
Development of a Rapid Throughput Assay for Identification of hNav1.7 Antagonist Using Unique Efficacious Sodium Channel Agonist, Antillatoxin

Abstract
:1. Introduction
2. Results
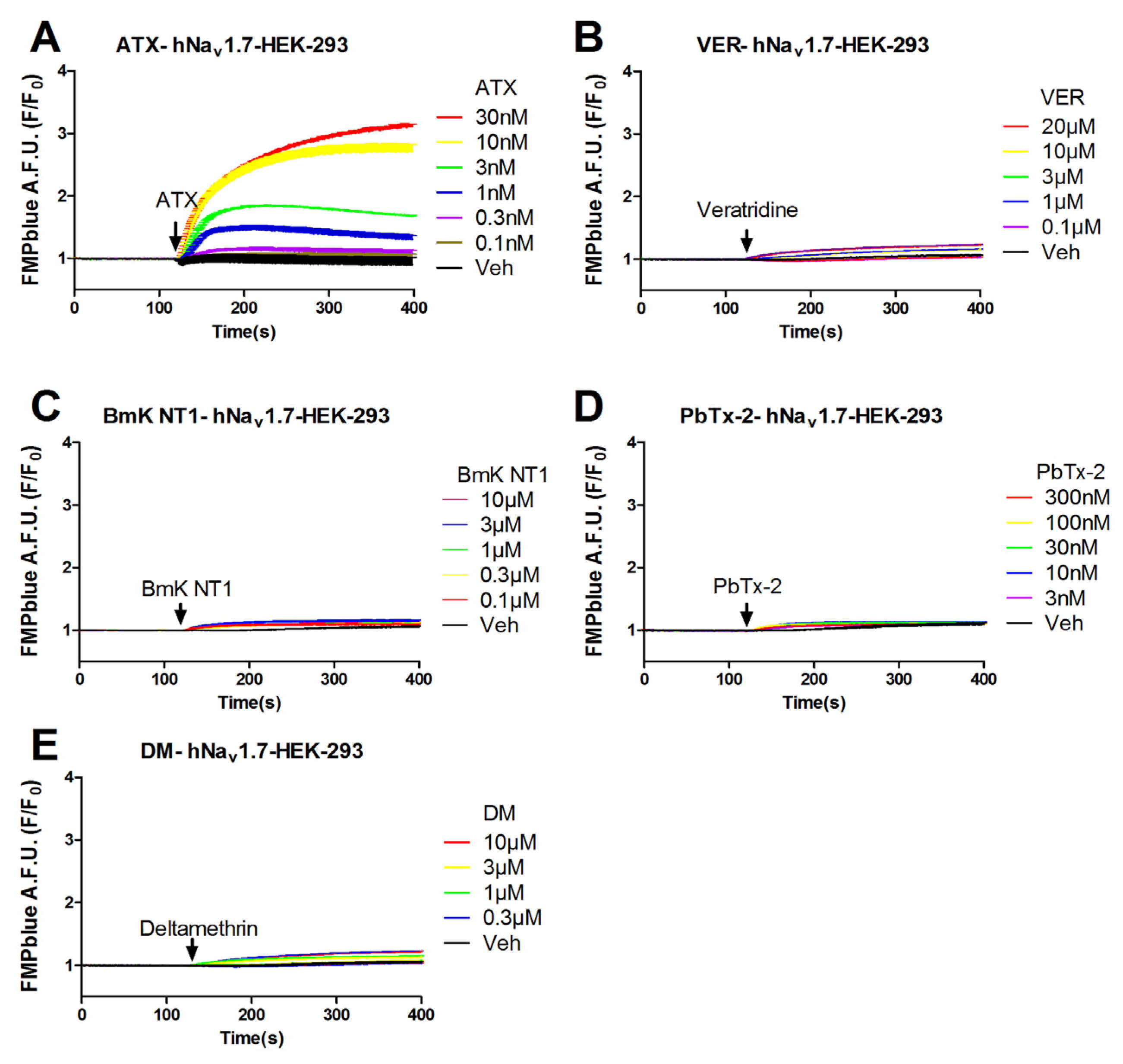
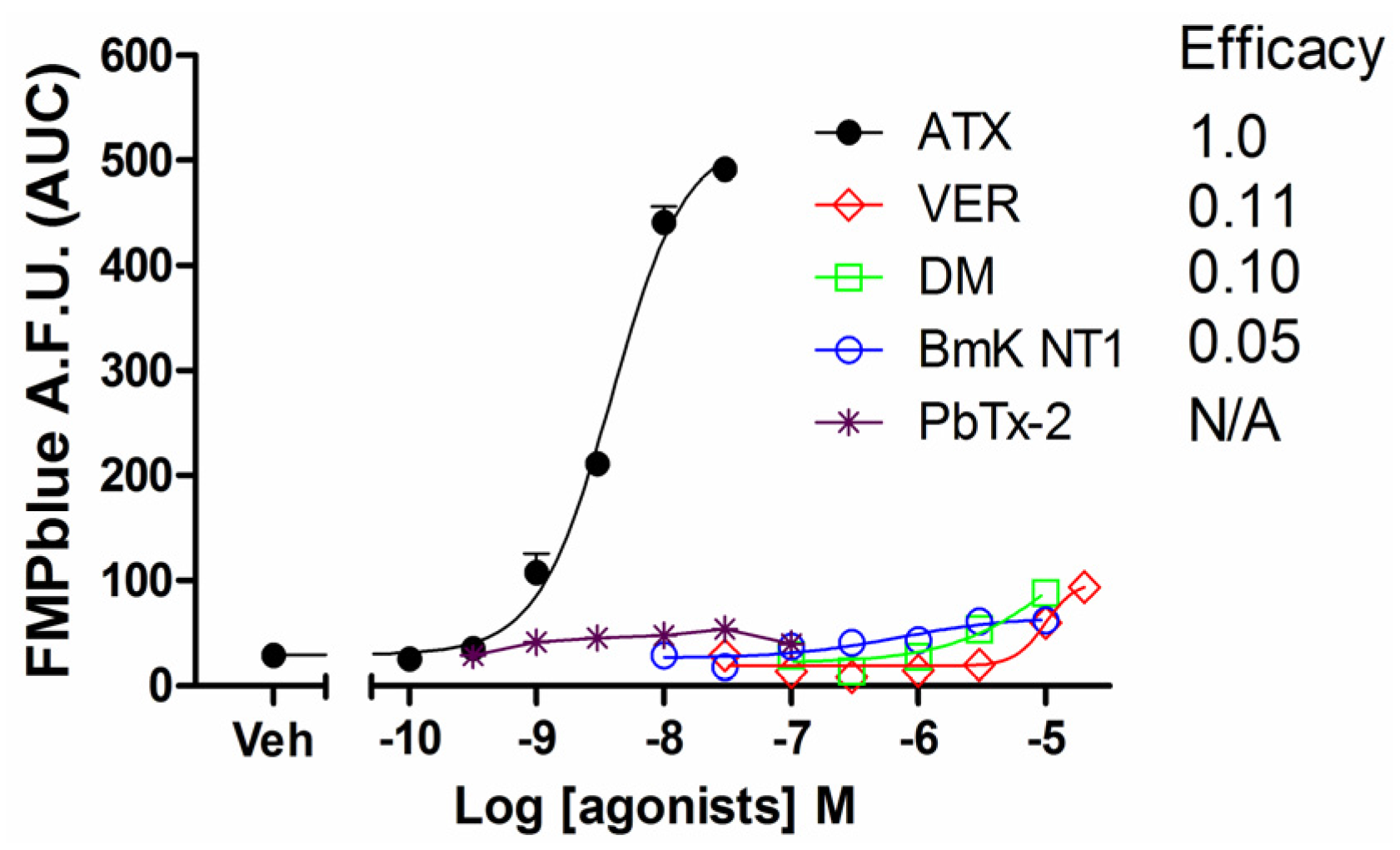
2.1. Influence of VGSC Agonists on Membrane Depolarization in HEK-293 Cells Stably Expressing hNav1.7


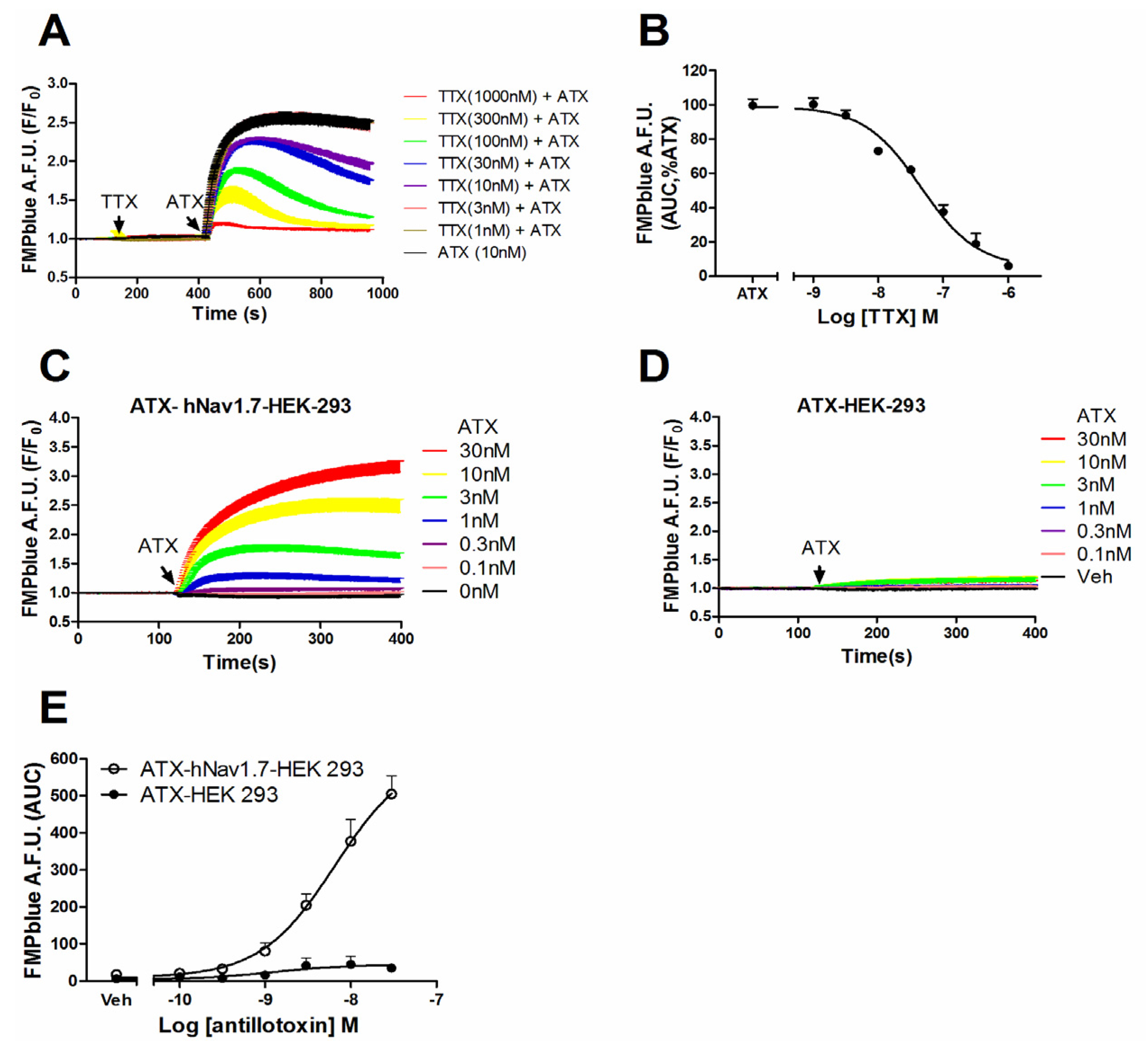
2.2. ATX-Induced Membrane Depolarization Was Dependent on the Activation of hNav1.7

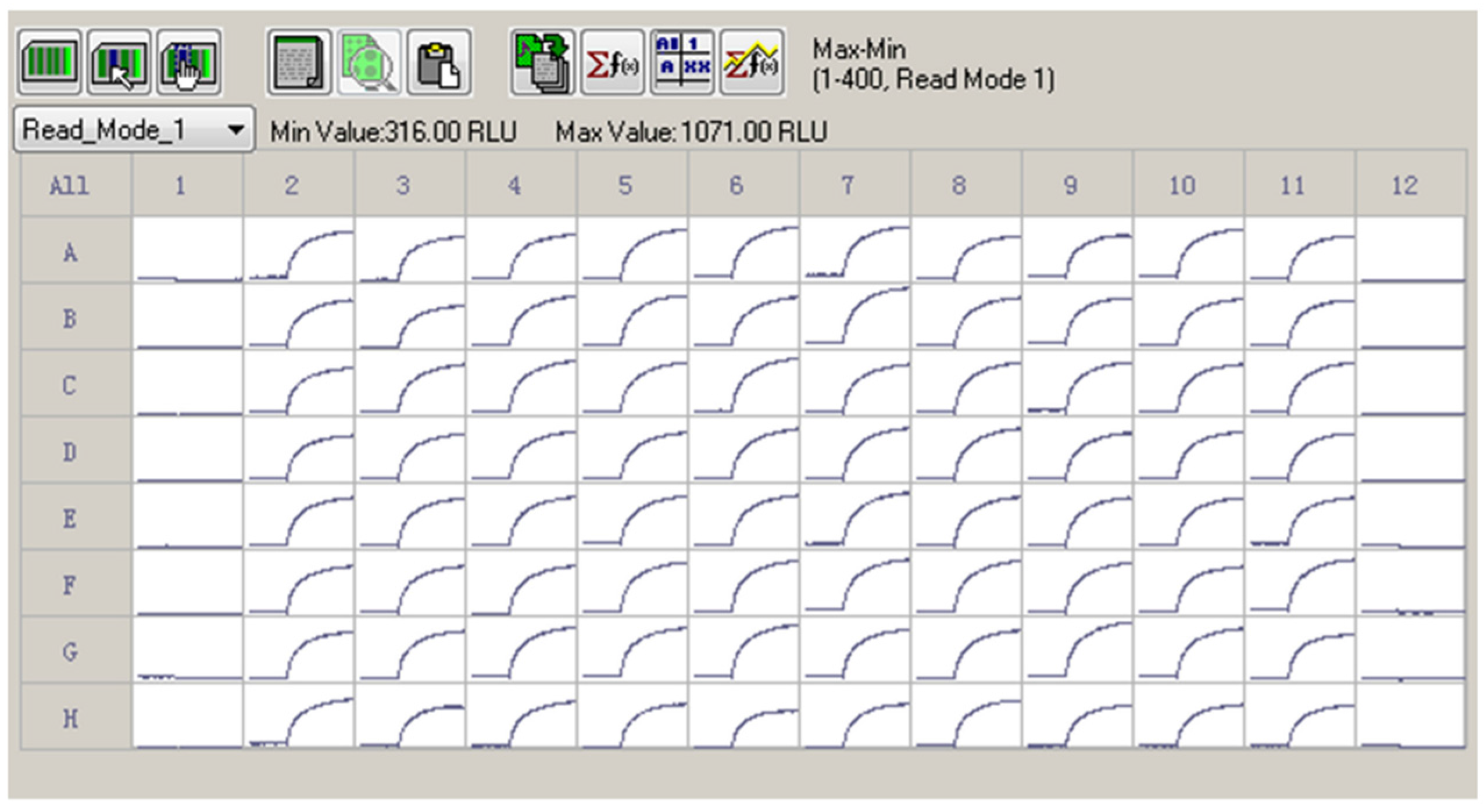
2.3. Z′ Factor Determination

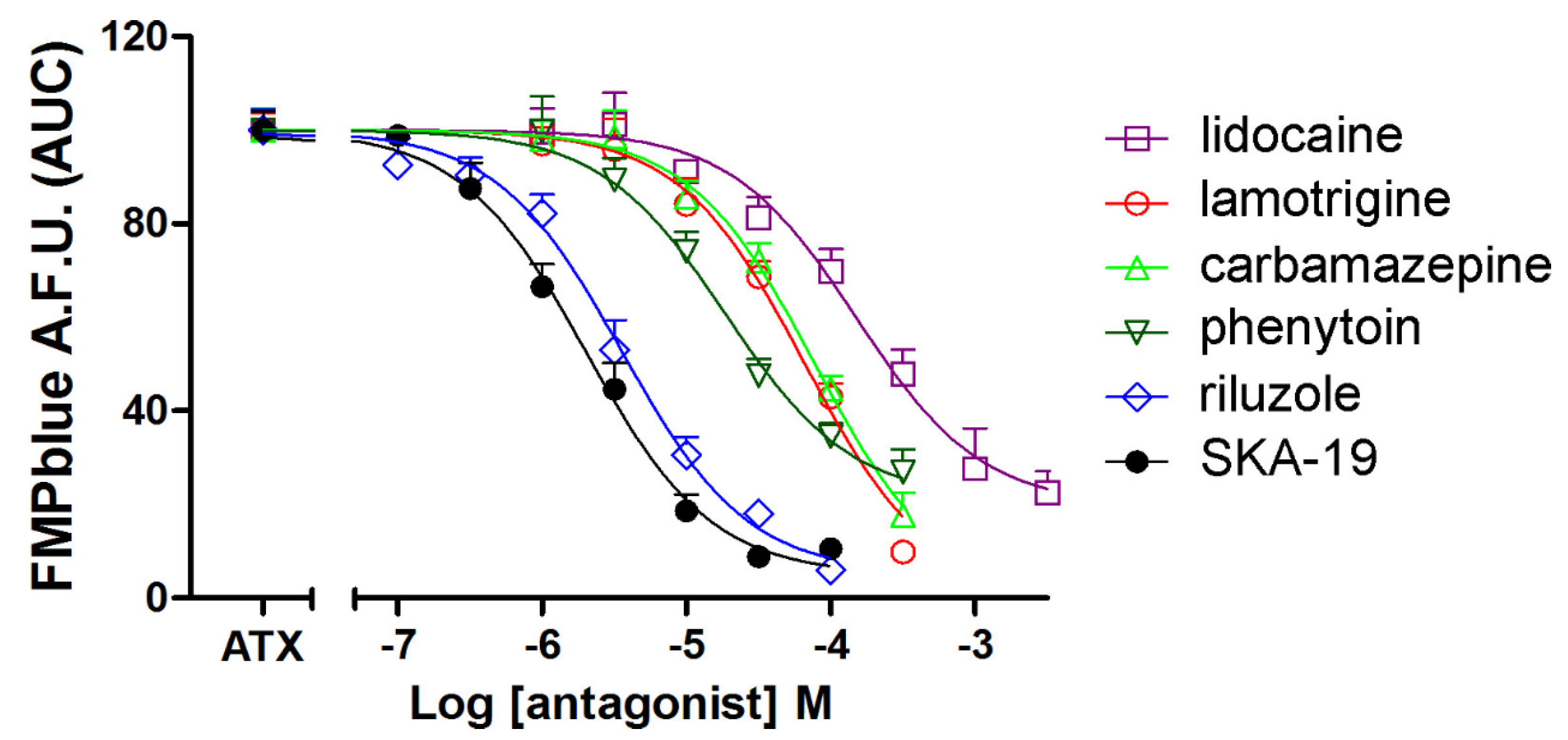
2.4. Influence of an Array of VGSC Antagonists on ATX-Induced Membrane Depolarization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (μM) (95% CI) | Patch-Clamp IC50 (μM) | Reference |
|---|---|---|---|
| SKA-19 | 2.02 (1.49–4.74) | 5.8 | [28] |
| Riluzole | 3.58 (2.67–4.80) | 2 | [29] |
| Phenytoin | 18.7 (11.8–29.7) | 31.6 | [24] |
| Lamotrigine | 66.3 (40.6–108.1) | 79 | [30] |
| Carbamazepine | 77.7 (49.9–121.0) | 101 | [30] |
| Lidocaine | 150.6 (92.9–244.0) | 110 | [31] |
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Membrane Potential Change Detection
4.4. Data Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ATX | antillatoxin |
| FBS | fetal bovine serum |
| FLIPR | fluorescence imaging plate reader |
| FMP | FLIPR membrane potential |
| HEK | human embryonic kidney |
| hNav1.7 | human voltage-gated sodium channel subtype 1.7 |
| HTS | high throughput screen |
| PbTxs | brevetoxins |
| SBFI/AM | sodium-binding benzofuran isophthalate/acetoxymethyl ester |
| SKA-19 | 2-Amino-6-trifluoromethylthiobenzothiazole |
| Tl+ | thallium |
| TTX | tetrodotoxin |
| VGSCs | voltage-gated sodium channels |
References
- Frank, H.Y.; Yarov-Yarovoy, V.; Gutman, G.A.; Catterall, W.A. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 2005, 57, 387–395. [Google Scholar]
- Murray, J.K.; Ligutti, J.; Liu, D.; Zou, A.; Poppe, L.; Li, H.; Andrews, K.L.; Moyer, B.D.; McDonough, S.I.; Favreau, P.; et al. Engineering Potent and Selective Analogues of GpTx-1, a Tarantula Venom Peptide Antagonist of the NaV1. 7 Sodium Channel. J. Med. Chem. 2015, 58, 2299–2314. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Frank, H.Y.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldin, A.L.; Barchi, R.L.; Caldwell, J.H.; Hofmann, F.; Howe, J.R.; Hunter, J.C.; Kallen, R.G.; Mandel, G.; Meisler, M.H.; Netter, Y.B.; et al. Nomenclature of voltage-gated sodium channels. Neuron 2000, 28, 365–368. [Google Scholar] [CrossRef]
- Cao, Z.; George, J.; Gerwick, W.H.; Baden, D.G.; Rainier, J.D.; Murray, T.F. Influence of lipid-soluble gating modifier toxins on sodium influx in neocortical neurons. J. Pharmacol. Exp. Ther. 2008, 326, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Ruigt, G.F.; Neyt, H.C.; Van der Zalm, J.M.; Van den Bercken, J. Increase of sodium current after pyrethroid insecticides in mouse neuroblastoma cells. Brain Res. 1987, 437, 309–322. [Google Scholar] [CrossRef]
- Estacion, M.; Han, C.; Choi, J.-S.; Hoeijmakers, J.; Lauria, G.; Drenth, J.; Gerrits, M.M.; Dib-Hajj, S.D.; Faber, C.G.; Merkies, I.; et al. Intra-and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1. 7. Mol. Pain 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Howe, J.R.; Waxman, S.G. Slow closed-state inactivation: A novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 1998, 18, 9607–9619. [Google Scholar] [PubMed]
- Toledo-Aral, J.J.; Moss, B.L.; He, Z.-J.; Koszowski, A.G.; Whisenand, T.; Levinson, S.R.; Wolf, J.J.; Silos-Santiago, I.; Halegoua, S.; Mandel, G. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc. Natl. Acad. Sci. USA 1997, 94, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Li, S.; Xu, Z.; Li, H.; Ma, L.; Fan, J.; Bu, D.; Liu, B.; Fan, Z.; et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004, 41, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Rush, A.M.; Cummins, T.R.; Hisama, F.M.; Novella, S.; Tyrrell, L.; Marshall, L.; Waxman, S.G. Gain-of-function mutation in Nav1. 7 in familial erythromelalgia induces bursting of sensory neurons. Brain 2005, 128, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Nassar, M.A.; Stirling, L.C.; Forlani, G.; Baker, M.D.; Matthews, E.A.; Dickenson, A.H.; Wood, J.N. Nociceptor-specific gene deletion reveals a major role for Nav1. 7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. USA 2004, 101, 12706–12711. [Google Scholar] [CrossRef] [PubMed]
- Minett, M.S.; Nassar, M.A.; Clark, A.K.; Passmore, G.; Dickenson, A.H.; Wang, F.; Malcangio, M.; Wood, J.N. Distinct Nav1. 7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, J.; Bowlby, M.; Peri, R.; Vasilyev, D.; Arias, R. High-throughput electrophysiology: An emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov. 2008, 7, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Farre, C.; Stoelzle, S.; Haarmann, C.; George, M.; Brüggemann, A.; Fertig, N. Automated ion channel screening: Patch clamping made easy. Expert Opin. Ther. Targets 2007, 11, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-P.; Mangano, T.; Hufeisen, S.; Setola, V.; Roth, B.L. Identification of human Ether-a-go-go related gene modulators by three screening platforms in an academic drug-discovery setting. Assay Drug Dev. Technol. 2010, 8, 727–742. [Google Scholar] [CrossRef] [PubMed]
- Niswender, C.M.; Johnson, K.A.; Luo, Q.; Ayala, J.E.; Kim, C.; Conn, P.J.; Weaver, C.D. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 2008, 73, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.M.; Bhave, G.; Chauder, B.A.; Banerjee, S.; Lornsen, K.A.; Redha, R.; Fallen, K.; Lindsley, C.W.; Weaver, C.D.; Denton, J.S. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7. 1. Mol. Pharmacol. 2009, 76, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Delpire, E.; Days, E.; Lewis, L.M.; Mi, D.; Kim, K.; Lindsley, C.W.; Weaver, C.D. Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. USA 2009, 106, 5383–5388. [Google Scholar] [CrossRef] [PubMed]
- Carmosino, M.; Rizzo, F.; Torretta, S.; Procino, G.; Svelto, M. High-throughput fluorescent-based NKCC functional assay in adherent epithelial cells. BMC Cell Biol. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Shafer, T.J.; Crofton, K.M.; Gennings, C.; Murray, T.F. Additivity of pyrethroid actions on sodium influx in cerebrocortical neurons in primary culture. Environ. Health Perspect. 2011, 119, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; He, Y.; Qiao, J.; Zhang, C.; Cao, Z. The natural scorpion peptide, BmK NT1 activates voltage-gated sodium channels and produces neurotoxicity in primary cultured cerebellar granule cells. Toxicon 2016, 109, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Gerwick, W.H.; Murray, T.F. Antillatoxin is a sodium channel activator that displays unique efficacy in heterologously expressed rNav1. 2, rNav1. 4 and rNav1. 5 alpha subunits. BMC Neurosci. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, S.; Dekermendjian, K.; Julien, R.; Huang, J.; Lund, P.-E.; Krupp, J.; Kronqvist, R.; Larsson, O.; Bostwick, R. Cellular HTS assays for pharmacological characterization of NaV1. 7 modulators. Assay Drug Dev. Technol. 2008, 6, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Felix, J.P.; Williams, B.S.; Priest, B.T.; Brochu, R.M.; Dick, I.E.; Warren, V.A.; Yan, L.; Slaughter, R.S.; Kaczorowski, G.J.; Smith, M.M.; et al. Functional assay of voltage-gated sodium channels using membrane potential-sensitive dyes. Assay Drug Dev. Technol. 2004, 2, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Berman, F.; Gerwick, W.; Murray, T. Antillatoxin and kalkitoxin, ichthyotoxins from the tropical cyanobacterium Lyngbya majuscula, induce distinct temporal patterns of NMDA receptor-mediated neurotoxicity. Toxicon 1999, 37, 1645–1648. [Google Scholar] [CrossRef]
- Li, W.; Berman, F.; Okino, T.; Yokokawa, F.; Shioiri, T.; Gerwick, W.; Murray, T. Antillatoxin is a marine cyanobacterial toxin that potently activates voltage-gated sodium channels. Proc. Natl. Acad. Sci. USA 2001, 98, 7599–7604. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Nguyen, H.M.; Cao, Z.; Brown, B.M.; Jenkins, D.P.; Zolkowska, D.; Chen, Y.-J.; Tanaka, B.S.; Goldin, A.L.; Rogawski, M.A.; et al. The riluzole derivative 2-amino-6-trifluoromethylthio-benzothiazole (SKA-19), a mixed KCa2 activator and NaV blocker, is a potent novel anticonvulsant. Neurotherapeutics 2015, 12, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-H.; Huang, C.-S.; Nagata, K.; Yeh, J.Z.; Narahashi, T. Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J. Pharmacol. Exp. Ther. 1997, 282, 707–714. [Google Scholar] [PubMed]
- Castle, N.; Printzenhoff, D.; Zellmer, S.; Antonio, B.; Wickenden, A.; Silvia, C. Sodium channel inhibitor drug discovery using automated high throughput electrophysiology platforms. Comb. Chem. High Throughput Screen. 2009, 12, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Sheets, P.L.; Jarecki, B.W.; Cummins, T.R. Lidocaine reduces the transition to slow inactivation in Nav1. 7 voltage-gated sodium channels. Br. J. Pharmacol. 2011, 164, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Shafer, T.J.; Meyer, D.A.; Crofton, K.M. Developmental neurotoxicity of pyrethroid insecticides: Critical review and future research needs. Environ. Health Perspect. 2005, 113, 123–136. [Google Scholar] [CrossRef] [PubMed]
- LePage, K.T.; Dickey, R.W.; Gerwick, W.H.; Jester, E.L.; Murray, T.F. On the use of neuro-2a neuroblastoma cells versus intact neurons in primary culture for neurotoxicity studies. Crit. Rev. Neurobiol. 2005, 17, 27–50. [Google Scholar] [CrossRef]
- Moran, O.; Nizzari, M.; Conti, F. Endogenous expression of the β1A sodium channel subunit in HEK-293 cells. FEBS Lett. 2000, 473, 132–134. [Google Scholar] [CrossRef]
- Chemin, J.; Monteil, A.; Briquaire, C.; Richard, S.; Perez-Reyes, E.; Nargeot, J.; Lory, P. Overexpression of T-type calcium channels in HEK-293 cells increases intracellular calcium without affecting cellular proliferation. FEBS Lett. 2000, 478, 166–172. [Google Scholar] [CrossRef]
- Ulbricht, W. Effects of veratridine on sodium currents and fluxes. In Reviews of Physiology Biochemistry and Pharmacology; Springer: Berlin, Germany; Heidelberg, Germany, 1998; Volume 133, pp. 1–54. [Google Scholar]
- He, B.; Soderlund, D.M. Human embryonic kidney (HEK293) cells express endogenous voltage-gated sodium currents and Na v 1.7 sodium channels. Neurosci. Lett. 2010, 469, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.Z.; Gilmore, E.S.; Estacion, M.; Eastman, E.; Taylor, S.; Melanson, M.; Dib-Hajj, S.D.; Waxman, S.G. A novel Nav1. 7 mutation producing carbamazepine-responsive erythromelalgia. Ann. Neurol. 2009, 65, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, P.; Vijayaragavan, K.; Chahine, M. Differential modulation of Nav1. 7 and Nav1. 8 peripheral nerve sodium channels by the local anesthetic lidocaine. Br. J. Pharmacol. 2004, 142, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-C. A common anticonvulsant binding site for phenytoin, carbamazepine, and lamotrigine in neuronal Na+ channels. Mol. Pharmacol. 1998, 54, 712–721. [Google Scholar] [PubMed]
- Clare, J.J.; Tate, S.N.; Nobbs, M.; Romanos, M.A. Voltage-gated sodium channels as therapeutic targets. Drug Discov. Today 2000, 5, 506–520. [Google Scholar] [CrossRef]
- Errington, A.C.; Stöhr, T.; Heers, C.; Lees, G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol. Pharmacol. 2008, 73, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-C.; Bean, B.P. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol. Pharmacol. 1994, 46, 716–725. [Google Scholar] [PubMed]
- Kuo, C.-C.; Chen, R.-S.; Lu, L.; Chen, R.-C. Carbamazepine inhibition of neuronal Na+ currents: Quantitative distinction from phenytoin and possible therapeutic implications. Mol. Pharmacol. 1997, 51, 1077–1083. [Google Scholar] [PubMed]
- Cardenas, C.A.; Cardenas, C.G.; de Armendi, A.J.; Scroggs, R.S. Carbamazepine interacts with a slow inactivation state of Na V 1.8-like sodium channels. Neurosci. Lett. 2006, 408, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Lenkowski, P.W.; Batts, T.W.; Smith, M.D.; Ko, S.-H.; Jones, P.J.; Taylor, C.H.; McCusker, A.K.; Davis, G.C.; Hartmann, H.A.; White, H.S.; et al. A pharmacophore derived phenytoin analogue with increased affinity for slow inactivated sodium channels exhibits a desired anticonvulsant profile. Neuropharmacology 2007, 52, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Franceschetti, S.; Avanzini, G.; Mantegazza, M. Phenytoin inhibits the persistent sodium current in neocortical neurons by modifying its inactivation properties. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Sun, G.; Clare, J.J.; Werkman, T.R.; Wadman, W.J. Properties of human brain sodium channel α-subunits expressed in HEK293 cells and their modulation by carbamazepine, phenytoin and lamotrigine. Br. J. Pharmacol. 2014, 171, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.; Hellier, J.L.; Uskert-Newsom, J.; Williams, P.A.; Staley, K.J.; Yee, A.S. Diphenytoin, riluzole and lidocaine: Three sodium channel blockers, with different mechanisms of action, decrease hippocampal epileptiform activity. Neuropharmacology 2013, 73, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Bean, B.P.; Cohen, C.J.; Tsien, R.W. Lidocaine block of cardiac sodium channels. J. Gen. Physiol. 1983, 81, 613–642. [Google Scholar] [CrossRef] [PubMed]
- Starmer, C.; Grant, A.; Strauss, H. Mechanisms of use-dependent block of sodium channels in excitable membranes by local anesthetics. Biophys. J. 1984, 46, 15–27. [Google Scholar] [CrossRef]
- Tunnicliff, G. Basis of the antiseizure action of phenytoin. Gen. Pharmacol. Vasc. Syst. 1996, 27, 1091–1097. [Google Scholar] [CrossRef]
- Bello, O.S.; Gonzalez, J.; Capani, F.; Barreto, G.E. In silico docking reveals possible Riluzole binding sites on Nav1. 6 sodium channel: Implications for amyotrophic lateral sclerosis therapy. J. Theor. Biol. 2012, 315, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 1977, 69, 497–515. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, D.S.; McPhee, J.C.; Scheuer, T.; Catterall, W.A. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. USA 1996, 93, 9270–9275. [Google Scholar] [CrossRef] [PubMed]
- McPhee, J.C.; Ragsdale, D.S.; Scheuer, T.; Catterall, W.A. A critical role for transmembrane segment IVS6 of the sodium channel α subunit in fast inactivation. J. Biol. Chem. 1995, 270, 12025–12034. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Whiteaker, K.L.; Gopalakrishnan, S.M.; Groebe, D.; Shieh, C.-C.; Warrior, U.; Burns, D.J.; Coghlan, M.J.; Scott, V.E.; Gopalakrishnani, M. Validation of FLIPR membrane potential dye for high throughput screening of potassium channel modulators. J. Biomol. Screen. 2001, 6, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Okura, K.; Matsuoka, S.; Goto, R.; Inoue, M. The twisted side chain of antillatoxin is important for potent toxicity: Total synthesis and biological evaluation of antillatoxin and analogues. Angew. Chem., Int. Ed. 2010, 49, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Wu, Z. Alternative statistical parameter for HTS assay quality assessment. J. Biomol. Screen. 2007, 12. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Li, X.; Jin, L.; Zhang, F.; Inoue, M.; Yu, B.; Cao, Z. Development of a Rapid Throughput Assay for Identification of hNav1.7 Antagonist Using Unique Efficacious Sodium Channel Agonist, Antillatoxin. Mar. Drugs 2016, 14, 36. https://0-doi-org.brum.beds.ac.uk/10.3390/md14020036
Zhao F, Li X, Jin L, Zhang F, Inoue M, Yu B, Cao Z. Development of a Rapid Throughput Assay for Identification of hNav1.7 Antagonist Using Unique Efficacious Sodium Channel Agonist, Antillatoxin. Marine Drugs. 2016; 14(2):36. https://0-doi-org.brum.beds.ac.uk/10.3390/md14020036
Chicago/Turabian StyleZhao, Fang, Xichun Li, Liang Jin, Fan Zhang, Masayuki Inoue, Boyang Yu, and Zhengyu Cao. 2016. "Development of a Rapid Throughput Assay for Identification of hNav1.7 Antagonist Using Unique Efficacious Sodium Channel Agonist, Antillatoxin" Marine Drugs 14, no. 2: 36. https://0-doi-org.brum.beds.ac.uk/10.3390/md14020036