Marine-Derived Natural Lead Compound Disulfide-Linked Dimer Psammaplin A: Biological Activity and Structural Modification

 ,
, 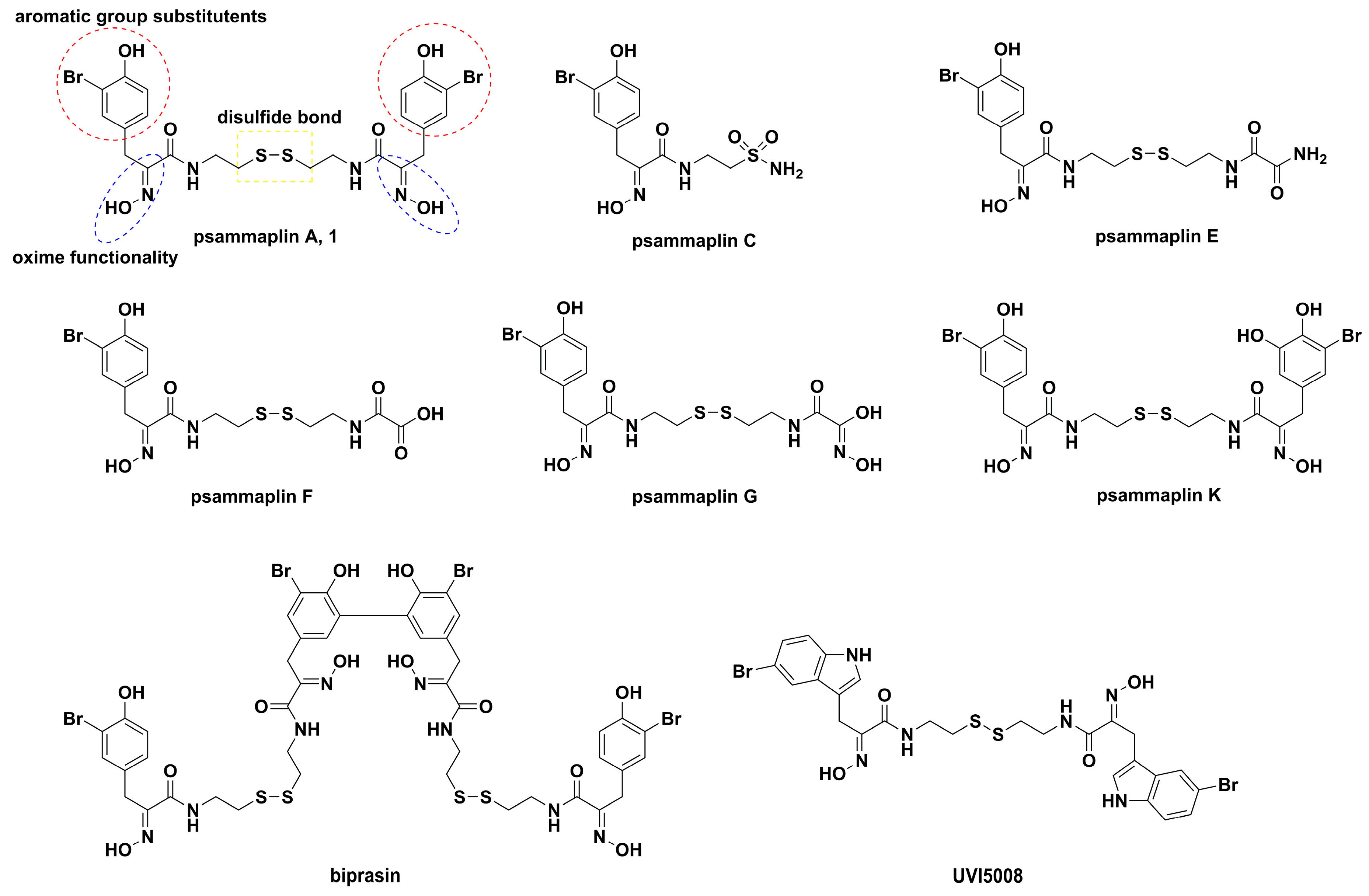{kind=link}
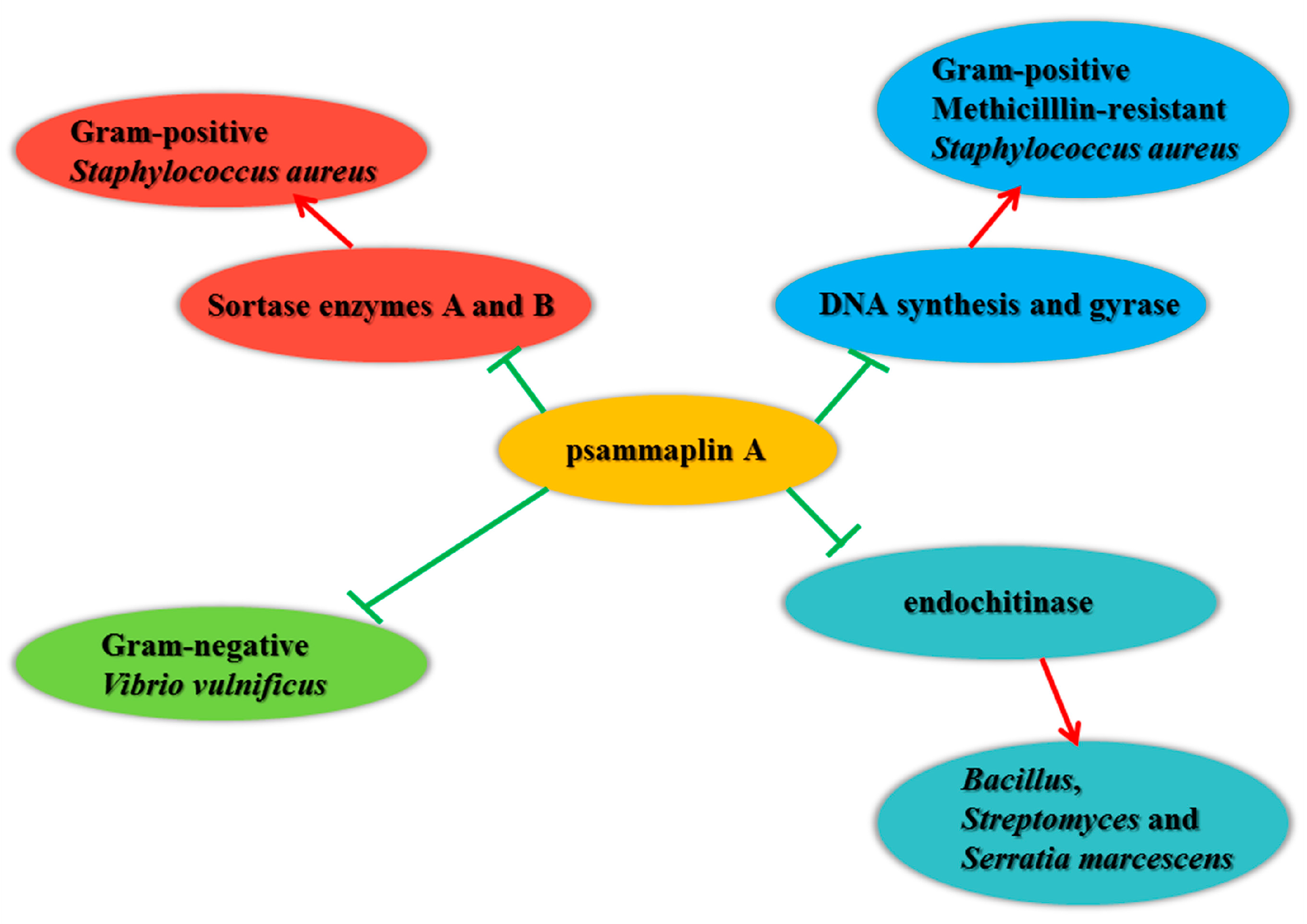{kind=link}
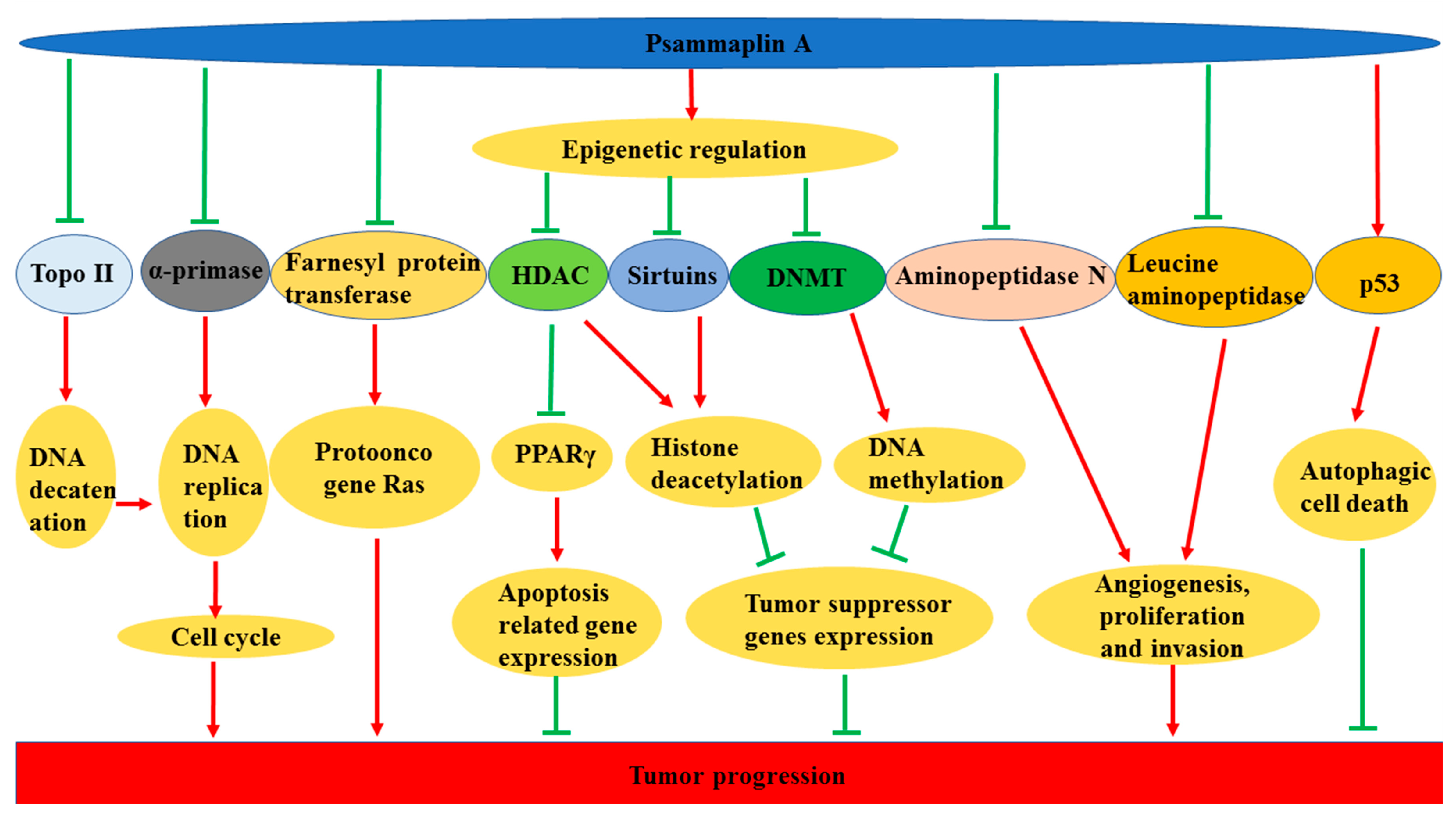{kind=link}
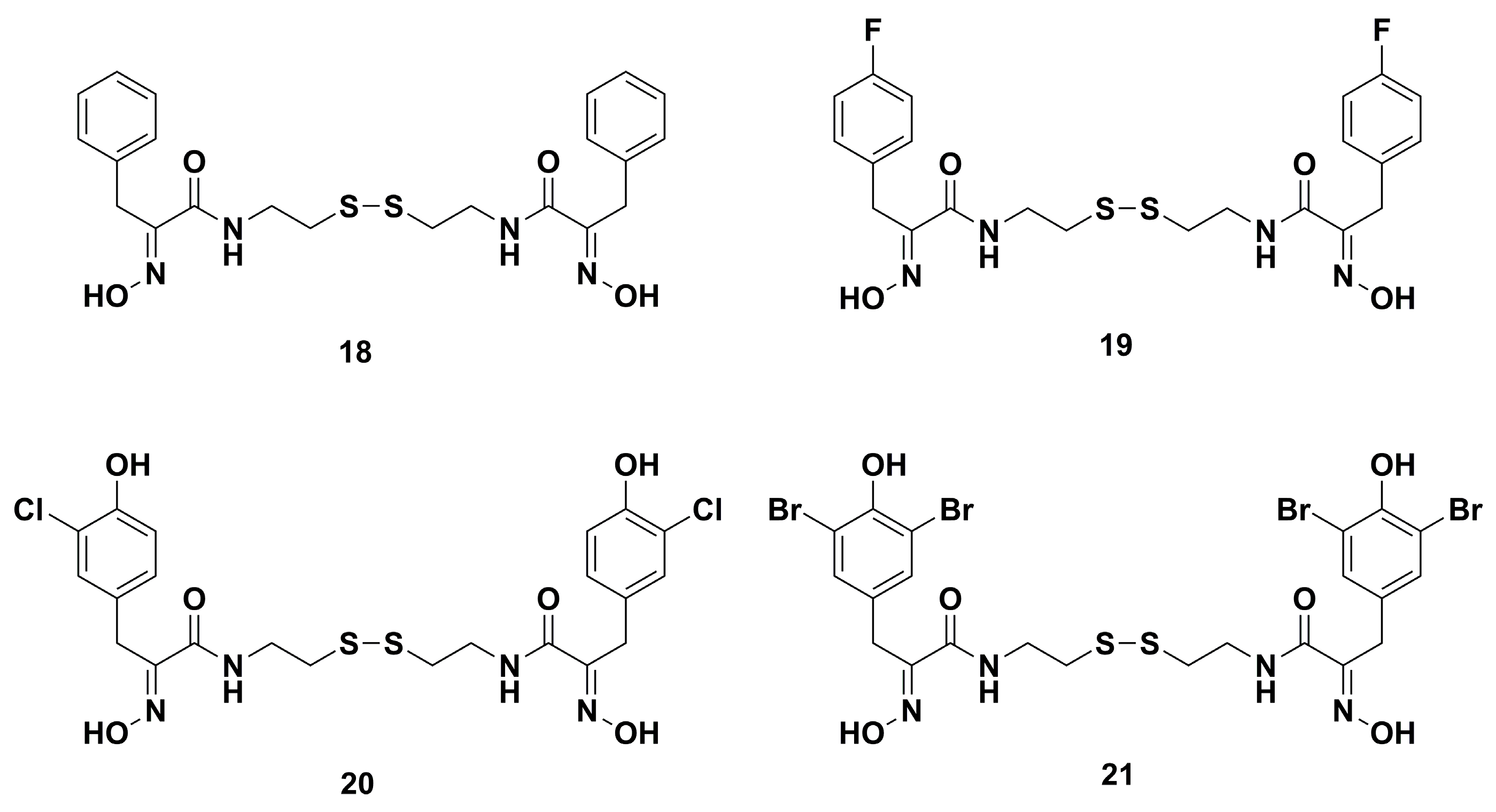{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
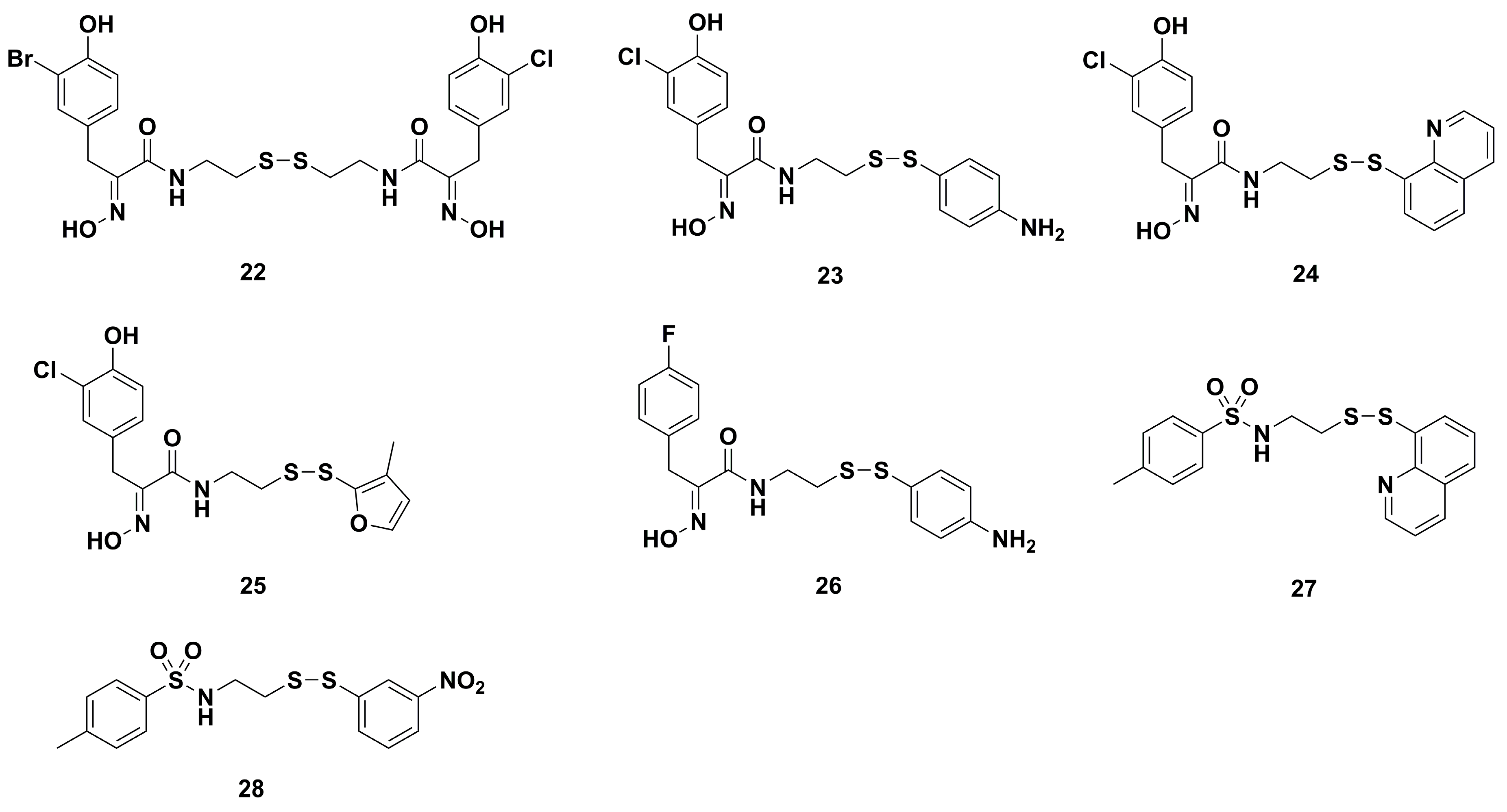
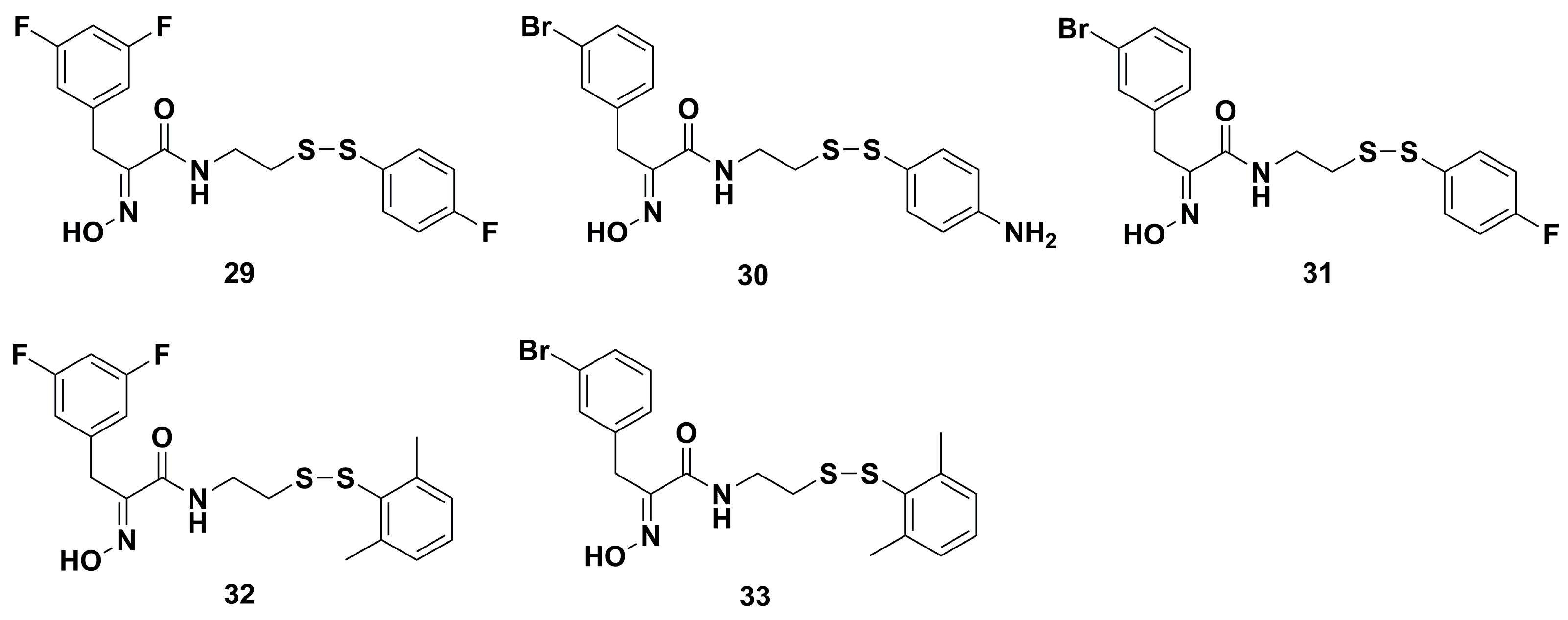
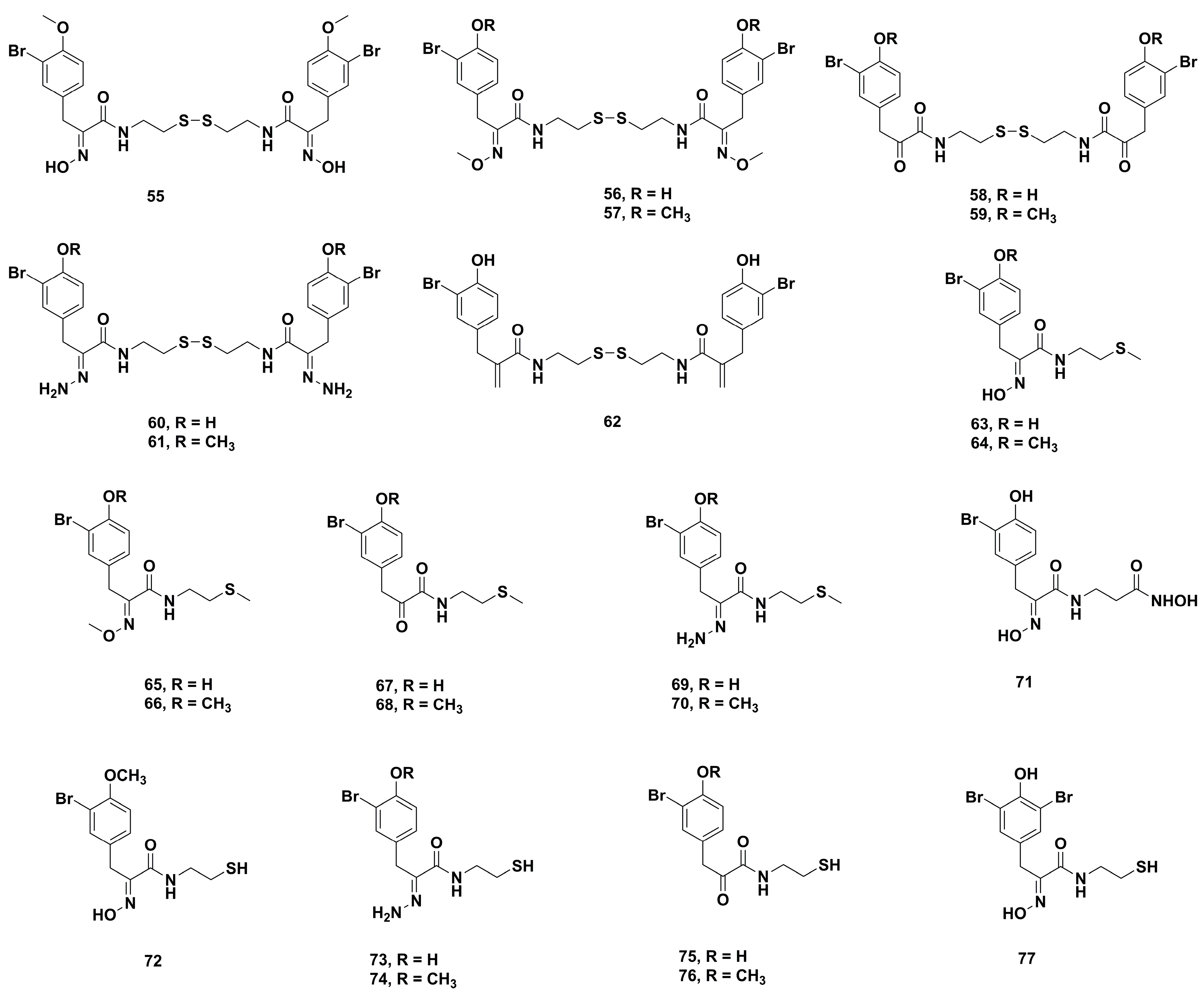
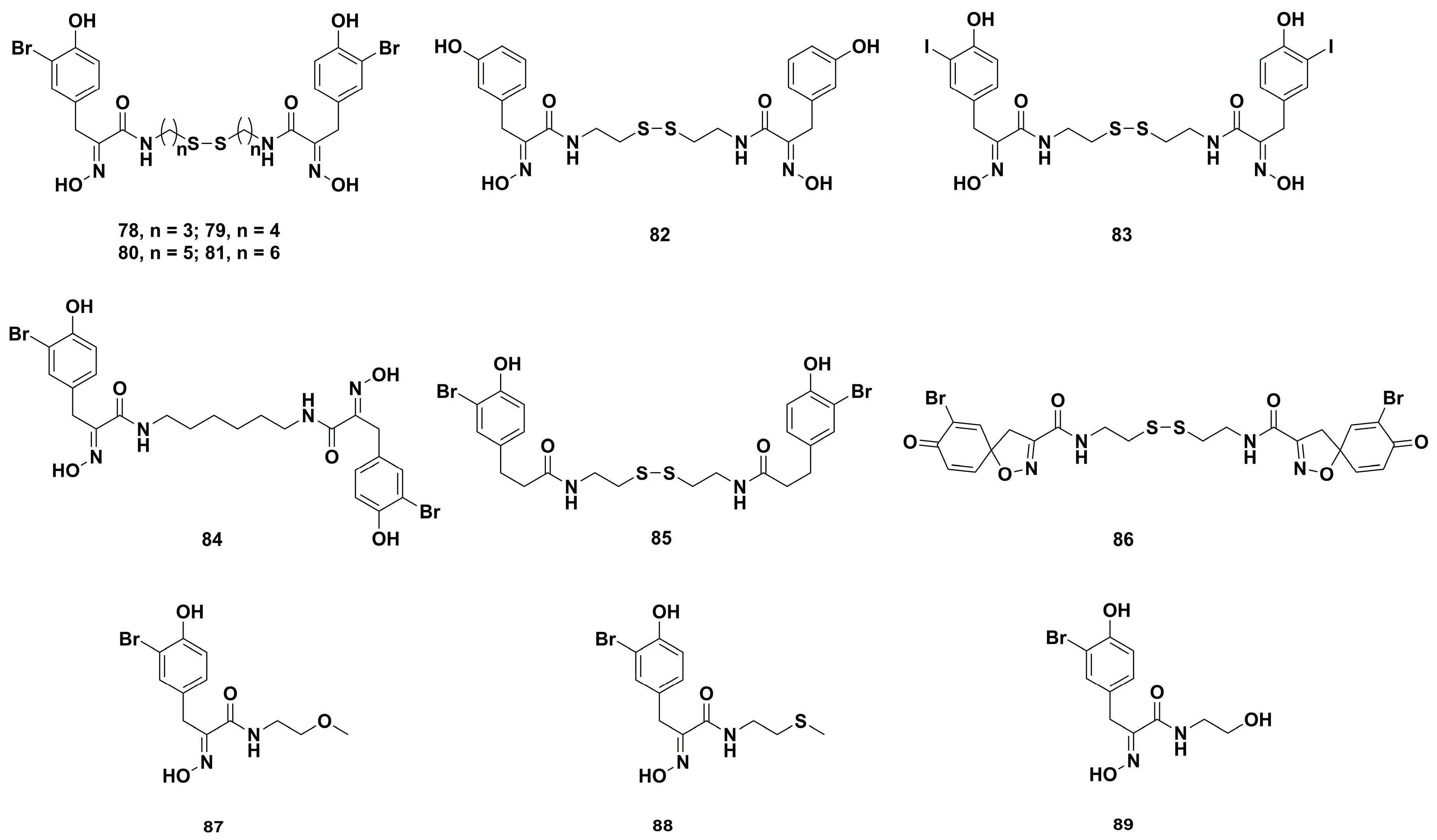
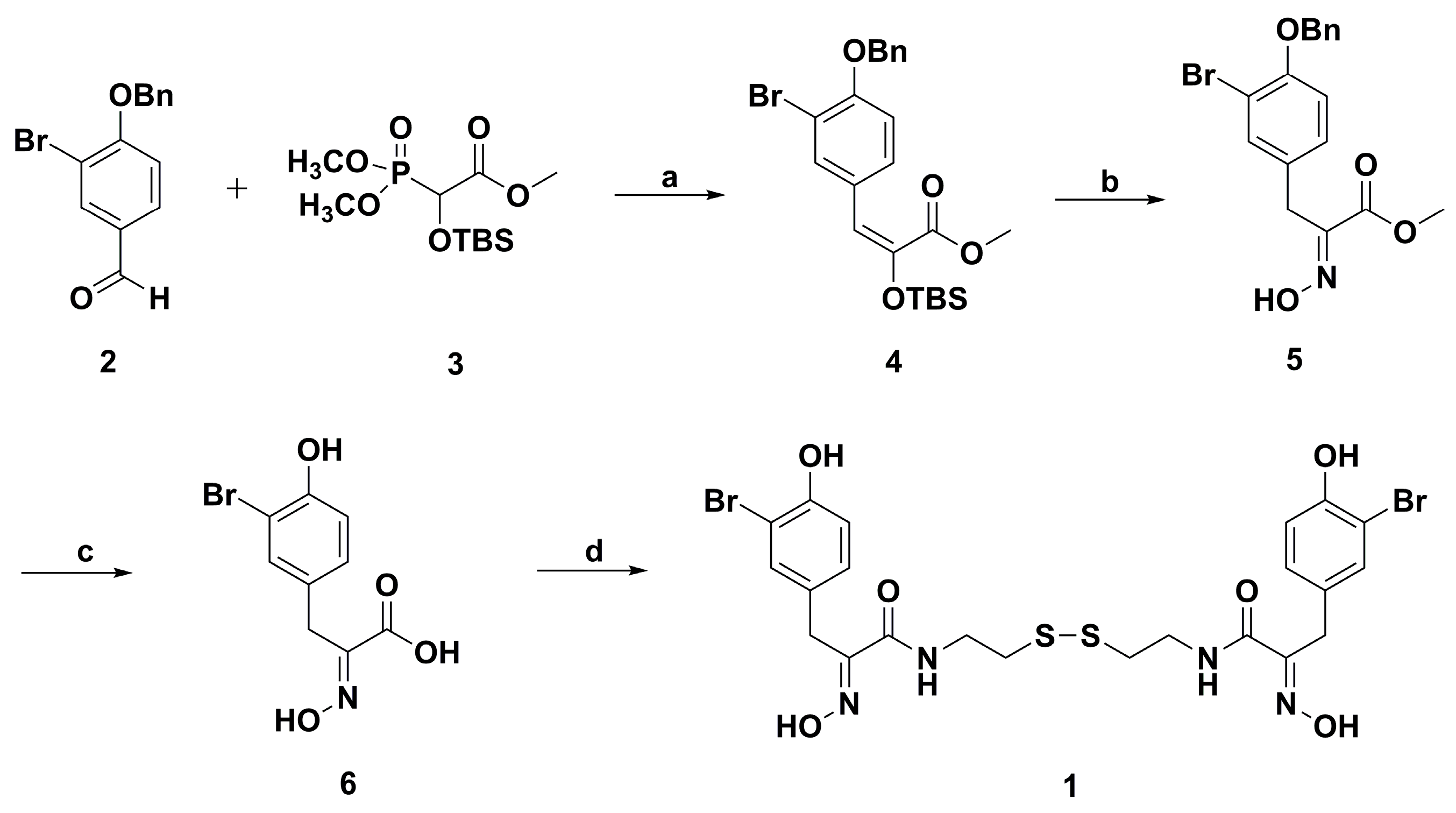
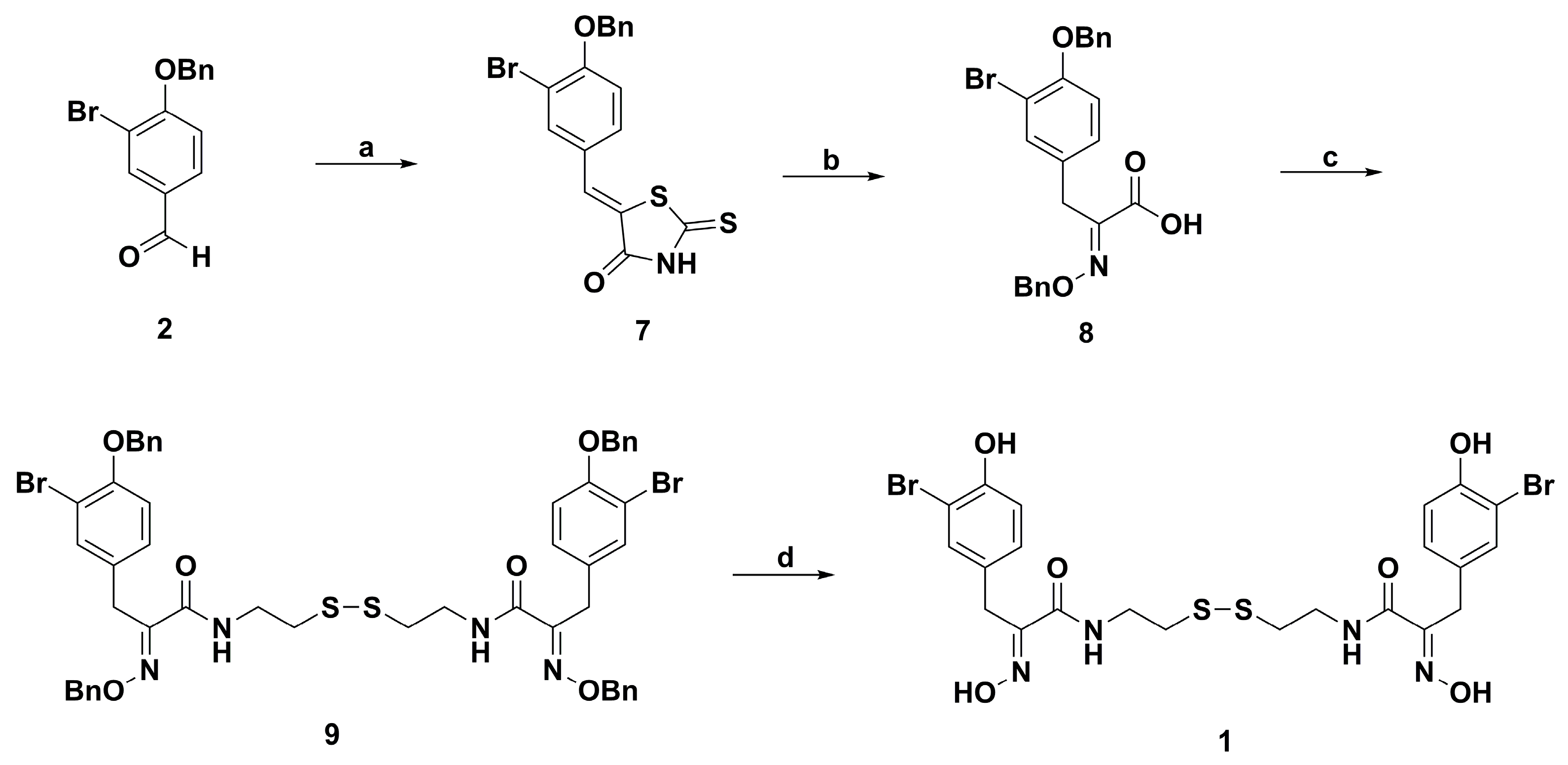
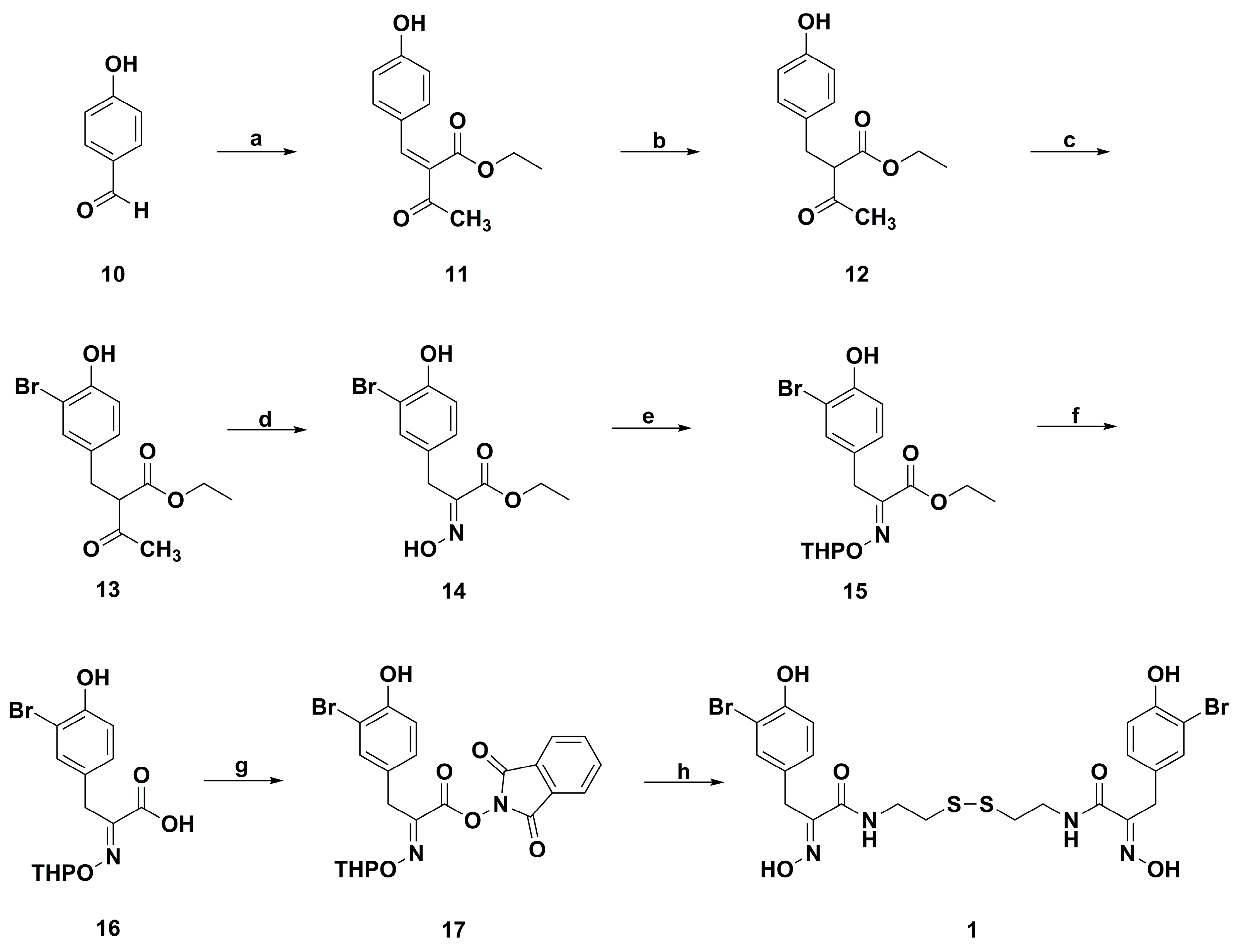
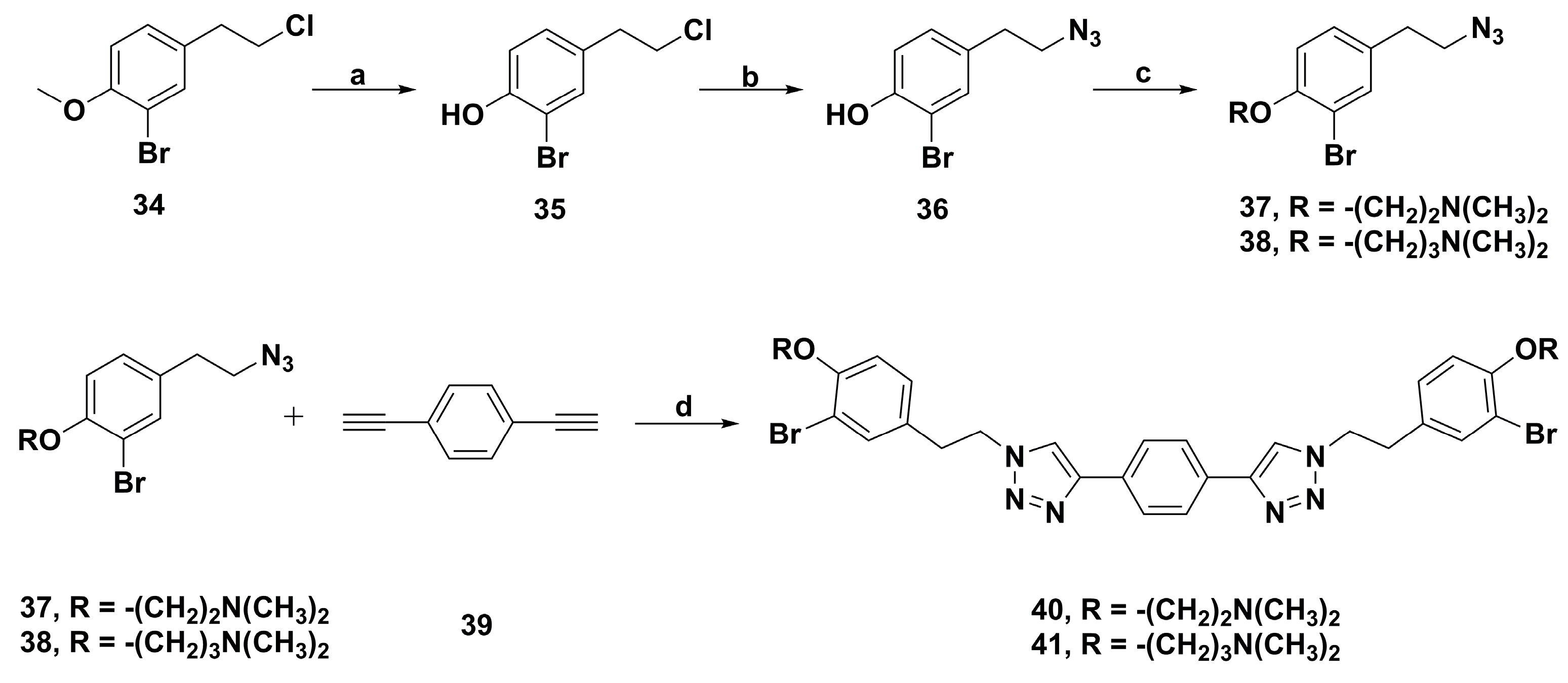
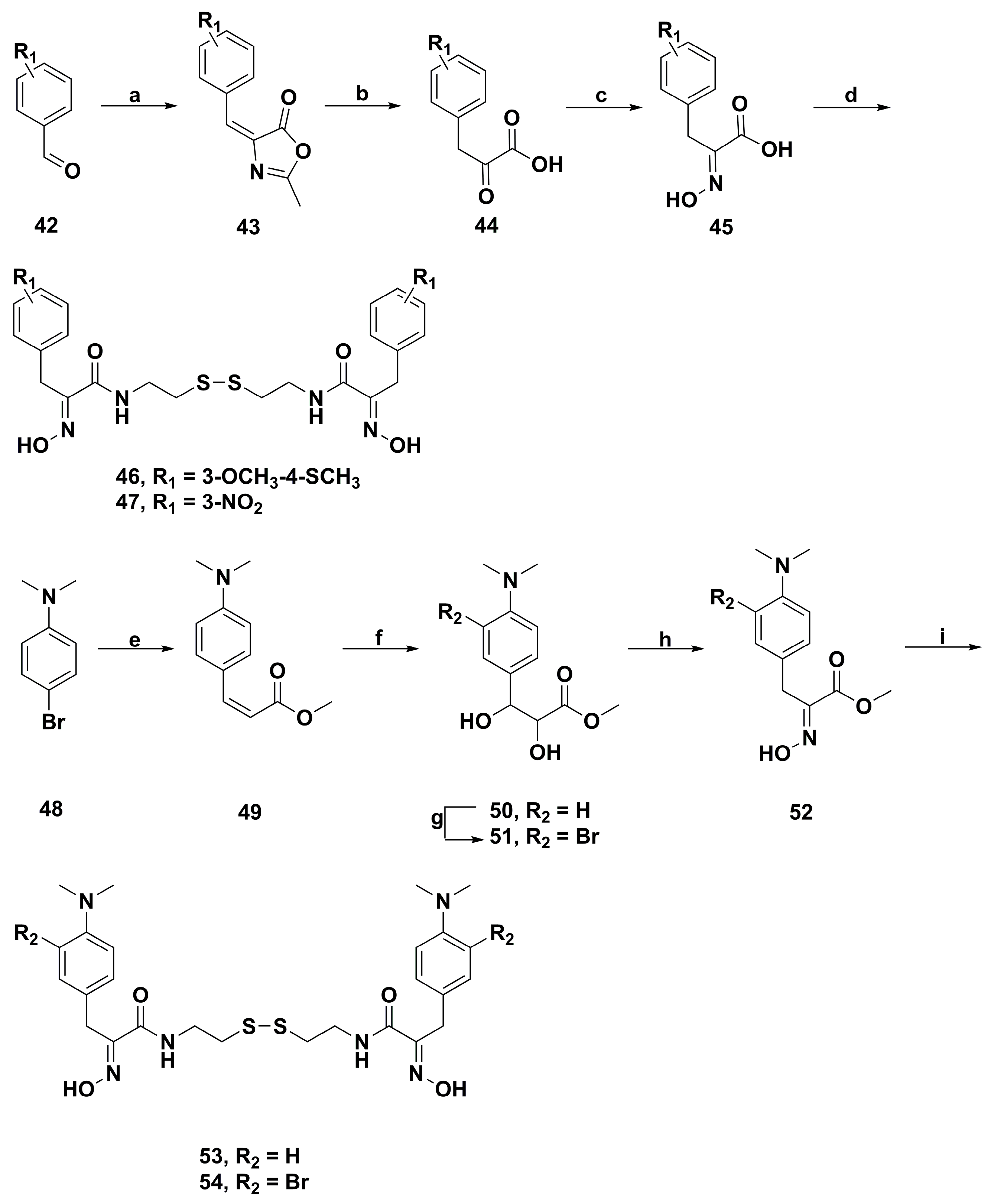
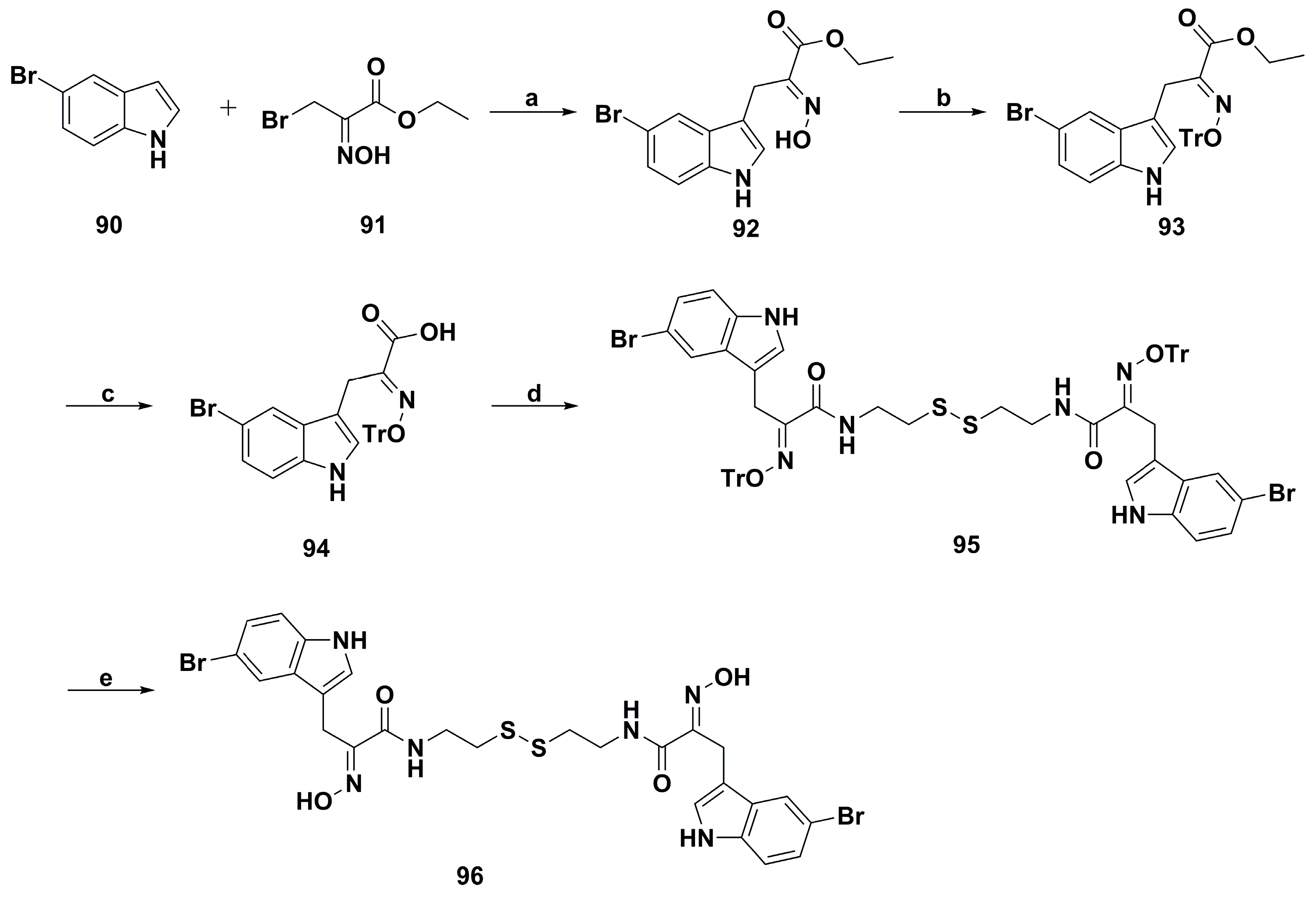
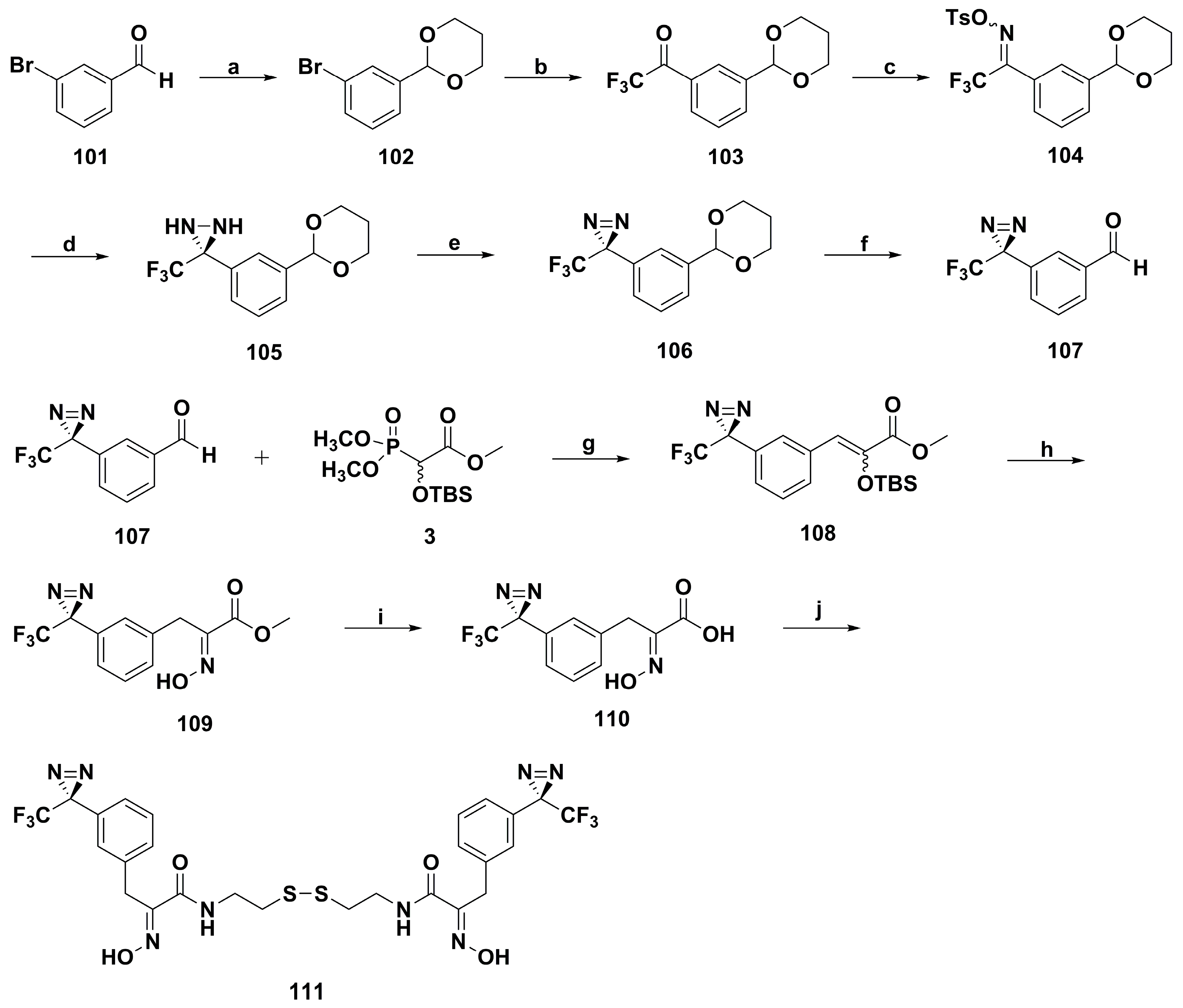
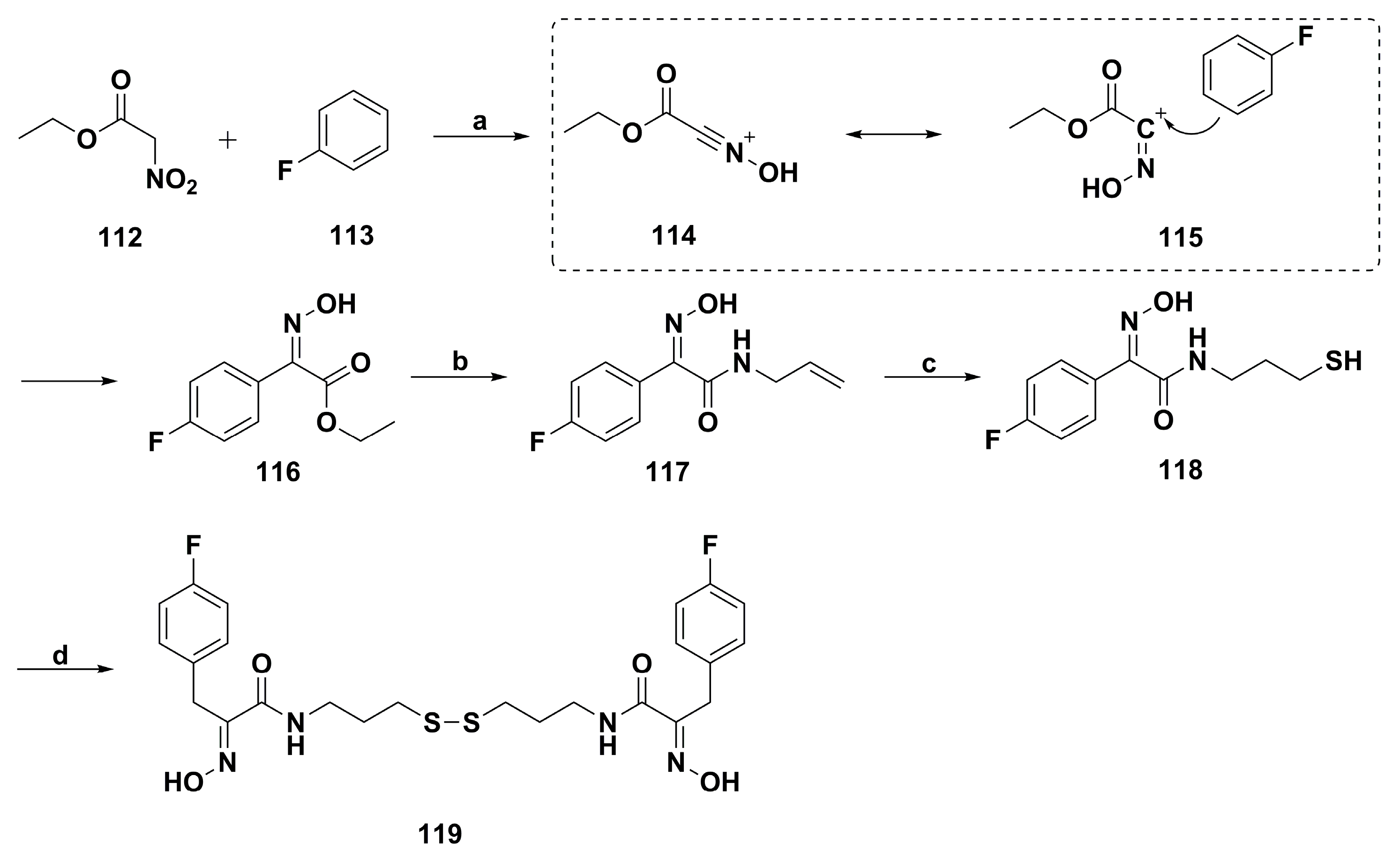
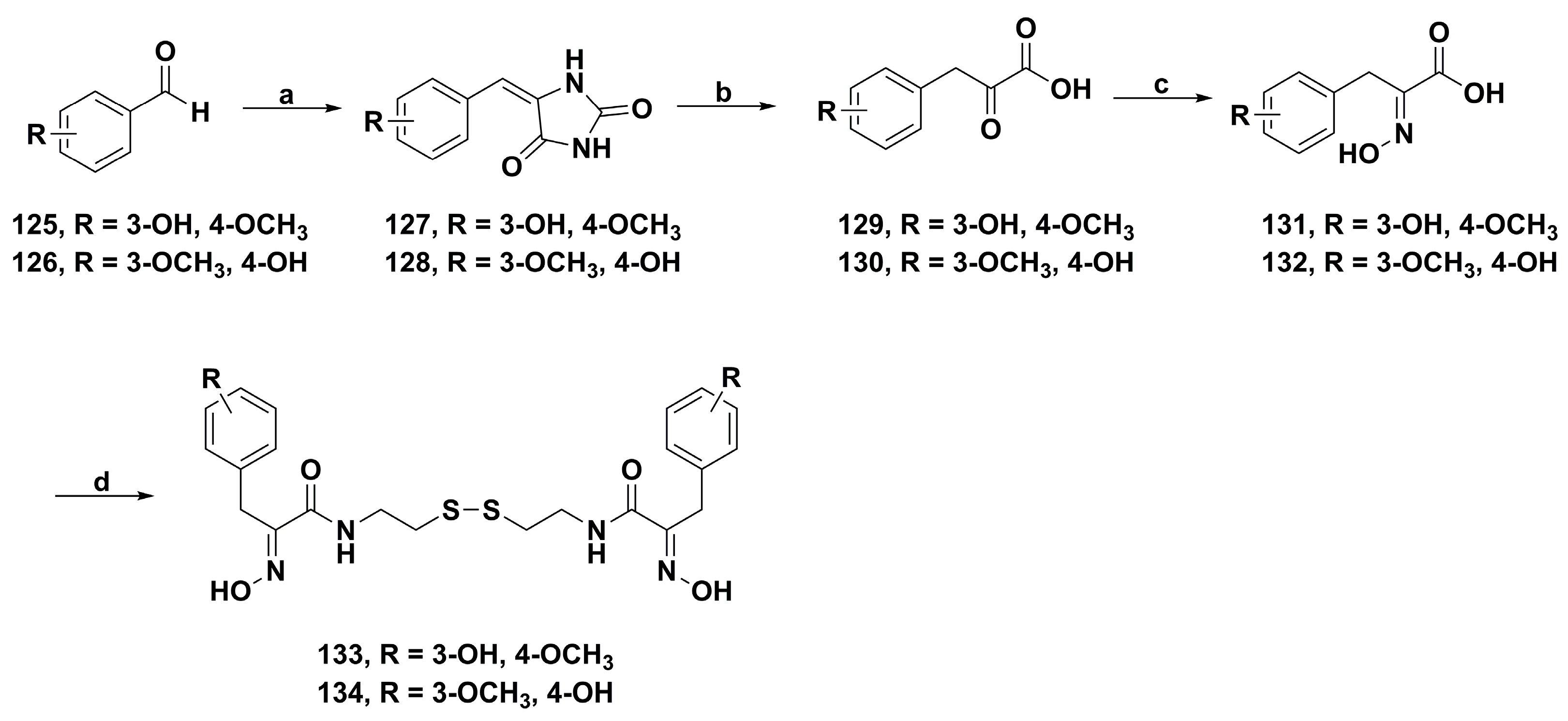
2. Synthetic Chemistry
3. Pharmacological Activity
3.1. Antimicrobial Activity
3.2. Antiviral Activity
3.3. Embryo Development Promotive Activity
3.4. Insecticidal Activity
3.5. Active Chemical Defense
3.6. Eryptosis Induction Acitivity
3.7. Anticancer Effects
4. Medicinal Chemistry
4.1. Antibacterial Derivatives
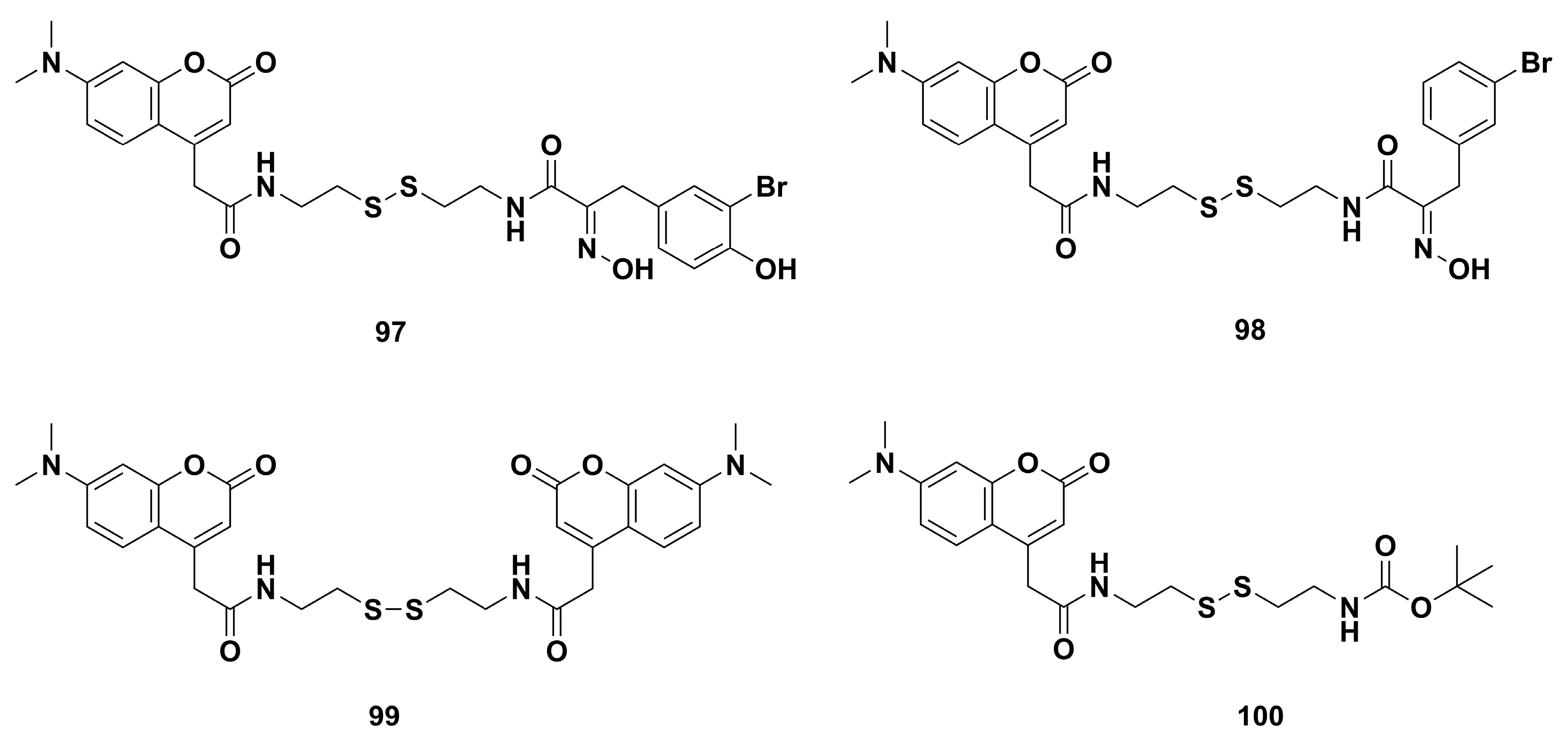
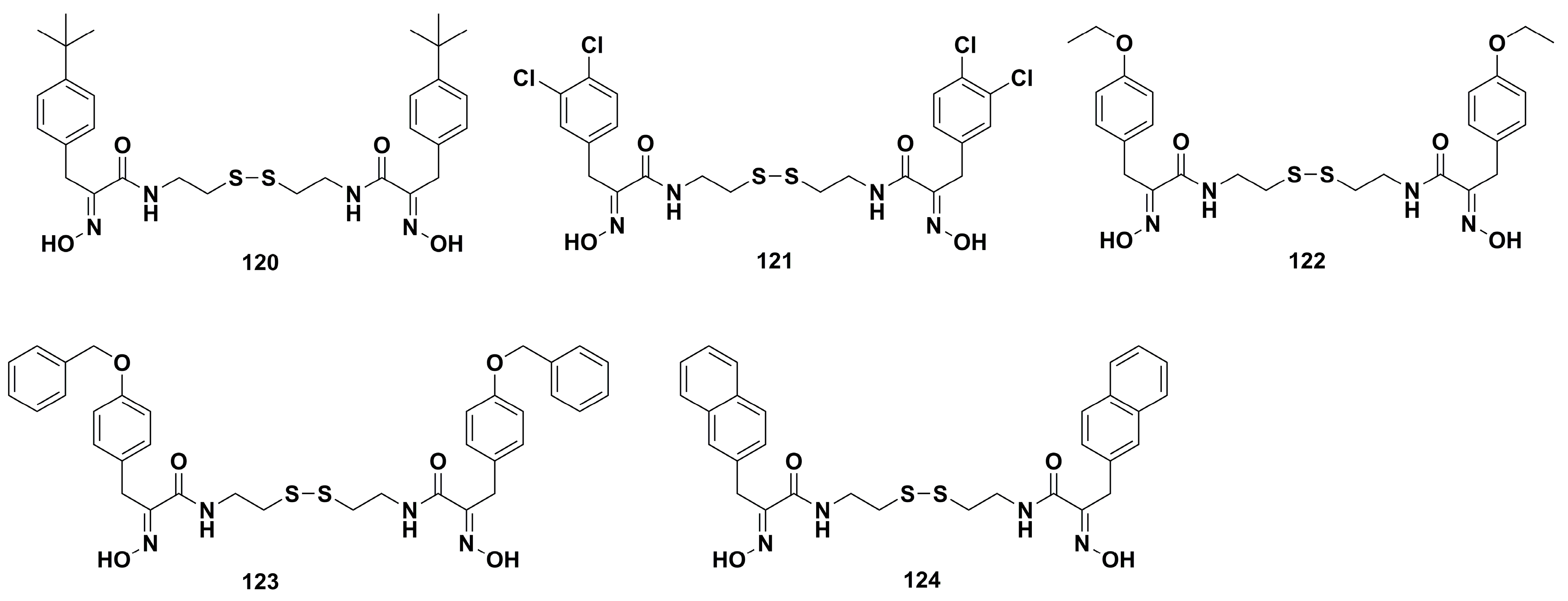
4.2. Anticancer Derivatives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Paterson, I.; Anderson, E.A. The renaissance of natural products as drug candidates. Science 2005, 310, 451. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzer, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [Green Version]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef]
- Sparks, T.C.; Hahn, D.R.; Garizi, N.V. Natural products, their derivatives, mimics and synthetic equivalents: Role in agrochemical discovery. Pest. Manag. Sci. 2017, 73, 700–715. [Google Scholar] [CrossRef]
- Mishra, B.B.; Tiwari, V.K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem. 2011, 46, 4769–4807. [Google Scholar] [CrossRef]
- Tang, C.; Wu, B.; Wu, J.; Zhang, Z.; Yu, B. Novel strategies using total gastrodin and gastrodigenin, or total gastrodigenin for quality control of gastrodia elata. Molecules 2018, 23, 270. [Google Scholar] [CrossRef]
- Soldatou, S.; Baker, B.J. Cold-water marine natural products, 2006 to 2016. Nat. Prod. Rep. 2017, 34, 585–626. [Google Scholar] [CrossRef]
- Chen, J.; Wang, B.; Lu, Y.; Guo, Y.; Sun, J.; Wei, B.; Zhang, H.; Wang, H. Quorum sensing inhibitors from marine microorganisms and their synthetic derivatives. Mar. Drugs 2019, 17, 80. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, A.; Naughton, L.M.; Montánchez, I.; Dobson, A.D.W.; Rai, D.K. Current status and future prospects of marine natural products (MNPs) as antimicrobials. Mar. Drugs 2017, 15, 272. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F. Have marine natural product drug discovery efforts been productive and how can we improve their efficiency? Expert Opin. Drug Discov. 2019, 15, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, Z.; Cui, W. Marine-derived natural compounds for the treatment of Parkinson’s disease. Mar. Drugs 2019, 157, 221. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Nay, B.; Yang, M.; Ni, Y.; Wang, H.; Yao, L.; Li, X. Marine sponges of the genus Stelletta as promising drug sources: Chemical and biological aspects. Acta. Pharm. Sin. B 2019, 9, 237–257. [Google Scholar] [CrossRef]
- Mudit, M.; El Sayed, K.A. Cancer control potential of marine natural product scaffolds through inhibition of tumor cell migration and invasion. Drug Discov. Today 2016, 21, 1745–1760. [Google Scholar] [CrossRef]
- Mayer, A.M.S.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef]
- Olivera, B.M.; Cruz, L.J.; de Santos, V.; LeCheminant, G.W.; Griffin, D.; Zeikus, R.; McIntosh, J.M.; Galyean, R.; Varga, J.; Gray, W.R. Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using omega-conotoxin from Conus magus venom. Biochemistry 1987, 26, 2086–2090. [Google Scholar] [CrossRef]
- Heydari, B.; Abdullah, S.; Pottala, J.V.; Shah, R.; Abbasi, S.; Mandry, D.; Francis, S.A.; Lumish, H.; Ghoshhajra, B.B.; Hoffmann, U.; et al. Effect of omega-3 acid ethyl esters on left ventricular remodeling after acute myocardial infarction: The OMEGA-REMODEL randomized clinical trial. Circulation 2016, 134, 378–391. [Google Scholar] [CrossRef]
- Newland, A.M.; Li, J.X.; Wasco, L.E.; Aziz, M.T.; Lowe, D.K. Brentuximab vedotin: A CD30-directed antibody-cytotoxic drug conjugate. Pharmacotherapy 2013, 33, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Kamath, K.; Manna, T.; Okouneva, T.; Miller, H.P.; Davis, C.; Littlefield, B.A.; Wilson, L. The primary antimitotic mechanism of action of the synthetic halichondrin e7389 is suppression of microtubule growth. Mol. Cancer Ther. 2005, 4, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Quiñoà, E.; Crews, P. Phenolic constituents of Psammaplysilla. Tetrahedron Lett. 1987, 28, 3229–3232. [Google Scholar] [CrossRef]
- Rodriguez, A.D.; Akee, R.K.; Scheuer, P.J. Two bromotyrosine-cysteine derived metabolites from a sponge. Tetrahedron Lett. 1987, 28, 4989–4992. [Google Scholar] [CrossRef]
- Arabshahi, L.; Schmitz, F.J. Brominated tyrosine metabolites from an unidentified sponge. J. Org. Chem. 1987, 52, 3584–3586. [Google Scholar] [CrossRef]
- Mujumdar, P.; Teruya, K.; Tonissen, K.F.; Vullo, D.; Supuran, C.T.; Peat, T.S.; Poulsen, S.A. An unusual natural product primary sulfonamide: Synthesis, carbonic anhydrase inhibition, and protein X-ray structures of psammaplin C. J. Med. Chem. 2016, 59, 5462–5470. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, D.; Sun, D.; Yang, S.; Hu, G.; Wu, Z.; Zhao, L. Synthesis of the marine bromotyrosine psammaplin F and crystal structure of a psammaplin A analogue. Molecules 2010, 15, 8784–8795. [Google Scholar] [CrossRef] [PubMed]
- Pina, I.C.; Gautschi, J.T.; Wang, G.Y.S.; Sanders, M.L.; Schmitz, F.J.; France, D.; Cornell-Kennon, S.; Sambucetti, L.C.; Remiszewski, S.W.; Perez, L.B.; et al. Psammaplins from the sponge Pseudoceratina purpurea: Inhibition of both histone deacetylase and DNA methyltransferase. J. Org. Chem. 2003, 68, 3866–3873. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Liu, Y.; Hong, J.; Lee, C.O.; Cho, H.; Kim, D.K.; Im, K.S.; Jung, J.H. New bromotyrosine derivatives from an association of two sponges, Jaspis wondoensis and Poecillastra wondoensis. J. Nat. Prod. 2003, 66, 1495–1498. [Google Scholar] [CrossRef]
- Kim, D.; Lee, I.S.; Jung, J.H.; Yang, S.I. Psammaplin A, a natural bromotyrosine derivative from a sponge, possesses the antibacterial activity against methicillin-resistant Staphylococcus aureus and the DNA gyrase-inhibitory activity. Arch. Pharm. Res. 1999, 22, 25–29. [Google Scholar] [CrossRef]
- Zhou, Y.D.; Li, J.; Du, L.; Mahdi, F.; Le, T.P.; Chen, W.L.; Swanson, S.M.; Watabe, K.; Nagle, D.G. Biochemical and anti-triple negative metastatic breast tumor cell properties of psammaplins. Mar. Drugs 2018, 16, 442. [Google Scholar] [CrossRef]
- Ahn, M.Y.; Jung, J.H.; Na, Y.J.; Kim, H.S. A natural histone deacetylase inhibitor, psammaplin A, induces cell cycle arrest and apoptosis in human endometrial cancer cells. Gynecol. Oncol. 2008, 108, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Kim, H.S.; Kang, Y.J.; Yoon, S.; Lee, J.; Choi, W.S.; Jung, J.H.; Kim, H.S. Psammaplin A induces sirtuin 1-dependent autophagic cell death in doxorubicin-resistant MCF-7/adr human breast cancer cells and xenografts. Biochim. Biophys. Acta. 2015, 1850, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, E.A. Tumor protein (TP)-p53 members as regulators of autophagy in tumor cells upon marine drug exposure. Mar. Drugs 2016, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, I.S.; Jung, J.H.; Lee, C.O.; Choi, S.U. Psammaplin A, a natural phenolic compound, has inhibitory effect on human topoisomerase II and is cytotoxic to cancer cells. Anticancer Res. 1999, 19, 4085–4090. [Google Scholar] [PubMed]
- Tabudravu, J.N.; Eijsink, V.G.H.; Gooday, G.W.; Jaspars, M.; Komander, D.; Legg, M.; Synstad, B.; van Aalten, D.M.F. Psammaplin A, a chitinase inhibitor isolated from the Fijian marine sponge Aplysinella rhax. Bioorg. Med. Chem. 2002, 10, 1123–1128. [Google Scholar] [CrossRef]
- Shin, J.; Lee, H.S.; Seo, Y.; Rho, J.R.; Cho, K.W.; Paul, V.J. New bromotyrosine metabolites from the sponge Aplysinella rhax. Tetrahedron 2000, 56, 9071–9077. [Google Scholar] [CrossRef]
- Nicholas, G.M.; Eckman, L.L.; Ray, S.; Hughes, R.O.; Pfefferkorn, J.A.; Barluenga, S.; Nicolaou, K.C.; Bewley, C.A. Bromotyrosine-derived natural and synthetic products as inhibitors of mycothiol-s-conjugate amidase. Bioorg. Med. Chem. Lett. 2002, 12, 2487–2490. [Google Scholar] [CrossRef]
- Jiang, Y.; Ahn, E.Y.; Ryu, S.H.; Kim, D.K.; Park, J.S.; Yoon, H.J.; Yoo, S.; Lee, B.J.; Lee, D.S.; Jung, J.H. Cytotoxicity of psammaplin A from a two-sponge association may correlate with the inhibition of DNA replication. BMC Cancer 2004, 4, 70. [Google Scholar] [CrossRef]
- Shim, J.S.; Lee, H.S.; Shin, J.; Kwon, H.J. Psammaplin A, a marine natural product, inhibits aminopeptidase N and suppresses angiogenesis in vitro. Cancer Lett. 2004, 203, 163–169. [Google Scholar] [CrossRef]
- Kim, D.H.; Shin, J.; Kwon, H.J. Psammaplin A is a natural prodrug that inhibits class I histone deacetylase. Exp. Mol. Med. 2007, 39, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Benedetti, R.; Pérez-Rodríguez, S.; Nebbioso, A.; García-Rodríguez, J.; Carafa, V.; Stuhldreier, M.; Conte, M.; Rodríguez-Barrios, F.; Stunnenberg, H.G.; et al. Indole-derived psammaplin A analogues as epigenetic modulators with multiple inhibitory activities. J. Med. Chem. 2012, 55, 9467–9491. [Google Scholar] [CrossRef]
- Hentschel, F.; Lindel, T. Synthesis of oximinotyrosine-derived marine natural products. Synthesis 2010, 2, 181–204. [Google Scholar]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [PubMed]
- Malve, H. Exploring the ocean for new drug developments: Marine pharmacology. J. Pharm. Bioallied. Sci. 2016, 8, 83–91. [Google Scholar] [CrossRef]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [Green Version]
- Datta, D.; Talapatra, S.N.; Swarnakar, S. Bioactive compounds from marine invertebrates for potential medicines-an overview. Int. Lett. Nat. Sci. 2015, 34, 42–61. [Google Scholar] [CrossRef]
- Lindequist, U. Marine-derived pharmaceuticals-challenges and opportunities. Biomol. Ther. 2016, 24, 561–571. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef]
- Kanase, H.R.; Singh, K.M. Marine pharmacology: Potential, challenges, and future in India. J. Med. Sci. 2018, 38, 49–53. [Google Scholar]
- Shinde, P.; Banerjee, P.; Mandhare, A. Marine natural products as source of new drugs: A patent review (2015–2018). Expert Opin. Ther. Pat. 2019, 29, 283–309. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Xiong, Y.; Qi, X.; Tang, W.; Dai, J.; Gu, Q.; Li, J. Molecular targets of active anticancer compounds derived from marine sources. Mar. Drugs 2018, 16, 175. [Google Scholar] [CrossRef] [PubMed]
- Calcabrini, C.; Catanzaro, E.; Bishayee, A.; Turrini, E.; Fimognari, C. Marine sponge natural products with anticancer potential: An updated review. Mar. Drugs 2017, 15, 310. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ding, T.; Li, J. Medicinal purposes: Bioactive metabolites from marine-derived organisms. Mini Rev. Med. Chem. 2019, 19, 138–164. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Sharma, M.; Joshi, P.; Rawat, D.S. Clinical status of anti-cancer agents derived from marine sources. Anticancer Agents Med. Chem. 2008, 8, 603–617. [Google Scholar] [CrossRef]
- Adrian, T.E. Novel marine-derived anti-cancer agents. Curr. Pharm. Des. 2007, 13, 3417–3426. [Google Scholar] [CrossRef]
- Jaiganesh, R.; Sampath Kumar, N.S. Marine bacterial sources of bioactive compounds. Adv. Food Nutr. Res. 2012, 65, 389–408. [Google Scholar]
- Peng, J.; Li, J.; Hamann, M.T. The marine bromotyrosine derivatives. Alkaloids Chem. Biol. 2005, 61, 59–262. [Google Scholar]
- Godert, A.M.; Angelino, N.; Read, A.W.; Morey, S.R.; James, S.R.; Karpf, A.R.; Sufrin, J.R. An improved synthesis of psammaplin A. Bioorg. Med. Chem. Lett. 2006, 16, 3330–3333. [Google Scholar] [CrossRef]
- García, J.; Franci, G.; Pereira, R.; Benedetti, R.; Nebbioso, A.; Rodríguez-Barrios, F.; Gronemeyer, H.; Altucci, L.; de Lera, A.R. Epigenetic profiling of the antitumor natural product psammaplin A and its analogues. Bioorg. Med. Chem. 2011, 19, 3637–3649. [Google Scholar] [CrossRef] [Green Version]
- Nicolaou, K.C.; Hughes, R.; Pfefferkorn, J.A.; Barluenga, S.; Roecker, A.J. Combinatorial synthesis through disulfide exchange: Discovery of potent psammaplin A type antibacterial agents active against methicillin-resistant Staphylococcus aureus (MRSA). Chem. Eur. J. 2001, 7, 4280–4295. [Google Scholar] [CrossRef]
- Hoshino, O.; Murakata, M.; Yamada, K. A convenient synthesis of a bromotyrosine derived metabolite, psammaplin A, from Psammaplysilla sp. Bioorg. Med. Chem. Lett. 1992, 2, 1561–1562. [Google Scholar] [CrossRef]
- Hentschel, F.; Sasse, F.; Lindel, T. Fluorescent analogs of the marine natural product psammaplin A: Synthesis and biological activity. Org. Biomol. Chem. 2012, 10, 7120–7133. [Google Scholar] [CrossRef] [PubMed]
- Kottakota, S.K.; Benton, M.; Evangelopoulos, D.; Guzman, J.D.; Bhakta, S.; McHugh, T.D.; Gray, M.; Groundwater, P.W.; Marrs, E.C.; Perry, J.D.; et al. Versatile routes to marine sponge metabolites through benzylidene rhodanines. Org. Lett. 2012, 14, 6310–6313. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Shin, Y.; Jung, M.; Ha, M.W.; Park, Y.; Lee, Y.J.; Shin, J.; Oh, K.B.; Lee, S.K.; Park, H.G. Efficient synthesis and biological activity of Psammaplin A and its analogues as antitumor agents. Eur. J. Med. Chem. 2015, 96, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Lee, M.; Jung, M.; Park, Y.; Kim, M.Y.; Park, H.G. Efficient synthetic method of Psammaplin, A. Tetrahedron Lett. 2012, 53, 4209–4211. [Google Scholar] [CrossRef]
- Oh, K.B.; Oh, M.N.; Kim, J.G.; Shin, D.S.; Shin, J. Inhibition of sortase-mediated Staphylococcus aureus adhesion to fibronectin via fibronectin-binding protein by sortase inhibitors. Appl. Microbiol. Biotechnol. 2006, 70, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Franci, G.; Folliero, V.; Cammarota, M.; Zannella, C.; Sarno, F.; Schiraldi, C.; Lera, A.R.; Altucci, L.; Galdiero, M. Epigenetic modulator UVI5008 inhibits MRSA by interfering with bacterial gyrase. Sci. Rep. 2018, 8, 13117. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Lee, A.; Jung, J.H.; Choi, S.H.; Kim, T.S. In vitro and in vivo anti-vibrio vulnificus activity of psammaplin a, a natural marine compound. Mol. Med. Rep. 2016, 14, 2691–2696. [Google Scholar] [CrossRef] [PubMed]
- Salam, K.A.; Furuta, A.; Noda, N.; Tsuneda, S.; Sekiguchi, Y.; Yamashita, A.; Moriishi, K.; Nakakoshi, M.; Tsubuki, M.; Tani, H.; et al. Psammaplin A inhibits hepatitis C virus NS3 helicase. J. Nat. Med. 2013, 67, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Richard, K.; Williams, D.E.; de Silva, E.D.; Brockman, M.A.; Brumme, Z.L.; Andersen, R.J.; Tietjen, I. Identification of novel HIV-1 latency-reversing agents from a library of marine natural products. Viruses 2018, 10, 348. [Google Scholar] [CrossRef]
- Mallol, A.; Santaló, J.; Ibáñez, E. Psammaplin A improves development and quality of somatic cell nuclear transfer mouse embryos. Cell Reprogram. 2014, 16, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Mallol, A.; Piqué, L.; Santaló, J.; Ibáñez, E. Morphokinetics of cloned mouse embryos treated with epigenetic drugs and blastocyst prediction. Reproduction 2016, 151, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiusen, T.J.; Kamble-Shripat, T. Delayed toxicity of two chitinolytic enzyme inhibitors (Psammaplin A and Pentoxifylline) against eastern subterranean termites (Isoptera: Rhinotermitidae). J. Entomol. Sci. 2013, 106, 1788–1793. [Google Scholar] [CrossRef]
- Husen, T.J.; Kamble, S.T. An evaluation of chitinase inhibitors, psammaplin A and pentoxifylline, treated diets against the eastern subterranean teimite (Isoptera: Rhinotermitidae). J. Entomol. Sci. 2014, 49, 228–245. [Google Scholar] [CrossRef]
- Saguez, J.; Hainez, R.; Cherqui, A.; Van Wuytswinkel, O.; Jeanpierre, H.; Lebon, G.; Noiraud, N.; Beaujean, A.; Jouanin, L.; Laberche, J.C.; et al. Unexpected effects of chitinases on the peach-potato aphid (Myzus persicae Sulzer) when delivered via transgenic potato plants (Solanum tuberosum Linné) and in vitro. Transgenic. Res. 2005, 14, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Francis, F.; Saguez, J.; Cherqui, A.; Vandermoten, S.; Vincent, C.; Versali, M.F.; Dommès, J.; De Pauw, E.; Giordanengo, P.; Haubruge, E. Purification and characterisation of a 31-kDa Chitinase from the myzus persicae aphid: A target for hemiptera biocontrol. Appl. Biochem. Biotechnol. 2012, 166, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Saguez, J.; Dubois, F.; Vincent, C.; Laberche, J.C.; Sangwan-Norreel, B.S.; Giordanengo, P. Differential aphicidal effects of chitinase inhibitors on the polyphagous homopteran Myzus persicae (Sulzer). Pest. Manag. Sci. 2006, 62, 1150–1154. [Google Scholar] [CrossRef]
- Thoms, C.; Schupp, P.J. Activated chemical defense in marine sponges—A case study on aplysinella rhax. J. Chem. Ecol. 2008, 34, 1242–1252. [Google Scholar] [CrossRef]
- Al Mamun Bhuyan, A.; Signoretto, E.; Lang, F. Triggering of suicidal erythrocyte death by psammaplin A. Cell. Physiol. Biochem. 2016, 39, 908–918. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, J.H.; Chie, E.K.; Young, P.D.; Kim, I.A.; Kim, I.H. DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radiat. Oncol. 2012, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Charkie, J. Psammaplin A: A putative adjuvant for DNA damaging therapies. J. Cancer Sci. Ther. 2014, 6, 505–509. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, T.H.; Seo, W.S.; Yoo, S.D.; Kim, I.H.; Joo, S.H.; Shin, S.; Park, E.S.; Ma, E.S.; Shin, B.S. Pharmacokinetics and tissue distribution of psammaplin A, a novel anticancer agent, in mice. Arch. Pharm. Res. 2012, 10, 1849–1854. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Pereira, R.; Khanwalkar, H.; Matarese, F.; García-Rodríguez, J.; Miceli, M.; Logie, C.; Kedinger, V.; Ferrara, F.; Stunnenberg, H.G.; et al. Death receptor pathway activation and increase of ROS production by the triple epigenetic inhibitor UVI5008. Mol. Cancer Ther. 2011, 10, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Mora, F.D.; Jones, D.K.; Desai, P.V.; Patny, A.; Avery, M.A.; Feller, D.R.; Smillie, T.; Zhou, Y.D.; Nagle, D.G. Bioassay for the identification of natural product-basedactivators of peroxisome proliferator-activated receptor-γ (PPARγ): The marine sponge metabolite psammaplin A activates PPARγ and induces apoptosis in human breast tumor cells. J. Nat. Prod. 2006, 69, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Baud, M.G.; Leiser, T.; Petrucci, V.; Gunaratnam, M.; Neidle, S.; Meyer-Almes, F.J.; Fuchter, M.J. Thioester derivatives of the natural product psammaplin A as potent histone deacetylase inhibitors. Beilstein. J. Org. Chem. 2013, 9, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Salaroglio, I.C.; Mujumdar, P.; Annovazzi, L.; Kopecka, J.; Mellai, M.; Schiffer, D.; Poulsen, S.A.; Riganti, C. Carbonic anhydrase XII inhibitors overcome P-glycoprotein-mediated resistance to temozolomide in glioblastoma. Mol. Cancer Ther. 2018, 17, 2598–2609. [Google Scholar] [CrossRef]
- Hall, D.G.; Manku, S.; Wang, F. Solution- and solid-phase strategies for the design, synthesis, and screening of libraries based on natural product templates: A comprehensive survey. J. Comb. Chem. 2001, 3, 125–150. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Hughes, R.; Pfefferkorn, J.A.; Barluenga, S. Optimization and mechanistic studies of psammaplin A type antibacterial agents active against methicillin-resistant Staphylococcus aureus (MRSA). Chem. Eur. J. 2001, 7, 4296–4310. [Google Scholar] [CrossRef]
- Andjouh, S.; Blache, Y. Parallel synthesis of a bis-triazoles library as psammaplin A analogues: A new wave of antibiofilm compounds? Bioorg. Med. Chem. Lett. 2019, 29, 614–618. [Google Scholar] [CrossRef]
- Baud, M.G.; Leiser, T.; Meyer-Almes, F.J.; Fuchter, M.J. New synthetic strategies towards psammaplin A, access to natural product analogues for biological evaluation. Org. Biomol. Chem. 2011, 9, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Baud, M.G.; Leiser, T.; Haus, P.; Samlal, S.; Wong, A.C.; Wood, R.J.; Petrucci, V.; Gunaratnam, M.; Hughes, S.M.; Buluwela, L.; et al. Defining the mechanism of action and enzymatic selectivity of psammaplin A against its epigenetic targets. J. Med. Chem. 2012, 55, 1731–1750. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; La Thangue, N.B. HDAC inhibitors in cancer biology: Emerging mechanisms and clinical applications. Immunol. Cell Biol. 2012, 90, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, F.; Raimer, B.; Kelter, G.; Fiebig, H.H.; Sasse, F.; Lindel, T. Synthesis and cytotoxicity of a diazirine-based photopsammaplin. Eur. J. Org. Chem. 2014, 2014, 2120–2127. [Google Scholar] [CrossRef]
- El Bahhaj, F.; Désiré, J.; Blanquart, C.; Martinet, N.; Zwick, V.; Simoes-Pires, C.; Cuendet, M.; Gregoire, M.; Bertrand, P. Superacid and thiol-ene reactions for access to psammaplin analogues with HDAC inhibition activities. Tetrahedron 2014, 70, 9702–9708. [Google Scholar] [CrossRef]
- Wen, J.; Bao, Y.; Niu, Q.; Liu, J.; Yang, J.; Wang, W.; Jiang, T.; Fan, Y.; Li, K.; Wang, J.; et al. Synthesis, biological evaluation and molecular modeling studies of psammaplin A and its analogs as potent histone deacetylases inhibitors and cytotoxic agents. Bioorg. Med. Chem. Lett. 2016, 26, 4372–4376. [Google Scholar] [CrossRef]



















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jing, Q.; Hu, X.; Ma, Y.; Mu, J.; Liu, W.; Xu, F.; Li, Z.; Bai, J.; Hua, H.; Li, D. Marine-Derived Natural Lead Compound Disulfide-Linked Dimer Psammaplin A: Biological Activity and Structural Modification. Mar. Drugs 2019, 17, 384. https://0-doi-org.brum.beds.ac.uk/10.3390/md17070384
Jing Q, Hu X, Ma Y, Mu J, Liu W, Xu F, Li Z, Bai J, Hua H, Li D. Marine-Derived Natural Lead Compound Disulfide-Linked Dimer Psammaplin A: Biological Activity and Structural Modification. Marine Drugs. 2019; 17(7):384. https://0-doi-org.brum.beds.ac.uk/10.3390/md17070384
Chicago/Turabian StyleJing, Qinxue, Xu Hu, Yanzi Ma, Jiahui Mu, Weiwei Liu, Fanxing Xu, Zhanlin Li, Jiao Bai, Huiming Hua, and Dahong Li. 2019. "Marine-Derived Natural Lead Compound Disulfide-Linked Dimer Psammaplin A: Biological Activity and Structural Modification" Marine Drugs 17, no. 7: 384. https://0-doi-org.brum.beds.ac.uk/10.3390/md17070384