The Inhibitory Effect of Protamine on Platelets is Attenuated by Heparin without Inducing Thrombocytopenia in Rodents

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
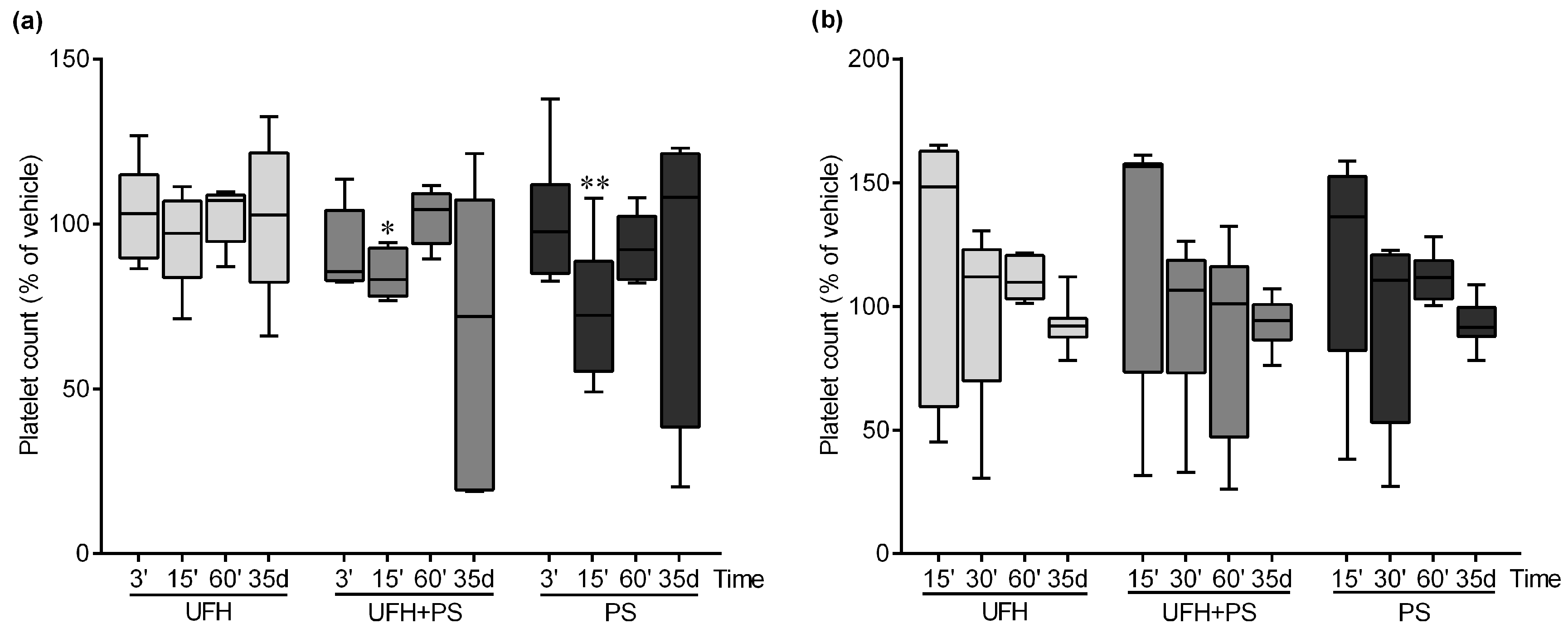
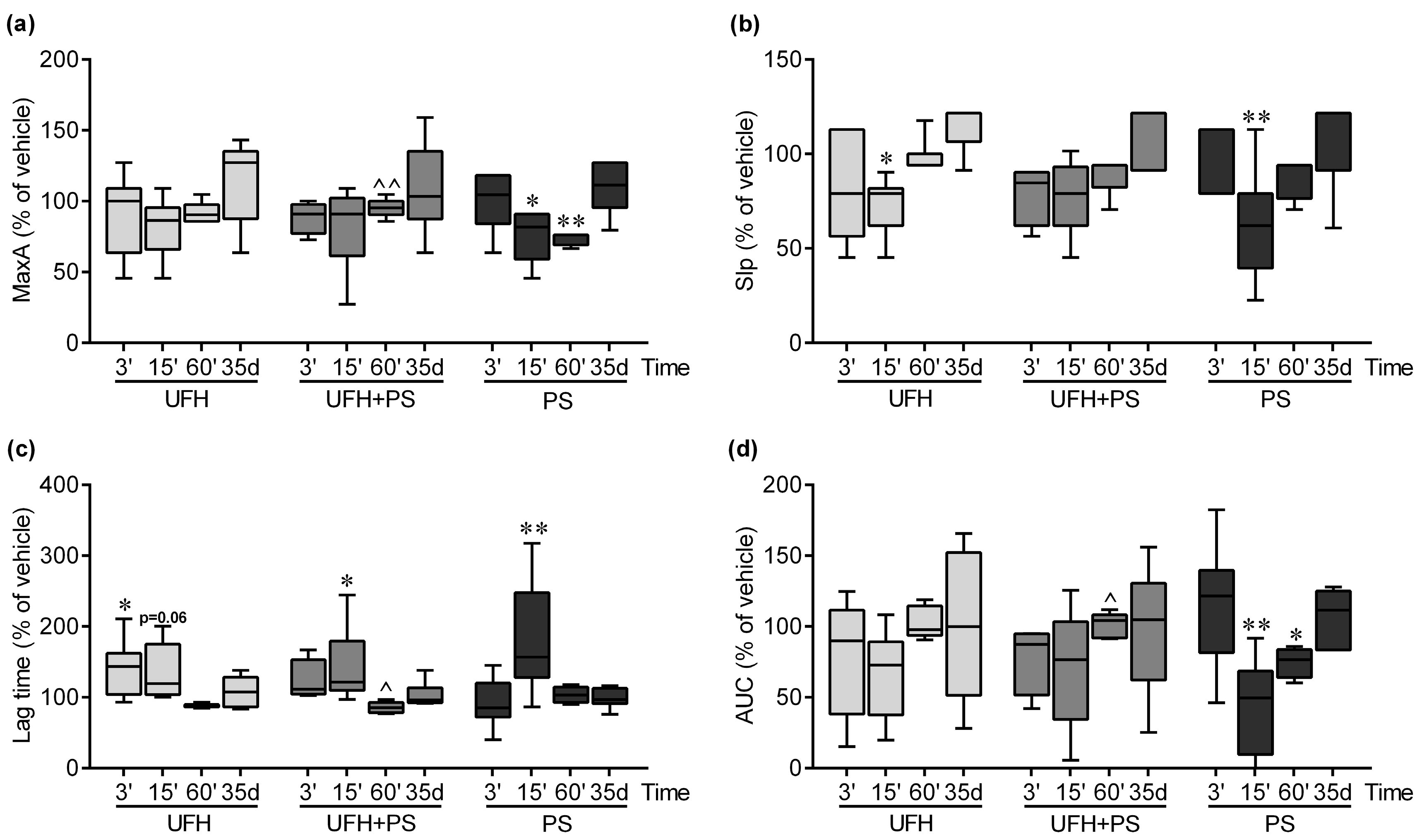
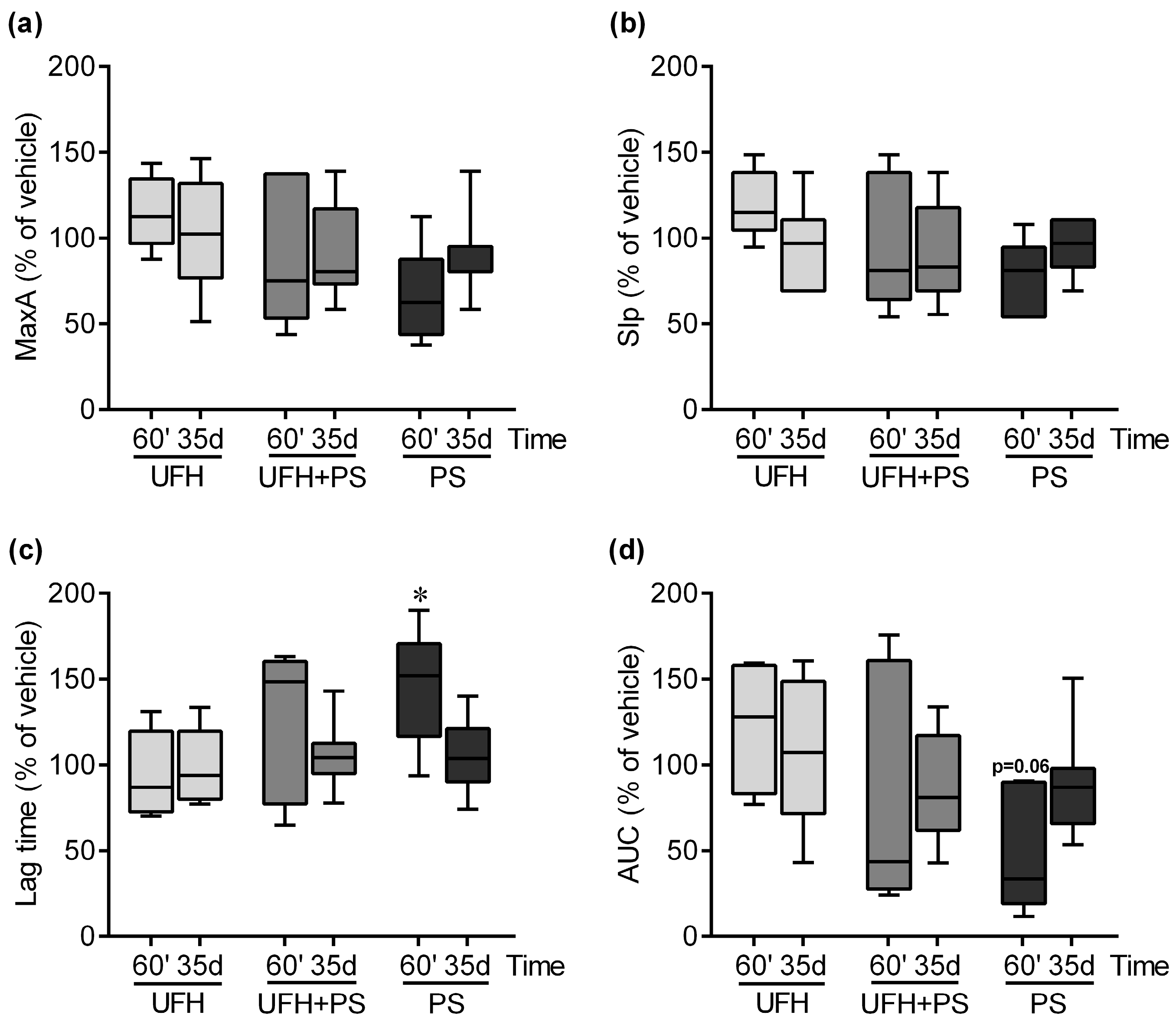
2.1. The Effect of UFH and PS on the Number of Platelets and Their Function in Mice and Rats
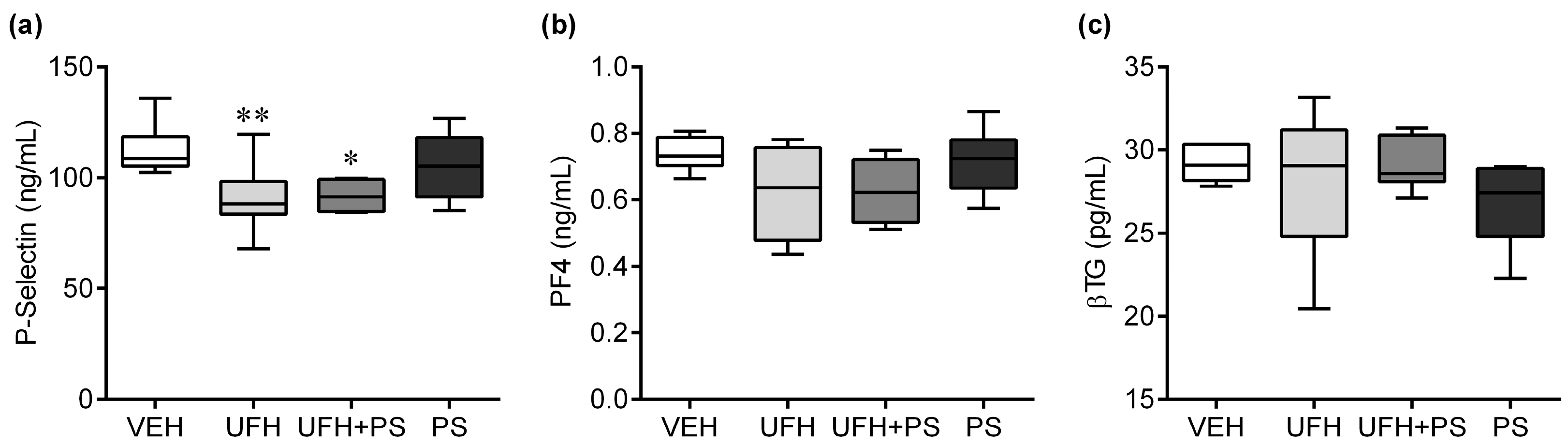
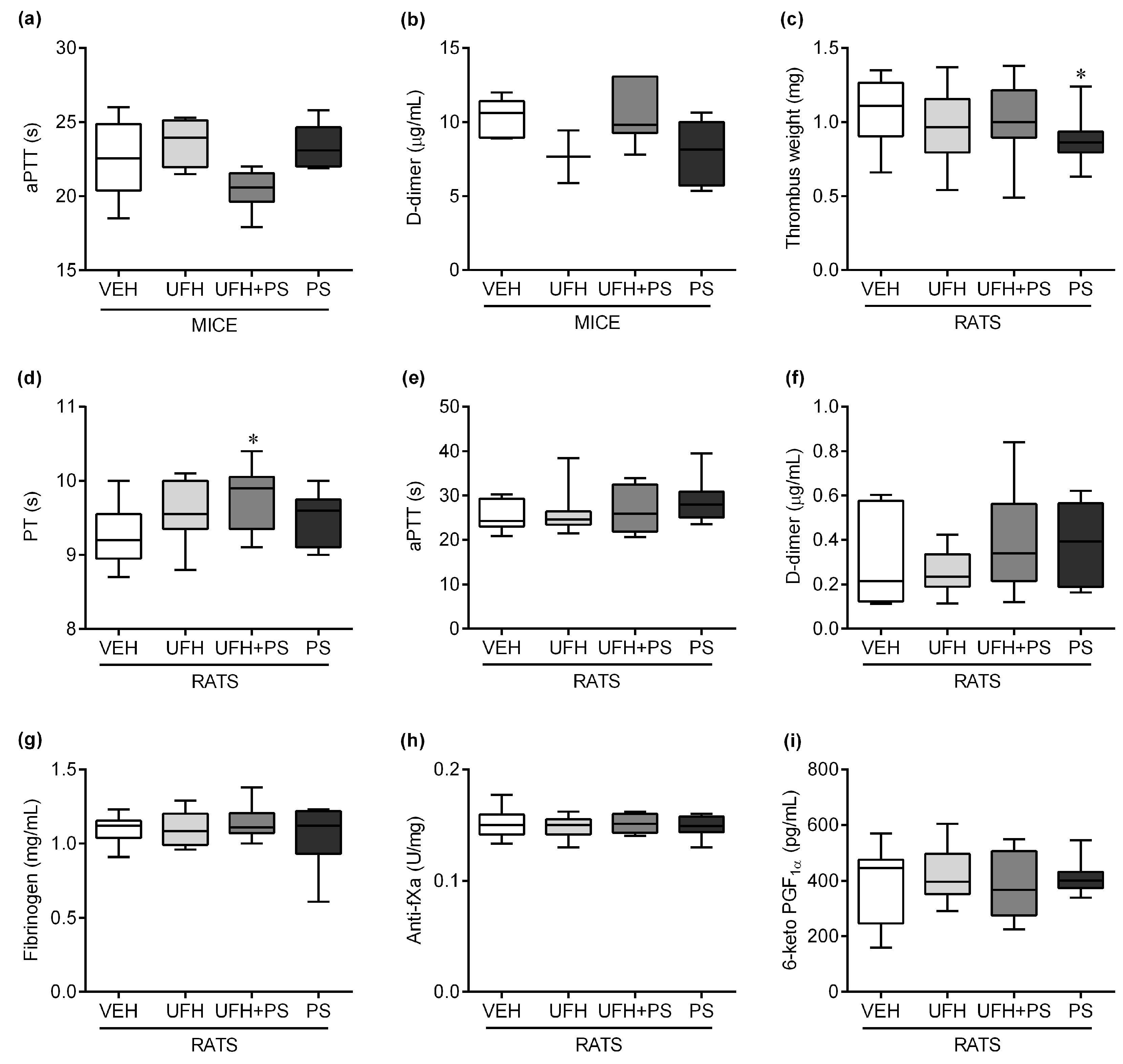
2.2. The Effect of UFH and PS on Thrombosis and Coagulation Parameters in Mice and Rats
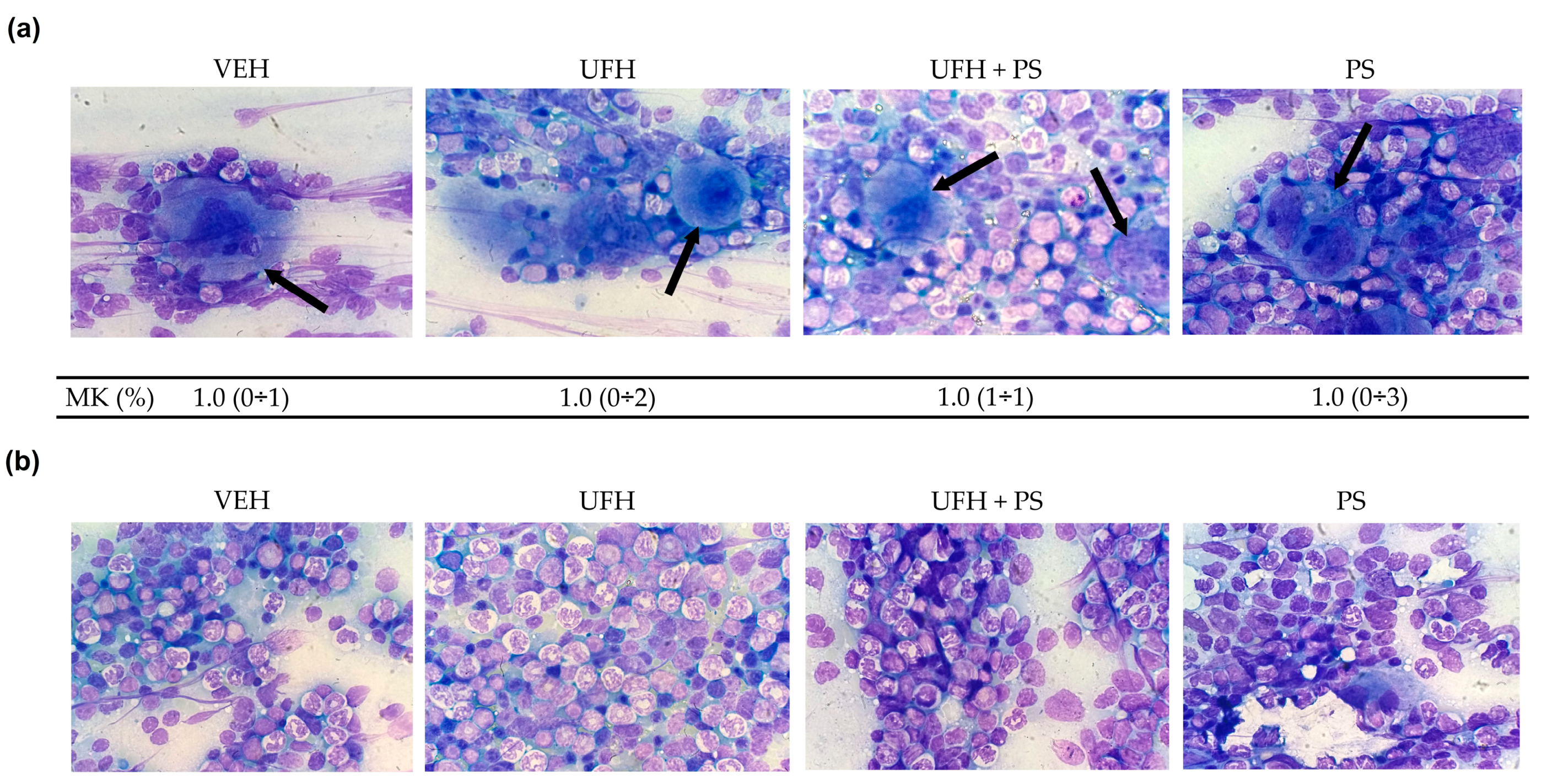
2.3. The Effect of UFH and PS on Megakaryocytopoiesis in Mice
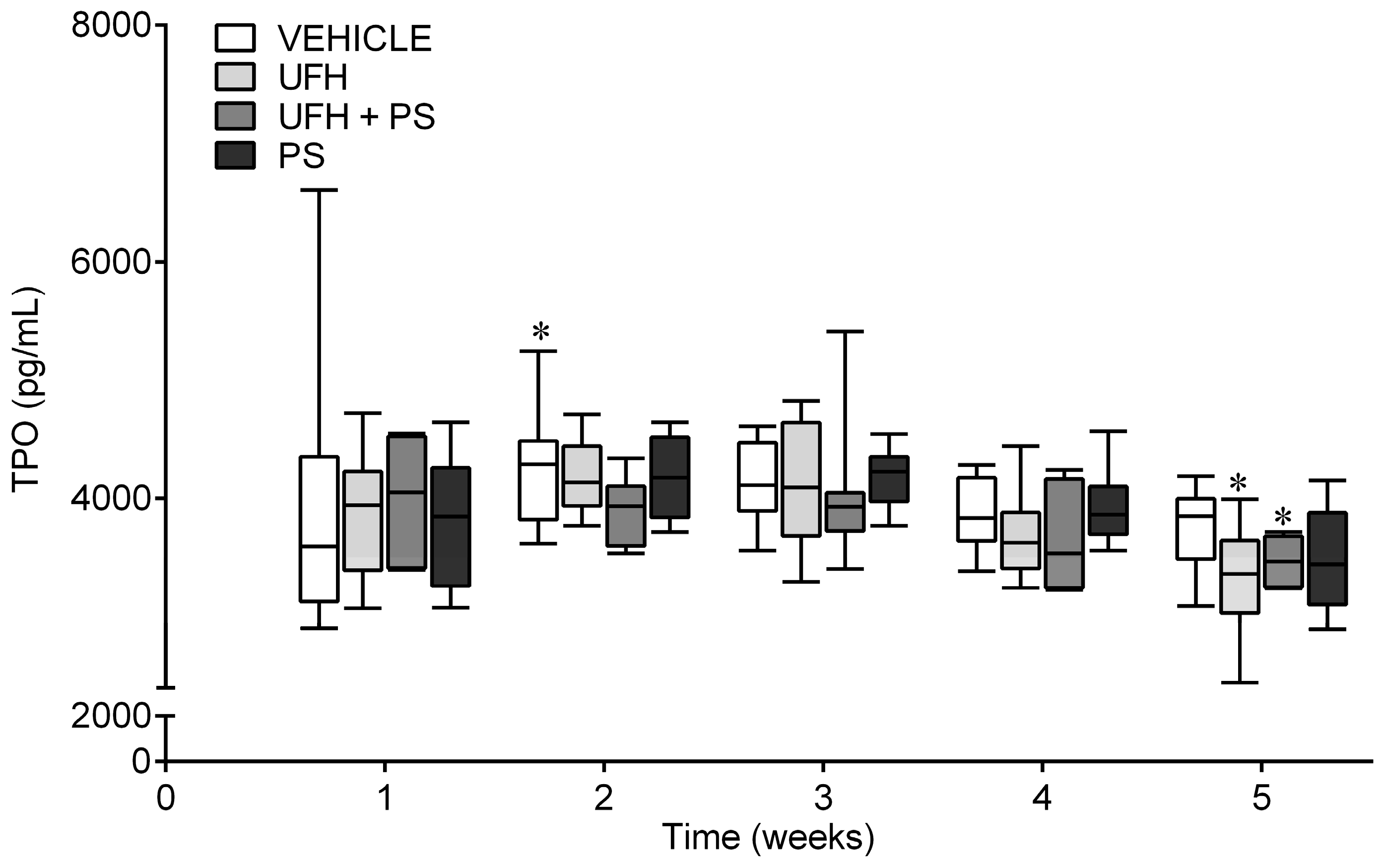
2.4. The Effect of UFH and PS on the Blood Count in the Mice and Rats
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals and Housing
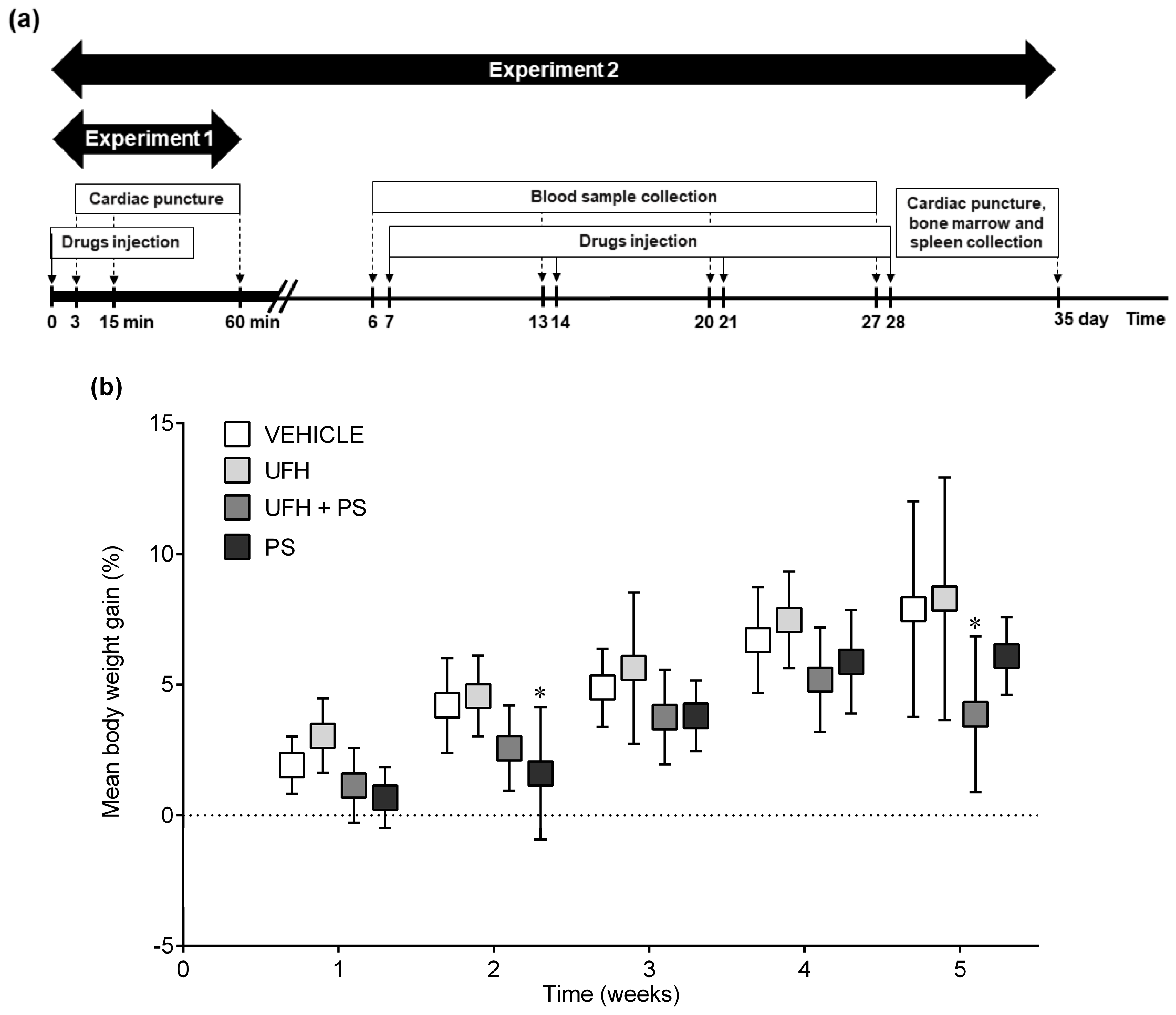
4.3. Experiment 1: The Number of Platelets and Their Aggregation up to 60 Min after a Single Injection of UFH and PS into Mice
4.4. Experiment 2: The Number of Platelets and Their Aggregation, Bone Marrow Cytology, and Coagulation Parameters 35 Days after the Repeated (Once a Week) Injection of UFH and PS into Mice
4.5. Experiment 3: The Number of Platelets and Their Aggregation up to 60 Min after a Single Injection of UFH and PS into Rats
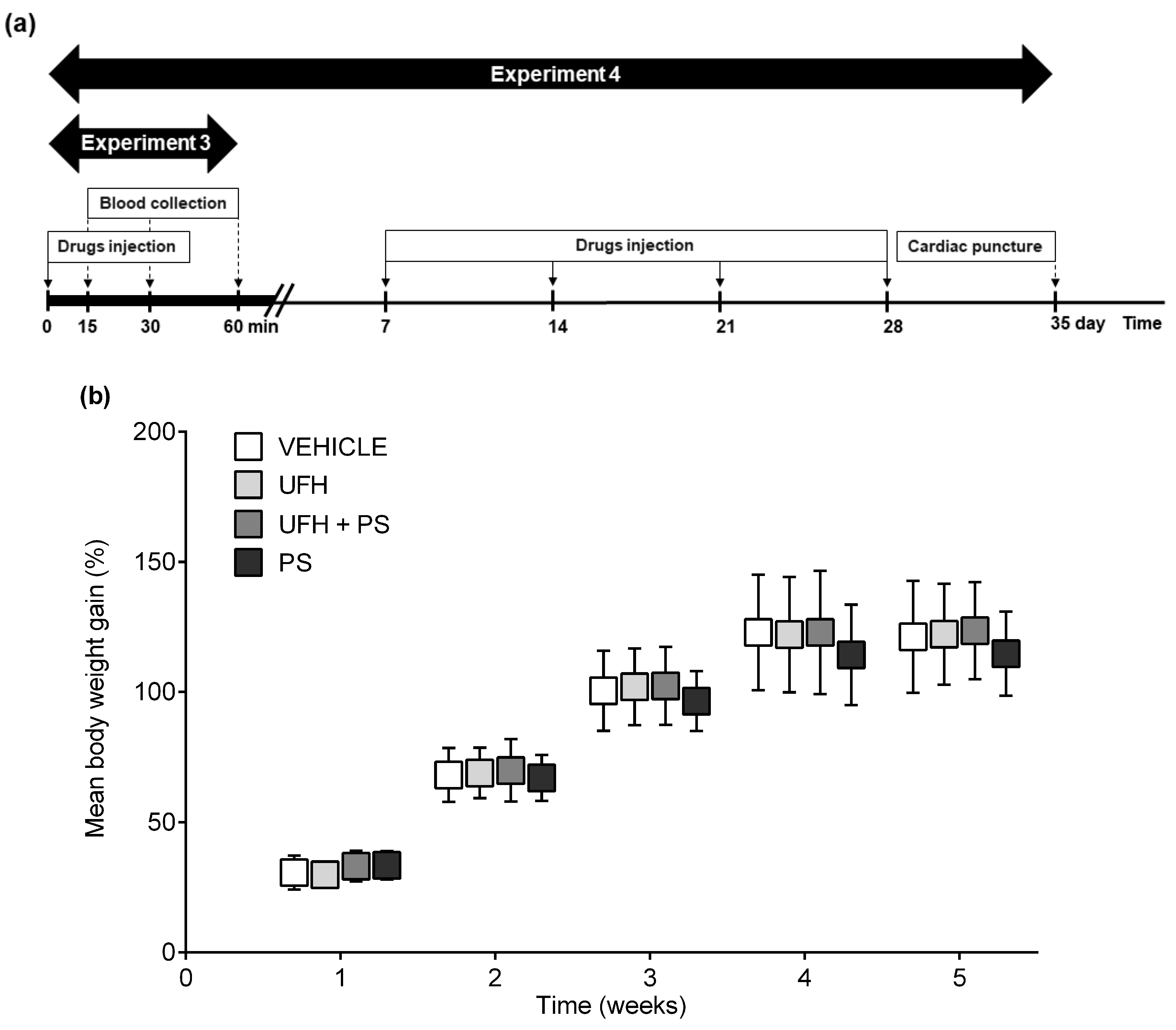
4.6. Experiment 4: The Number of Platelets and Their Aggregation, Arterial Thrombosis, and Coagulation Parameters 35 Days after the Repeated (Once a Week) Injection of UFH and PS into Rats
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| anti-fXa | anti-factor Xa |
| aPTT | activated partial thromboplastin time |
| βTG | β-thromboglobulin |
| CPB | cardiopulmonary bypass |
| ELISA | enzyme-linked immunosorbent assay |
| GPIb | glycoprotein Ib |
| IgG | immunoglobulin G |
| NO | nitric oxide |
| NPH | neutral protamine Hagedorn insulin |
| PF4 | platelet factor 4 |
| PolyP | polyphosphates |
| PS | protamine sulfate |
| PT | prothrombin time |
| TPO | thrombopoietin |
| UFH | unfractionated heparin |
| vWF | von Willebrand factor |
| VEH | vehicle |
References
- Horrow, J.C. Protamine: A review of its toxicity. Anesth. Analg. 1985, 64, 248–261. [Google Scholar] [CrossRef]
- Teoh, K.H.; Young, E.; Blackall, M.H.; Roberts, R.S.; Hirsh, J. Can extra protamine eliminate heparin rebound following cardiopulmonary bypass surgery? J. Thorac. Cardiovasc. Surg. 2004, 128, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drugs. com. Protamine Side Effects. Available online: https://www.drugs.com/sfx/protamine-side-effects.html (accessed on 14 March 2019).
- Mochizuki, T.; Olson, P.J.; Szlam, F.; Ramsay, J.G.; Levy, J.H. Protamine reversal of heparin affects platelet aggregation and activated clotting time after cardiopulmonary bypass. Anesth. Analg. 1998, 87, 781–785. [Google Scholar] [PubMed]
- Boer, C.; Meesters, M.I.; Veerhoek, D.; Vonk, A.B.A. Anticoagulant and side-effects of protamine in cardiac surgery: A narrative review. Br. J. Anaesth. 2018, 120, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef]
- Cook, J.J.; Niewiarowski, S.; Yan, Z.; Schaffer, L.; Lu, W.; Stewart, G.J.; Mosser, D.M.; Myers, J.A.; Maione, T.E. Platelet factor 4 efficiently reverses heparin anticoagulation in the rat without adverse effects of heparin-protamine complexes. Circulation 1992, 85, 1102–1109. [Google Scholar] [CrossRef]
- Lindblad, B.; Borgstrom, A.; Wakefield, T.W.; Whitehouse, W.M., Jr.; Stanley, J.C. Protamine reversal of anticoagulation achieved with a low molecular weight heparin. The effects on eicosanoids, clotting and complement factors. Thromb. Res. 1987, 48, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Al-Mondhiry, H.; Pierce, W.S.; Basarab, R.M. Protamine-induced thrombocytopenia and leukopenia. Thromb. Haemost. 1985, 53, 60–64. [Google Scholar] [CrossRef]
- Heyns, A.D.; Lötter, M.G.; Badenhorst, P.N.; van Reenen, O.R.; Pieters, H.; Minnaar, P.C.; Retief, F.P. Kinetics and in vivo redistribution of (111)Indium-labelled human platelets after intravenous protamine sulphate. Thromb. Haemost. 1980, 44, 65–68. [Google Scholar]
- Eika, C. On the mechanism of platelet aggregation induced by heparin, protamine and polybrene. Scand. J. Haematol. 1972, 9, 248–257. [Google Scholar] [CrossRef]
- Barstad, R.M.; Stephens, R.W.; Hamers, M.J.; Sakariassen, K.S. Protamine sulphate inhibits platelet membrane glycoprotein Ib-von Willebrand factor activity. Thromb. Haemost. 2000, 83, 334–337. [Google Scholar] [PubMed]
- Chen, W.; Liang, X.; Syed, A.K.; Jessup, P.; Church, W.R.; Ware, J.; Josephson, C.D.; Li, R. Inhibiting GPIbα Shedding Preserves Post-Transfusion Recovery and Hemostatic Function of Platelets After Prolonged Storage. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1821–1828. [Google Scholar] [CrossRef]
- Kirklin, J.K.; Chenoweth, D.E.; Naftel, D.C.; Blackstone, E.H.; Kirklin, J.W.; Bitran, D.D.; Curd, J.G.; Reves, J.G.; Samuelson, P.N. Effects of protamine administration after cardiopulmonary bypass on complement, blood elements, and the hemodynamic state. Ann. Thorac. Surg. 1986, 41, 193–199. [Google Scholar] [CrossRef]
- Radegran, K.; Drugge, U.; Olsson, P. Pulmonary vasoconstriction by induced platelet aggregation. Acta Anaesth. Scand. 1974, 18, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Rådegran, K.; Bergentz, S.E.; Lewis, D.H.; Ljungqvist, U.; Olsson, P. Pulmonary effects of induced platelet aggregation. Intravascular obstruction or vasoconstriction? Scand. J. Clin. Lab. Investig. 1971, 28, 423–427. [Google Scholar] [CrossRef]
- Velders, A.J.; Wildevuur, C.R. Platelet damage by protamine and the protective effect of prostacyclin: An experimenta1 study in dogs. Ann. Thorac. Surg. 1986, 42, 168–171. [Google Scholar] [CrossRef]
- Radegran, K.; Taylor, G.A.; Olsson, P. Mode of action of protamine in regard to its circulatory and respiratory side effects. Eur. Surg. Res. 1971, 3, 139–143. [Google Scholar] [CrossRef]
- Wakefield, T.W.; Whitehouse, W.M., Jr.; Stanley, J.C. Depressed cardiovascular function and altered platelet kinetics following protamine sulfate reversal of heparin activity. J. Vasc. Surg. 1984, 1, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Rent, R.; Ertel, N.; Eisenstein, R.; Gewurz, H. Complement Activation by Interaction of Polyanions and Polycations. J. Immunol. 1975, 114, 120–124. [Google Scholar]
- Cuker, A.; Cines, D.B. Protamine-induced thrombocytopenia? Blood 2013, 121, 2818–2819. [Google Scholar] [CrossRef] [Green Version]
- Bakchoul, T.; Jouni, R.; Warkentin, T.E. Protamine (heparin)-induced thrombocytopenia: A review of the serological and clinical features associated with anti-protamine/heparin antibodies. J. Thromb. Haemost. 2016, 14, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Grieshaber, P.; Bakchoul, T.; Wilhelm, J.; Wagner, A.; Wollbrück, M.; Böning, A.; Sachs, U. Platelet-activating protamine-heparin-antibodies lead to higher protamine demand in patients undergoing cardiac surgery. J. Thorac. Cardiovasc. Surg. 2015, 150, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.M.; Welsby, I.J.; Phillips-Bute, B.; Ortel, T.L.; Arepally, G.M. High incidence of antibodies to protamine and protamine/heparin complexes in patients undergoing cardiopulmonary bypass. Blood 2013, 121, 2828–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakchoul, T.; Zöllner, H.; Amiral, J.; Panzer, S.; Selleng, S.; Kohlmann, T.; Brandt, S.; Delcea, M.; Warkentin, T.E.; Sachs, U.J.; et al. Anti-protamine-heparin antibodies: Incidence, clinical relevance, and pathogenesis. Blood 2013, 121, 2821–2827. [Google Scholar] [CrossRef] [PubMed]
- Zöllner, H.; Jouni, R.; Panzer, S.; Khadour, A.; Janzen, L.; Wesche, J.; Ten, B.M.; Schellong, S.; Heinken, A.; Greinacher, A.; et al. Platelet activation in the presence of Neutral Protamine Hagedorn insulin: A new feature of antibodies against protamine/heparin complexes. J. Thromb. Haemost. 2017, 15, 176–184. [Google Scholar] [CrossRef]
- Pouplard, C.; Leroux, D.; Rollin, J.; Amiral, J.; May, M.A.; Gruel, Y. Incidence of antibodies to protamine sulfate/heparin complexes incardiac surgery patients and impact on platelet activation and clinical outcome. Thromb. Haemost. 2013, 109, 1141–1147. [Google Scholar]
- Arman, M.; Krauel, K. Human platelet IgG Fc receptor FcγRIIA in immunity and thrombosis. J. Thromb. Haemost. 2015, 13, 893–908. [Google Scholar] [CrossRef]
- Quach, M.E.; Chen, W.; Li, R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood 2018, 131, 1512–1521. [Google Scholar] [CrossRef]
- Kalaska, B.; Sokolowska, E.; Kaminski, K.; Szczubialka, K.; Kramkowski, K.; Mogielnicki, A.; Nowakowska, M.; Buczko, W. Cationic derivative of dextran reverses anticoagulant activity of unfractionated heparin in animal models of arterial and venous thrombosis. Eur. J. Pharmacol. 2012, 686, 81–89. [Google Scholar] [CrossRef]
- Olsson, A.; Alfredsson, J.; Håkansson, E.; Svedjeholm, R.; Berglund, J.; Berg, S. Protamine reduces whole blood platelet aggregation after cardiopulmonary bypass. Scand. Cardiovasc. J. 2016, 50, 58–63. [Google Scholar] [CrossRef]
- Ellison, N.; Edmunds, L.H., Jr.; Colman, R.W. Platelet aggregation following heparin and protamine administration. Anesthesiology 1978, 48, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Ammar, T.; Fisher, C.F. The effects of heparinase 1 and protamine on platelet reactivity. Anesthesiology 1997, 86, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, B.; Wakefield, T.W.; Whitehouse, W.M., Jr.; Stanley, J.C. The effect of protamine sulfate on platelet function. Scand. J. Thorac. Cardiovasc. Surg. 1988, 22, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Storck, J.; Hollger, N.; Zimmermann, R.E. The influence of heparin and protamine sulfate on platelet ADP and platelet factor 4 release and the expression of glycoprotein IIb/IIIa. Haemostasis 1994, 24, 358–363. [Google Scholar] [PubMed]
- Griffin, M.J.; Rinder, H.M.; Smith, B.R.; Tracey, J.B.; Kriz, N.S.; Li, C.K.; Rinder, C.S. The effects of heparin, protamine, and heparin/protamine reversal on platelet function under conditions of arterial shear stress. Anesth. Analg. 2001, 93, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, E.; Kalaska, B.; Miklosz, J.; Mogielnicki, A. The toxicology of heparin reversal with protamine: Past, present and future. Expert Opin. Drug Metab. 2016, 12, 897–909. [Google Scholar] [CrossRef]
- Despotis, G.J.; Joist, J.H.; Hogue, C.W., Jr.; Alsoufiev, A.; Kater, K.; Goodnough, L.T.; Santoro, S.A.; Spitznagel, E.; Rosenblum, M.; Lappas, D.G. The impact of heparin concentration and activated clotting time monitoring on blood conservation: A prospective, randomized evaluation in patients under-going cardiac operation. J. Thorac. Cardiovasc. Surg. 1995, 110, 46–54. [Google Scholar] [CrossRef]
- Kalaska, B.; Kaminski, K.; Sokolowska, E.; Czaplicki, D.; Kujdowicz, M.; Stalinska, K.; Bereta, J.; Szczubialka, K.; Pawlak, D.; Nowakowska, M.; et al. Nonclinical evaluation of novel cationically modified polysaccharide antidotes for unfractionated heparin. PLoS ONE 2015, 10, e0119486. [Google Scholar] [CrossRef]
- Chang, S.W.; Voelkel, N.F. Charge-related lung microvascular injury. Am. Rev. Respir. Dis. 1989, 139, 534–545. [Google Scholar] [CrossRef]
- Chang, S.W.; Westcott, Y.; Henson, E.; Voelkel, N.E. Pulmonary vascular injury by polycations in perfused rat lungs. J. Appl. Physiol. 1987, 62, 1932–1943. [Google Scholar] [CrossRef]
- Fairman, R.P.; Sessler, C.N.; Bierman, M.; Glauser, F.L. Protamine sulfate causes pulmonary hypertension and edema in isolated rat lungs. J. Appl. Physiol. 1987, 62, 1363–1367. [Google Scholar] [CrossRef]
- Koster, A.; Börgermann, J.; Gummert, J.; Rudloff, M.; Zittermann, A.; Schirmer, U. Protamine overdose and its impact on coagulation, bleeding, and transfusions after cardiopulmonary bypass: Results of a randomized double-blind controlled pilot study. Clin. Appl. Thromb. Hemost. 2014, 20, 290–295. [Google Scholar] [CrossRef]
- Khan, N.U.; Wayne, C.K.; Barker, J.; Strang, T. The effects of protamine overdose on coagulation parameters as measured by the thrombelastograph. Eur. J. Anaesthesiol. 2010, 27, 624–627. [Google Scholar] [CrossRef]
- Kalaska, B.; Kaminski, K.; Miklosz, J.; Yusa, S.I.; Sokolowska, E.; Blazejczyk, A.; Wietrzyk, J.; Kasacka, I.; Szczubialka, K.; Pawlak, D.; et al. Heparin-binding copolymer reverses effects of unfractionated heparin, enoxaparin, and fondaparinux in rats and mice. Transl. Res. 2016, 177, 98–112. [Google Scholar] [CrossRef]
- Kalathottukaren, M.T.; Abraham, L.; Kapopara, P.R.; Lai, B.F.; Shenoi, R.A.; Rosell, F.I.; Conway, E.M.; Pryzdial, E.L.; Morrissey, J.H.; Haynes, C.A.; et al. Alteration of blood clotting and lung damage by protamine are avoided using the heparin and polyphosphate inhibitor UHRA. Blood 2017, 129, 1368–1379. [Google Scholar] [CrossRef] [Green Version]
- Ansell, J.E.; Bakhru, S.H.; Laulicht, B.E.; Steiner, S.S.; Grosso, M.A.; Brown, K.; Dishy, V.; Lanz, H.J.; Mercuri, M.F.; Noveck, R.J.; et al. Single-dose ciraparantag safely and completely reverses anticoagulant effects of edoxaban. Thromb. Haemost. 2017, 117, 238–245. [Google Scholar] [CrossRef]
- Ponnusamy, V.; Sinha, A.; Kupfer, Y. Andexanet Alfa for Bleeding with Factor Xa Inhibitors. N. Engl. J. Med. 2019, 381, 191–192. [Google Scholar]
- Gurney, D.; Lip, G.Y.; Blann, A.D. A reliable plasma marker of platelet activation: Does it exist? Am. J. Hematol. 2002, 70, 139–144. [Google Scholar] [CrossRef]
- Koenig, A.; Norgard-Sumnicht, K.; Linhardt, R.; Varki, A. Differential interactions of heparin and heparan sulfate glycosaminoglycans with the selectins. Implications for the use of unfractionated and low molecular weight heparins as therapeutic agents. J. Clin. Investig. 1998, 101, 877–889. [Google Scholar] [CrossRef]
- Greinacher, A.; Fuerll, B.; Zinke, H.; Müllejans, B.; Krüger, W.; Michetti, N.; Motz, W.; Schwertz, H. Megakaryocyte impairment by eptifibatide-induced antibodies causes prolonged thrombocytopenia. Blood 2009, 114, 1250–1253. [Google Scholar] [CrossRef]
- Bakchoul, T.; Sachs, U.J. Platelet destruction in immune thrombocytopenia. Understanding the mechanisms. Hamostaseologie 2016, 36, 187–194. [Google Scholar]
- Reagan, W.J.; Irizarry-Rovira, A.; Poitout-Belissent, F.; Bolliger, A.P.; Ramaiah, S.K.; Travlos, G.; Walker, D.; Bounous, D.; Walter, G. Bone Marrow Working Group of ASVCP/STP. Best practices for evaluation of bone marrow in nonclinical toxicity studies. Toxicol. Pathol. 2011, 39, 435–448. [Google Scholar] [CrossRef]
- Sokolowska, E.; Kalaska, B.; Kaminski, K.; Lewandowska, A.; Blazejczyk, A.; Wietrzyk, J.; Kasacka, I.; Szczubialka, K.; Pawlak, D.; Nowakowska, M.; et al. The Toxicokinetic Profile of Dex40-GTMAC3-a Novel Polysaccharide Candidate for Reversal of Unfractionated Heparin. Front. Pharmacol. 2016, 7, 60. [Google Scholar]
- Tripodi, A.; Caldwell, S.H.; Hoffman, M.; Trotter, J.F.; Sanyal, A.J. Review article: The prothrombin time test as a measure of bleeding risk and prognosis in liver disease. Aliment Pharmacol. Ther. 2007, 26, 141–148. [Google Scholar] [CrossRef]
- Ni Ainle, F.; Preston, R.J.; Jenkins, P.V.; Nel, H.J.; Johnson, J.A.; Smith, O.P.; White, B.; Fallon, P.G.; O’Donnell, J.S. Protamine sulfate down-regulates thrombin generation by inhibiting factor V activation. Blood 2009, 114, 1658–1665. [Google Scholar] [CrossRef] [Green Version]
- Bolliger, D.; Szlam, F.; Azran, M.; Koyama, K.; Levy, J.H.; Molinaro, R.J.; Tanaka, K.A. The anticoagulant effect of protamine sulfate is attenuated in the presence of platelets or elevated factor VIII concentrations. Anesth. Analg. 2010, 111, 601–608. [Google Scholar] [CrossRef]
- Nielsen, V.G. Protamine enhances fibrinolysis by decreasing clot strength: Role of tissue factorinitiated thrombin generation. Ann. Thorac. Surg. 2006, 81, 1720–1727. [Google Scholar] [CrossRef]
- Cylwik, D.; Mogielnicki, A.; Kramkowski, K.; Stokowski, J.; Buczko, W. Antithrombotic effect of L-arginine in hypertensive rats. J. Physiol. Pharmacol. 2004, 55, 563–574. [Google Scholar]
- Mogielnicki, A.; Kramkowski, K.; Hermanowicz, J.M.; Buczko, W. N-methylnicotinamide failed to induce endothelial prostacyclin release in perfused rat hindquarters. Pharmacol. Rep. 2008, 60, 1025–1029. [Google Scholar]
- Mogielnicki, A.; Kramkowski, K.; Pietrzak, L.; Buczko, W. N-methylnicotinamide inhibits arterial thrombosis in hypertensive rats. J. Physiol. Pharmacol. 2007, 58, 515–527. [Google Scholar]
- Chlopicki, S.; Swies, J.; Mogielnicki, A.; Buczko, W.; Bartus, M.; Lomnicka, M.; Adamus, J.; Gebicki, J. 1-Methylnicotinamide (MNA), a primary metabolite of nicotinamide, exerts anti-thrombotic activity mediated by a cyclooxygenase-2/prostacyclin pathway. Br. J. Pharmacol. 2007, 152, 230–239. [Google Scholar] [CrossRef]
- Travers, R.J.; Shenoi, R.A.; Kalathottukaren, M.T.; Kizhakkedathu, J.N.; Morrissey, J.H. Nontoxic polyphosphate inhibitors reduce thrombosis while sparing hemostasis. Blood 2014, 124, 3183–3190. [Google Scholar] [CrossRef] [Green Version]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miklosz, J.; Kalaska, B.; Kaminski, K.; Rusak, M.; Szczubialka, K.; Nowakowska, M.; Pawlak, D.; Mogielnicki, A. The Inhibitory Effect of Protamine on Platelets is Attenuated by Heparin without Inducing Thrombocytopenia in Rodents. Mar. Drugs 2019, 17, 539. https://0-doi-org.brum.beds.ac.uk/10.3390/md17090539
Miklosz J, Kalaska B, Kaminski K, Rusak M, Szczubialka K, Nowakowska M, Pawlak D, Mogielnicki A. The Inhibitory Effect of Protamine on Platelets is Attenuated by Heparin without Inducing Thrombocytopenia in Rodents. Marine Drugs. 2019; 17(9):539. https://0-doi-org.brum.beds.ac.uk/10.3390/md17090539
Chicago/Turabian StyleMiklosz, Joanna, Bartlomiej Kalaska, Kamil Kaminski, Malgorzata Rusak, Krzysztof Szczubialka, Maria Nowakowska, Dariusz Pawlak, and Andrzej Mogielnicki. 2019. "The Inhibitory Effect of Protamine on Platelets is Attenuated by Heparin without Inducing Thrombocytopenia in Rodents" Marine Drugs 17, no. 9: 539. https://0-doi-org.brum.beds.ac.uk/10.3390/md17090539