
The Electronic Structures and Optical Properties of Alkaline-Earth Metals Doped Anatase TiO2: A Comparative Study of Screened Hybrid Functional and Generalized Gradient Approximation

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
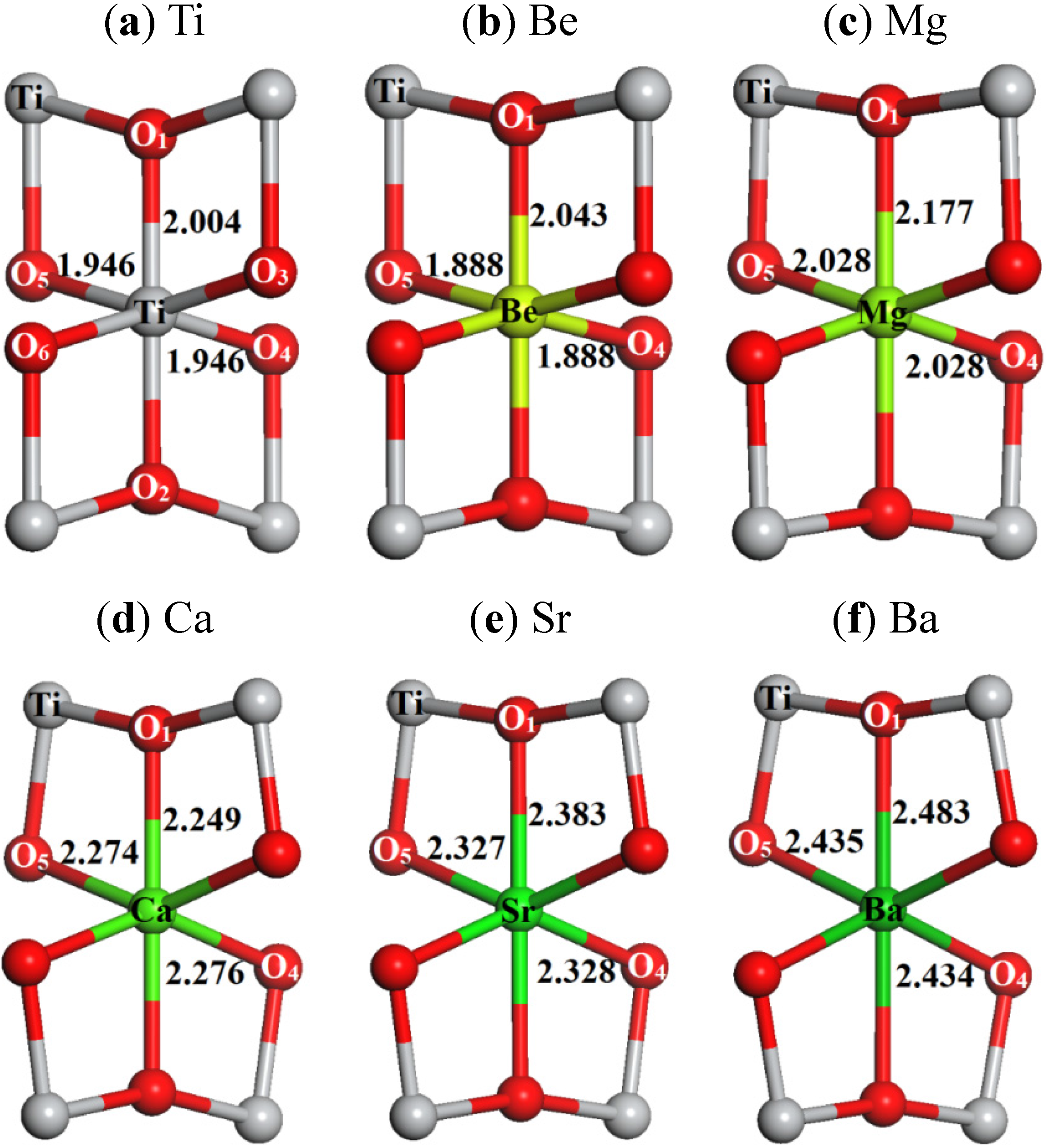
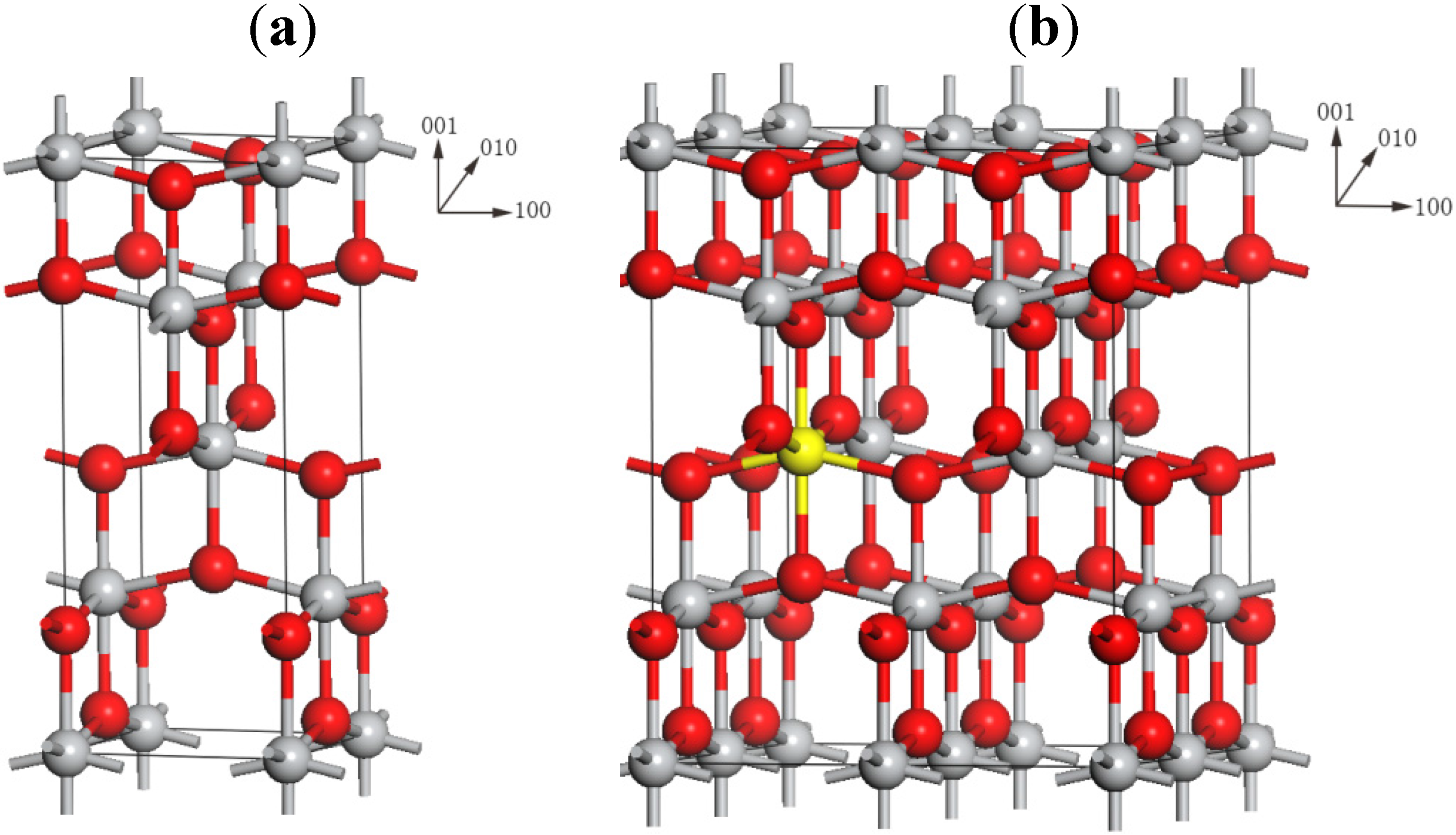
2.1. Local Structures

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quantity | Ti | Be | Mg | Ca | Sr | Ba |
|---|---|---|---|---|---|---|
| AEM-O1 | 2.004 | 2.043 | 2.177 | 2.249 | 2.383 | 2.483 |
| AEM-O2 | 2.004 | 2.042 | 2.178 | 2.248 | 2.383 | 2.483 |
| AEM-O3 | 1.946 | 1.888 | 2.028 | 2.274 | 2.327 | 2.435 |
| AEM-O4 | 1.946 | 1.888 | 2.028 | 2.276 | 2.328 | 2.434 |
| AEM-O5 | 1.946 | 1.888 | 2.028 | 2.274 | 2.327 | 2.435 |
| AEM-O6 | 1.946 | 1.888 | 2.028 | 2.276 | 2.328 | 2.434 |
| Δ | 0 | –0.025 | 0.126 | 0.301 | 0.380 | 0.485 |
| Ionic-radius | 0.53 | 0.31 | 0.65 | 0.99 | 1.13 | 1.35 |

2.2. Formation Energy
| Dopant Atom | GGA | HSE06 | ||
|---|---|---|---|---|
| O-Rich | Ti-Rich | O-Rich | Ti-Rich | |
| Be | 3.50 | 8.74 | 5.89 | 11.66 |
| Mg | 1.80 | 7.04 | 3.38 | 9.15 |
| Ca | 2.20 | 7.44 | 1.75 | 7.52 |
| Sr | 2.76 | 8.00 | 2.31 | 8.08 |
| Ba | 3.68 | 8.92 | 2.74 | 8.51 |
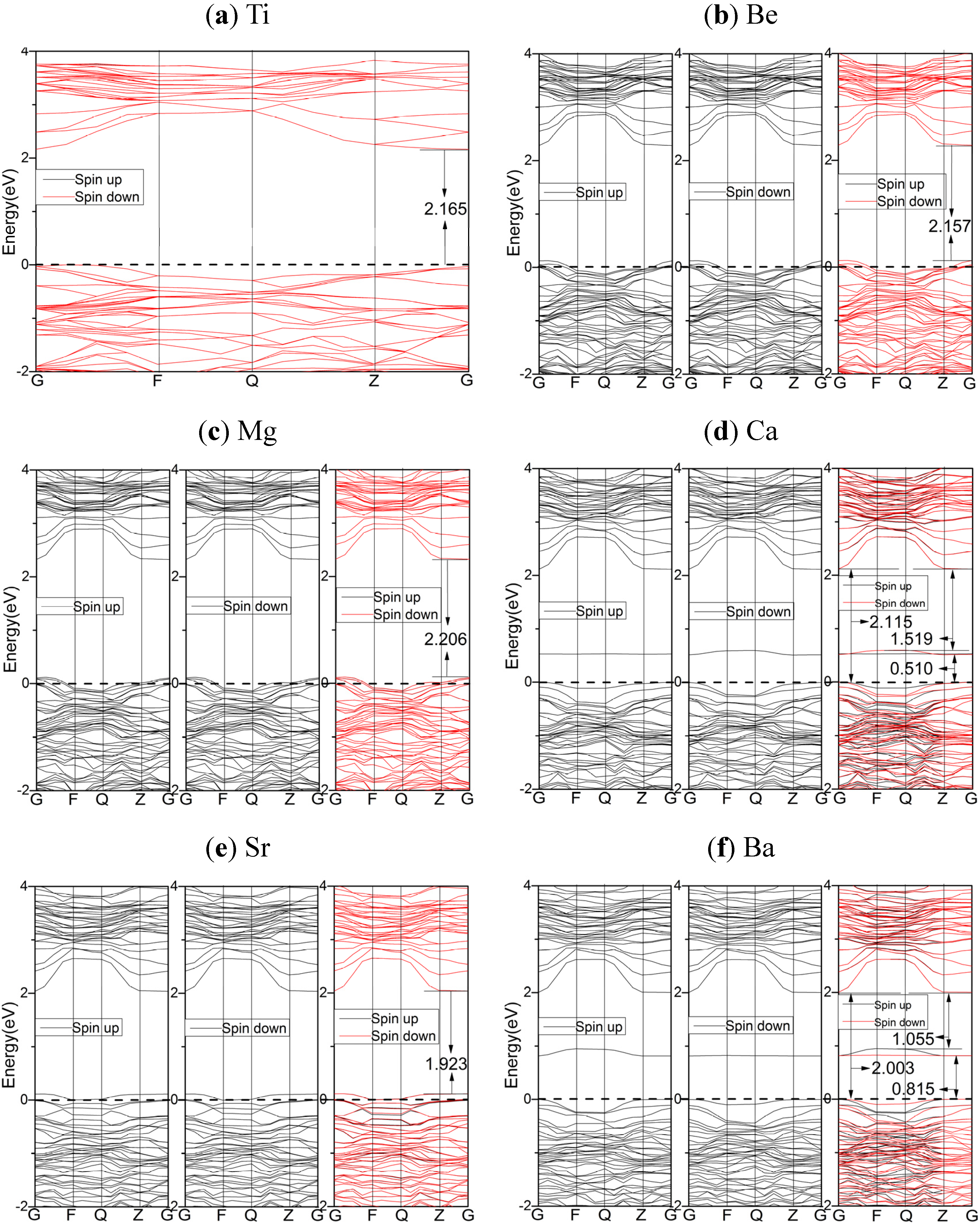
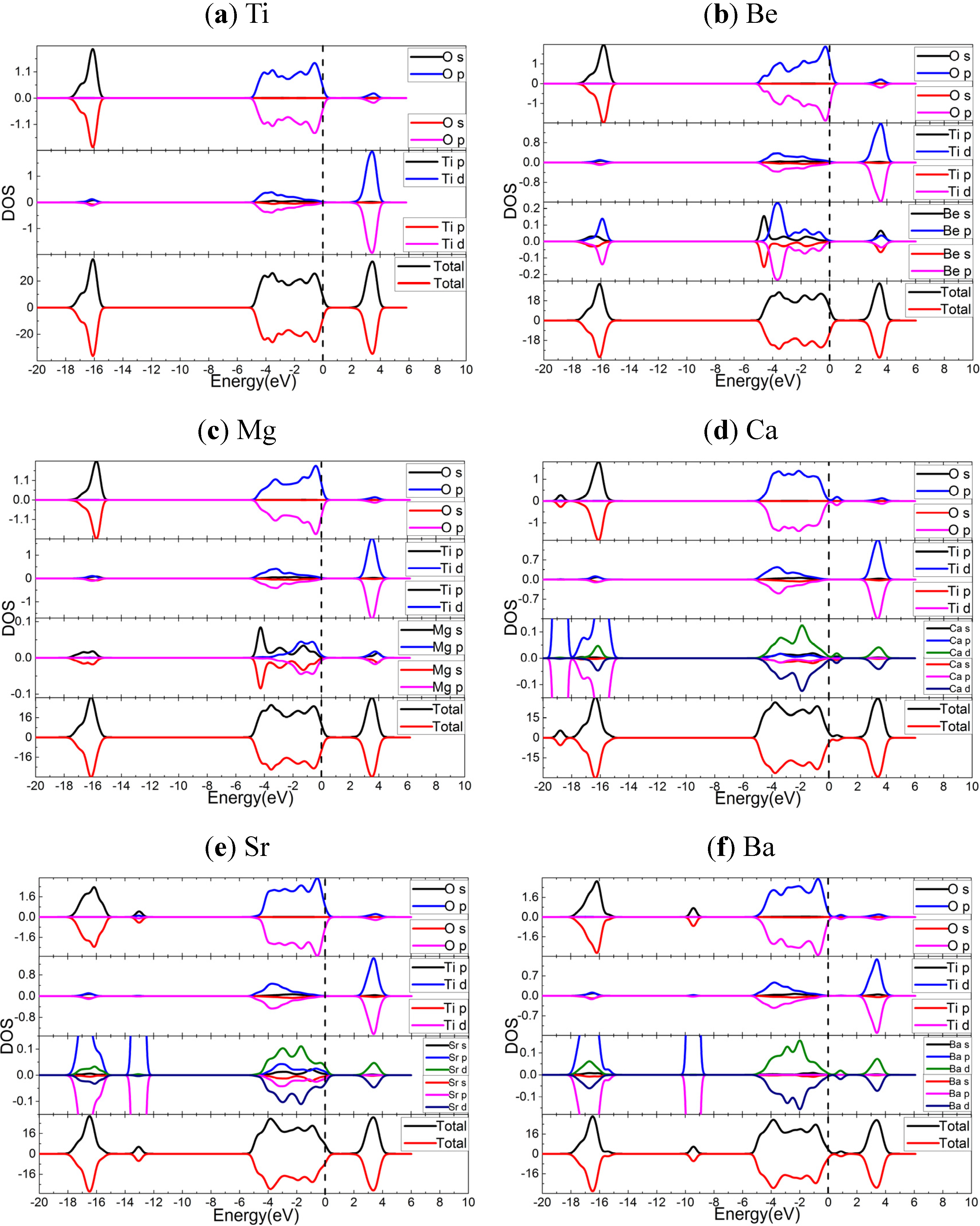
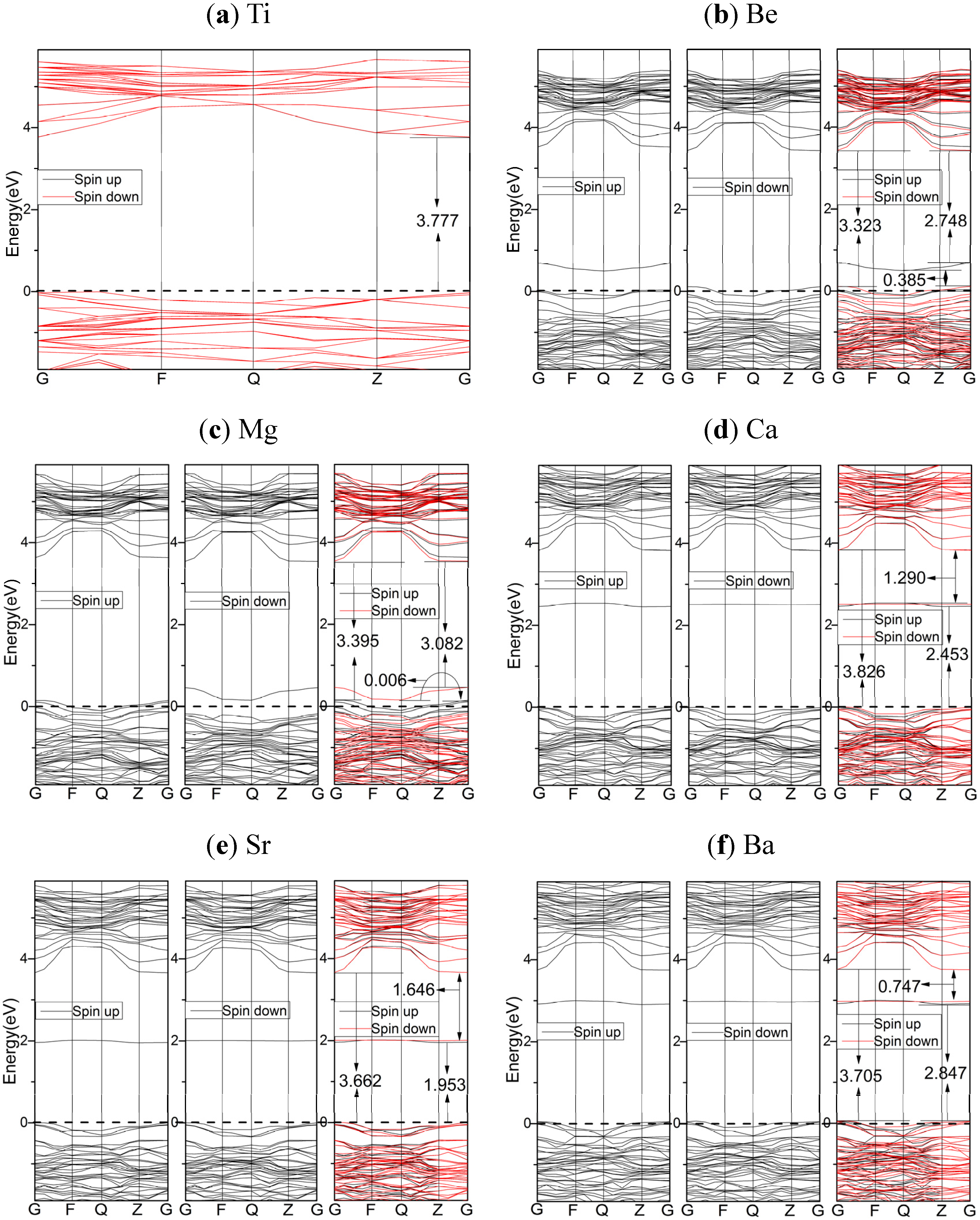
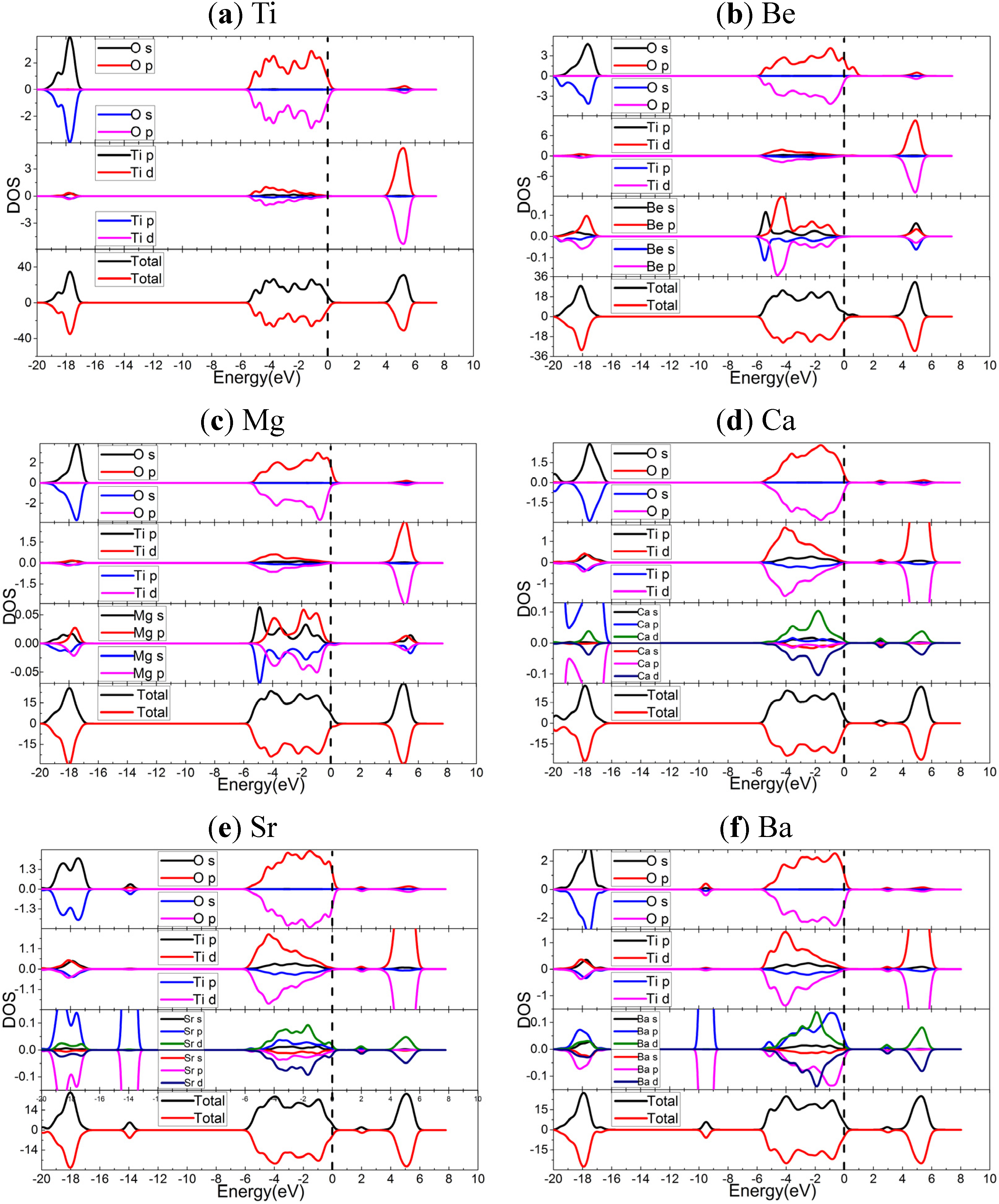
2.3. Electronic Properties
2.3.1. PBE Results


2.3.2. HSE06 Results


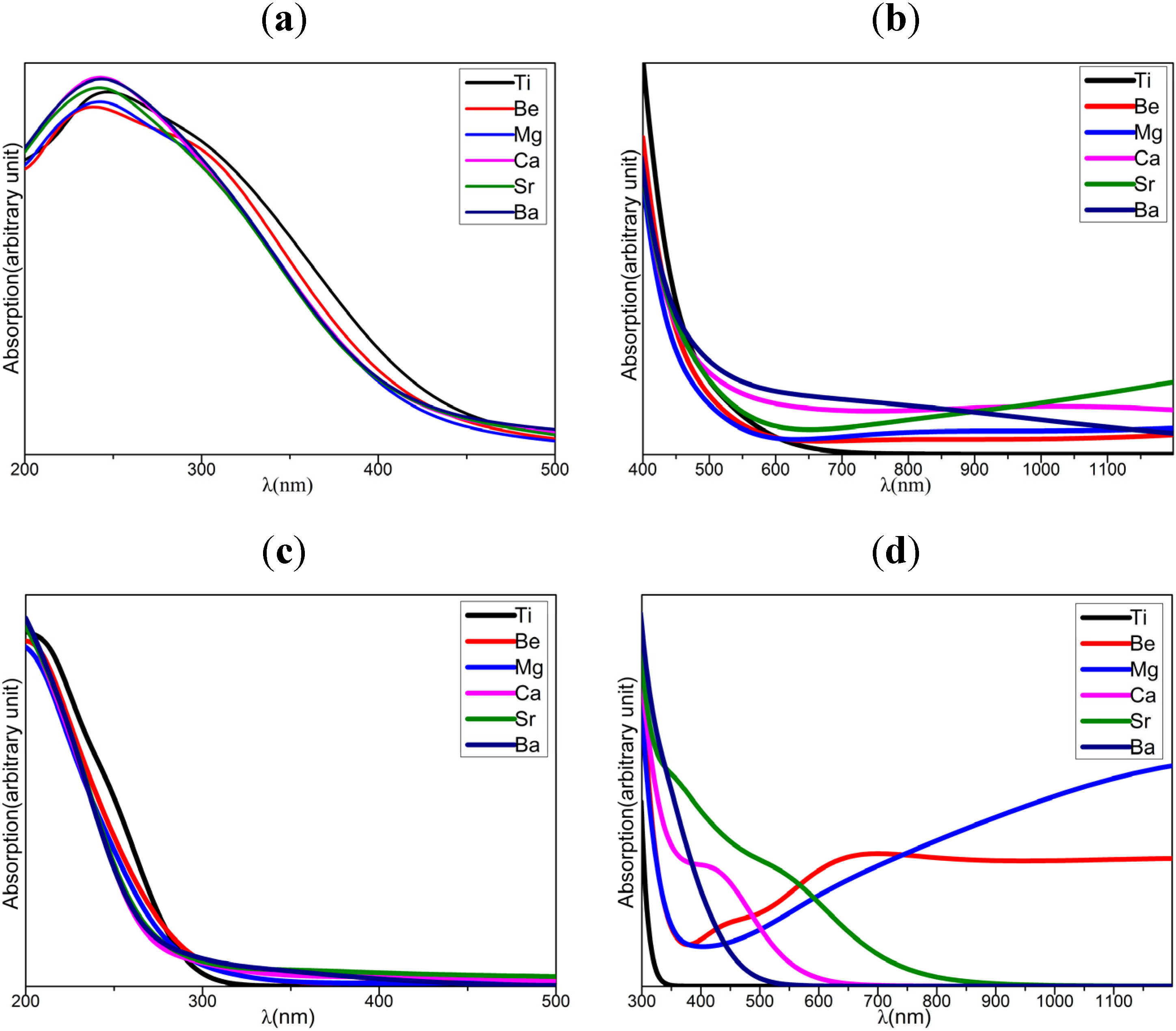
2.4. Optical Properties

3. Computational Methods

| Functional | PBE0 | B3LYP | HSE03 | HSE06 | Experiment |
|---|---|---|---|---|---|
| Band gap (eV) | 4.46 | 4.00 | 3.71 | 3.69 | 3.20 |
4. Conclusions
- Viewing from methodology, by combining the previous studies with our results, including band gaps and the dopant states, HSE06 provides a better description of the electronic structures.
- The formation energies indicate that the substitution of a lattice Ti atom with an AEM atom is more energetically favorable under O-rich growth conditions than those of Ti-rich cases.
- Based upon the HSE06 results, the analysis of electronic structures suggest that, AEM dopants shift the VBs to higher energy, and the more important is that the energies of dopant states for the cases of Ca, Sr, and Ba are quite higher than the top of VBs, while the Be and Mg dopants result into the spin polarized dopant states near Fermi levels. The components of VBs, CBs and dopant states support that the AEM dopants are active in inter-band transitions induced by lower energy excitations, which is important for the application of solar energy.
- Compared with anatase TiO2, the AEM dopants shift the absorption to longer wavelength and improve optical absorbance in visible and near-IR region. The Ca, Sr, and Ba dopants are superior to Be and Mg dopants to enhance absorbance in visible region, inversely, the Be and Mg dopants are better than Ca, Sr and Ba dopants for the improvement of absorbance in near-IR region. The compensating optical absorbance of Be/Mg and Ca/Sr/Ba dopants in different wavelength region present that the Be/Mg and Ca/Sr/Ba co-doped anatase TiO2 may possess prominent optical absorption properties.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Linsebigler, A.L.; Lu, G.; Yates, J.T. Photocatalysis on TiO2 surfaces: Principles, mechanisms, and selected results. Chem. Rev. 1995, 95, 735–758. [Google Scholar] [CrossRef]
- Henderson, M.A.; Lyubinetsky, I. Molecular-level insights into photocatalysis from scanning probe microscopy studies on TiO2(110). Chem. Rev. 2013, 113, 4428–4455. [Google Scholar] [CrossRef] [PubMed]
- Gratzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Grätzel, M. Dye-sensitized solar cells. J. Photochem. Photobiol. C 2003, 4, 145–153. [Google Scholar] [CrossRef]
- Li, B.; Wang, L.; Kang, B.; Wang, P.; Qiu, Y. Review of recent progress in solid-state dye-sensitized solar cells. Sol. Energy Mater. Sol. Cells 2006, 90, 549–573. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Klein, C.; Liska, P.; Grätzel, M. Synthesis of novel ruthenium sensitizers and their application in dye-sensitized solar cells. Coordin. Chem. Rev. 2005, 249, 1460–1467. [Google Scholar] [CrossRef]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Chen, X.; Shen, S.; Guo, L.; Mao, S.S. Semiconductor-based photocatalytic hydrogen generation. Chem. Rev. 2010, 110, 6503–6570. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mao, S.S. Titanium dioxide nanomaterials: Synthesis, properties, modifications, and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef] [PubMed]
- Aschauer, U.; Selloni, A. Hydrogen interaction with the anatase TiO2(101) surface. Phys. Chem. Chem. Phys. 2012, 14, 16595–16602. [Google Scholar] [CrossRef] [PubMed]
- Ulrike, D. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar]
- de Angelis, F.; di Valentin, C.; Fantacci, S.; Vittadini, A.; Selloni, A. Theoretical studies on anatase and less common TiO2 phases: Bulk, surfaces, and nanomaterials. Chem. Rev. 2014, 19, 9708–9753. [Google Scholar] [CrossRef] [PubMed]
- Muscat, J.; Swamy, V.; Harrison, N. First-principles calculations of the phase stability of TiO2. Phys. Rev. B 2002, 65. [Google Scholar] [CrossRef]
- Osorio-Guillén, J.; Lany, S.; Zunger, A. Atomic control of conductivity versus ferromagnetism in wide-gap oxides via selective doping: V, Nb, Ta in anatase TiO2. Phys. Rev. Lett. 2008, 100. [Google Scholar] [CrossRef]
- Avasarala, B.K.; Tirukkovalluri, S.R.; Bojja, S. Enhanced photocatalytic activity of beryllium doped titania in visible light on the degradation of methyl orange dye. Int. J. Mater. Res. 2010, 101, 1563–1570. [Google Scholar] [CrossRef]
- Avasarala, B.K.; Tirukkovalluri, S.R.; Bojja, S. Synthesis, characterization and photocatalytic activity of alkaline earth metal doped titania. Indian J. Chem. Sect. A-Inorg. Bio-Inorg. Phys. Theor. Anal. Chem. 2010, 49, 1189–1196. [Google Scholar]
- Akpan, U.G.; Hameed, B.H. Enhancement of the photocatalytic activity of TiO2 by doping it with calcium ions. J. Colloid Interface Sci. 2011, 357, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, D.L.; Jin, M.Z. Preparation and photocatalytic performance of Cs+/Sr2+-doped nano-sized TiO2. J. Cent. South. Univ. Technol. 2009, 16, 282–285. [Google Scholar]
- Kakiage, K.; Tokutome, T.; Iwamoto, S.; Kyomen, T.; Hanaya, M. Fabrication of a dye-sensitized solar cell containing a Mg-doped TiO2 electrode and a Br3−/Br− redox mediator with a high open-circuit photovoltage of 1.21 V. Chem. Commun. 2013, 49, 179–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qin, M.; Tao, H.; Ke, W.; Chen, Z.; Wan, J.; Qin, P.; Xiong, L.; Lei, H.; Yu, H.; et al. Performance enhancement of perovskite solar cells with Mg-doped TiO2 compact film as the hole-blocking layer. Appl. Phys. Lett. 2015, 106. [Google Scholar] [CrossRef]
- Manseki, K.; Ikeya, T.; Tamura, A.; Ban, T.; Sugiura, T.; Yoshida, T. Mg-doped TiO2 nanorods improving open-circuit voltages of ammonium lead halide perovskite solar cells. RSC Adv. 2014, 4, 9652–9655. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, S.; Mo, L.E; Huang, Y.; Tian, H.; Hu, L.; Huo, Z.; Dai, S.; Kong, F.; Pan, X. Charge recombination and band-edge shift in the dye-sensitized Mg2+-doped TiO2 solar cells. J. Phys. Chem. C 2011, 115, 16418–16424. [Google Scholar] [CrossRef]
- Liu, Q. Photovoltaic performance improvement of dye-sensitized solar cells based on Mg-doped TiO2 thin films. Electrochim. Acta 2014, 129, 459–462. [Google Scholar] [CrossRef]
- Liao, P.; Carter, E.A. New concepts and modeling strategies to design and evaluate photo-electro-catalysts based on transition metal oxides. Chem. Soc. Rev. 2013, 42, 2401–2422. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Tran, V.N.; Bach, T.C. Influences of metallic doping on anatase crystalline titanium dioxide: From electronic structure aspects to efficiency of TiO2-based dye sensitized solar cell (DSSC). Mater. Chem. Phys. 2014, 144, 114–121. [Google Scholar] [CrossRef]
- Morgan, B.J.; Scanlon, D.O.; Watson, G.W. Small polarons in Nb- and Ta-doped rutile and anatase TiO2. J. Mater. Chem. 2009, 19, 5175–5178. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, W.; Wu, P. Room-temperature ferromagnetism and optical properties in Mg-doped TiO2: A density functional theory investigation. J. Appl. Phys. 2014, 115. [Google Scholar] [CrossRef]
- Selcuk, S.; Selloni, A. DFT + U study of the surface structure and stability of Co3O4(110): Dependence on U. J. Phys. Chem. C 2015, 119, 9973–9979. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened coulomb potential. J. Chem. Phys. 2003, 118. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lu, Z.-Y. First-principles study of Fe-based superconductors: A comparison of screened hybrid functional with gradient corrected functional. Comput. Mater. Sci. 2012, 55, 284–294. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Clark, S.J.; Robertson, J. Calculation of point defects in rutile TiO2 by the screened-exchange hybrid functional. Phys. Rev. B 2012, 86. [Google Scholar] [CrossRef]
- Wu, P.; Zhou, Z.; Shi, J.; Guo, L. First-principles calculations of Cd1−xZnxS doped with alkaline earth metals for photocatalytic hydrogen generation. Int. J. Hydrogen Energy 2012, 37, 13074–13081. [Google Scholar] [CrossRef]
- Peyghan, A.A.; Noei, M. The alkali and alkaline earth metal doped ZnO nanotubes: DFT studies. Phys. B: Condens. Matter 2014, 432, 105–110. [Google Scholar] [CrossRef]
- Zhou, Z.H.; Li, M.T.; Guo, L.J. A first-principles theoretical simulation on the electronic structures and optical absorption properties for O vacancy and Ni impurity in TiO2 photocatalysts. J. Phys. Chem. Solids 2010, 71, 1707–1712. [Google Scholar] [CrossRef]
- Çelik, V.; Mete, E. Range-separated hybrid exchange-correlation functional analyses of anatase TiO2 doped with W, N, S, W/N, or W/S. Phys. Rev. B 2012, 86. [Google Scholar] [CrossRef]
- Long, R.; English, N.J. Synergistic effects of Bi/S codoping on visible light-activated anatase TiO2 photocatalysts from first principles. J. Phys. Chem. C 2009, 113, 8373–8377. [Google Scholar] [CrossRef]
- Van de Walle, C.G. First-principles calculations for defects and impurities: Applications to III-nitrides. J. Appl. Phys. 2004, 95. [Google Scholar] [CrossRef]
- Di Valentin, C.; Pacchioni, G.; Selloni, A. Theory of carbon doping of titanium dioxide. Chem. Mater. 2005, 17, 6656–6665. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B. Understanding photocatalytic activity of S- and P-doped TiO2 under visible light from first-principles. J. Phys. Chem. C 2007, 111, 18985–18994. [Google Scholar] [CrossRef]
- Sun, H.; Fan, W.; Li, Y.; Cheng, X.; Li, P.; Hao, J.; Zhao, X. Origin of improved visible photocatalytic activity of nitrogen/hydrogen codoped cubic In2O3: First-principles calculations. Phys. Chem. Chem. Phys. 2011, 13, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Gai, Y.; Li, J.; Li, S.-S.; Xia, J.-B.; Wei, S.-H. Design of narrow-gap TiO2: A passivated codoping approach for enhanced photoelectrochemical activity. Phys. Rev. Lett. 2009, 102. [Google Scholar] [CrossRef]
- Yu, H.; Irie, H.; Hashimoto, K. Conduction band energy level control of titanium dioxide: Toward an efficient visible-light-sensitive photocatalyst. J. Am. Chem. Soc. 2010, 132, 6898–6899. [Google Scholar] [CrossRef] [PubMed]
- Chandiran, A.K.; Sauvage, F.; Casas-Cabanas, M.; Comte, P.; Zakeeruddin, S.M.; Graetzel, M. Doping a TiO2 photoanode with Nb5+ to enhance transparency and charge collection efficiency in dye-sensitized solar cells. J. Phys. Chem. C 2010, 114, 15849–15856. [Google Scholar] [CrossRef]
- Okoye, C.M.I. Theoretical study of the electronic structure, chemical bonding and optical properties of KNbO3 in the paraelectric cubic phase. J. Phys. Condens. Matter 2003, 15. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the castep code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Janisch, R.; Spaldin, N. Understanding ferromagnetism in co-doped TiO2 anatase from first principles. Phys. Rev. B 2006, 73. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B. Origin of the photoactivity in boron-doped anatase and rutile TiO2 calculated from first principles. Phys. Rev. B 2007, 76, 195201. [Google Scholar] [CrossRef]
- Cromer, D.T.; Herrington, K. The structures of anatase and rutile. J. Am. Chem. Soc. 1955, 77, 4708–4709. [Google Scholar] [CrossRef]
- Howard, C.J.; Sabine, T.M.; Dickson, F. Structural and thermal parameters for rutile and anatase. Acta Crystallogr. Sect. B Struct. Sci. 1991, 47, 462–468. [Google Scholar] [CrossRef]
- Rubio-Ponce, A.; Conde-Gallardo, A.; Olguín, D. First-principles study of anatase and rutile TiO2 doped with Eu ions: A comparison of GGA and LDA + U calculations. Phys. Rev. B 2008, 78. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98. [Google Scholar] [CrossRef]
- Long, R.; English, N.J. Tailoring the electronic structure of TiO2 by cation codoping from hybrid density functional theory calculations. Phys. Rev. B 2011, 83. [Google Scholar] [CrossRef]
- Tang, H.; Berger, H.; Schmid, P.E.; Lévy, F.; Burri, G. Photoluminescence in TiO2 anatase single crystals. Solid State Commun. 1993, 87, 847–850. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.-G.; Zhang, C.-R.; Gong, J.-J.; Wu, Y.-Z.; Kou, S.-Z.; Yang, H.; Chen, Y.-H.; Liu, Z.-J.; Chen, H.-S. The Electronic Structures and Optical Properties of Alkaline-Earth Metals Doped Anatase TiO2: A Comparative Study of Screened Hybrid Functional and Generalized Gradient Approximation. Materials 2015, 8, 5508-5525. https://0-doi-org.brum.beds.ac.uk/10.3390/ma8085257
Ma J-G, Zhang C-R, Gong J-J, Wu Y-Z, Kou S-Z, Yang H, Chen Y-H, Liu Z-J, Chen H-S. The Electronic Structures and Optical Properties of Alkaline-Earth Metals Doped Anatase TiO2: A Comparative Study of Screened Hybrid Functional and Generalized Gradient Approximation. Materials. 2015; 8(8):5508-5525. https://0-doi-org.brum.beds.ac.uk/10.3390/ma8085257
Chicago/Turabian StyleMa, Jin-Gang, Cai-Rong Zhang, Ji-Jun Gong, You-Zhi Wu, Sheng-Zhong Kou, Hua Yang, Yu-Hong Chen, Zi-Jiang Liu, and Hong-Shan Chen. 2015. "The Electronic Structures and Optical Properties of Alkaline-Earth Metals Doped Anatase TiO2: A Comparative Study of Screened Hybrid Functional and Generalized Gradient Approximation" Materials 8, no. 8: 5508-5525. https://0-doi-org.brum.beds.ac.uk/10.3390/ma8085257