal water of Fe2−xYxMo3O12. J. Phys. Chem. C 2011, 115, 17806–17811. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Truitt, R.; Hermes, I.; Main, A.; Sendecki, A.; Lind, C. Low Temperature Synthesis and Characterization of AlScMo3O12. Materials 2015, 8, 700-716. https://0-doi-org.brum.beds.ac.uk/10.3390/ma8020700
Truitt R, Hermes I, Main A, Sendecki A, Lind C. Low Temperature Synthesis and Characterization of AlScMo3O12. Materials. 2015; 8(2):700-716. https://0-doi-org.brum.beds.ac.uk/10.3390/ma8020700
Chicago/Turabian StyleTruitt, Rebecca, Ilka Hermes, Alyssa Main, Anne Sendecki, and Cora Lind. 2015. "Low Temperature Synthesis and Characterization of AlScMo3O12" Materials 8, no. 2: 700-716. https://0-doi-org.brum.beds.ac.uk/10.3390/ma8020700