Screening Marine Natural Products for New Drug Leads against Trypanosomatids and Malaria

 ,
,  , ,
, ,
Abstract
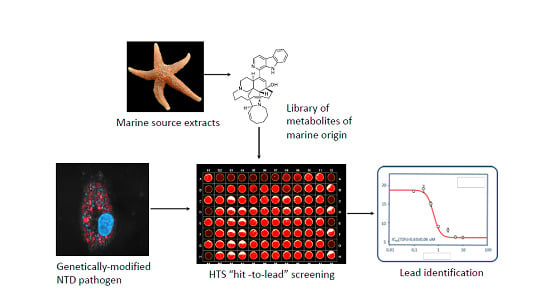
:
1. Introduction
2. Trypanosomatids-Borne NTDs and Malaria
2.1. Human African Trypanosomiasis
2.2. American Trypanosomiasis or Chagas Disease
2.3. Leishmaniasis
2.4. Malaria or Paludism
3. Prevention and Lack of Vaccines Against NTDs
4. The Pharmacological Approach
4.1. Treatment of African Trypanosomiasis
4.2. Treatment of American Trypanosomiasis
4.3. Treatment of Leishmaniasis
4.4. Treatment of Malaria
5. Current tools for Drug Screening
5.1. Phenotypic vs. Target-Based Screening in Trypanosomatids
5.2. Phenotypic vs. Target-Based Screening in Malaria
6. Marine Based Compounds for NTDs and Malaria
6.1. Algae-Derived Compounds
6.2. Sponge-Derived Compounds
6.3. Metabolites Derived from Other Invertebrates
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NTDs | Neglected Tropical Diseases; |
| WHO | World Health Organization; |
| HTS | High-Throughput Screening; |
| FDA | Food and Drug and Administration; |
| HAT | Human African Trypanosomiasis; |
| DALY | Disability Adjusted Life Year; |
| NECT | Nifurtimox-Eflornithine Combination Therapy; |
| CNS | Central Nervous System; |
| ODC | Ornithine Decarboxylase; |
| NTR-1 | type 1 nitroreductase; |
| PKDL | Post-Kala-azar Dermal Leishmaniasis; |
| AmpB | Amphotericin B; |
| ACT | Artemisinin-based Combined Therapies; |
| DNDi | Drugs for Neglected Diseases Initiative; |
| MMV | Medicine for Malaria Venture; |
| NGOs | Non-Governmental Organizations; |
| NCI | National Cancer Institute; |
| NPNPD | Program for the Discovery of Natural Products; |
| QSAR | Quantitative Structure-Activity Relationship; |
| CRK12 | Cdc2-related kinase 12; |
| PTR1 | Pteridine Reductase 1; |
| DHFR-TS | bifunctional Dihydrofolate Reductase-Thymidylate Synthase; |
| HCS | High Content Screening; |
| CCD | charge-coupled device; |
| SI | Selective Index; |
| LTopIB | Leishmania DNA topoisomerase IB. |
References
- Lindequist, U. Marine-derived pharmaceuticals—Challenges and opportunities. Biomol. Ther. 2016, 24, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faulkner, D.J.; Harper, M.K.; Haygood, M.G.; Salomon, C.E.; Schmidt, E.W. Symbiotic bacteria in sponges: Sources of bioactive substances. In Drugs from the Sea; Fusetani, N., Ed.; Karger: Basel, Switzerland, 2000; pp. 107–119. [Google Scholar]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, R.B.; Evdokimov, N.M.; Lefranc, F.; Valentão, P.; Kornienko, A.; Pereira, D.M.; Andrade, P.B.; Gomes, N.G.M. Marine-derived anticancer agents: Clinical benefits, innovative mechanisms, and new targets. Mar. Drugs 2019, 17, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, L.J. Brentuximab Vedotin: A review in CD30-positive Hodgkin lymphoma. Drugs 2017, 77, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Aseyev, O.; Ribeiro, J.M.; Cardoso, F. Review on the clinical use of eribulin mesylate for the treatment of breast cancer. Expert Opin. Pharmacother. 2016, 17, 589–600. [Google Scholar] [CrossRef]
- Schöffski, P.; Chawla, S.; Maki, R.G.; Italiano, A.; Gelderblom, H.; Choy, E.; Grignani, G.; Camargo, V.; Bauer, S.; Rha, S.Y.; et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: A randomised, open–label, multicentre, phase 3 trial. Lancet 2016, 387, 1629–1637. [Google Scholar] [CrossRef]
- Rinerhart, K.L.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.; Sun, F.; Li, H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumour agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: Results of a phase III randomized multicenter clinical trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef]
- Schwartsmann, G.; Brondani da Rocha, A.; Berlinck, R.G.; Jimeno, J. Marine organisms as a source of new anticancer agents. Lancet Oncol. 2001, 2, 221–225. [Google Scholar] [CrossRef]
- Molyneux, D.H.; Savioli, L.; Engels, D. Neglected tropical diseases: Progress towards addressing the chronic pandemic. Lancet 2017, 389, 312–325. [Google Scholar] [CrossRef]
- Mitra, A.K.; Mawson, A.R. Neglected tropical diseases: Epidemiology and global burden. Trop. Med. Infect. Dis. 2017, 2, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, M.B.; Zhou, X.N. Global burden on neglected tropical diseases. Lancet 2016, 16, P1113–P1114. [Google Scholar] [CrossRef]
- White, N.J.; Pukrittayakamee, S.; Hien, T.T.; Faiz, M.A.; Mokuolu, O.A.; Dondorp, A.M. Malaria. Lancet 2014, 383, 723–735. [Google Scholar] [CrossRef]
- Klohe, K.; Amuasi, J.; Moriku Kaducu, J.; Haavardsson, I.; Bogatyreva, E.; Husøy Onarheim, K.; Harrison, W.; Kristensen, F.; Prazeres da Costa, C.; Winkler, A.S. The 2017 Oslo conference report on neglected tropical diseases and emerging/re-emerging infectious diseases—Focus on populations underserved. Infect. Dis. Poverty 2019, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef]
- Dunn, N.; Wang, S.; Adigun, R. African Trypanosomiasis (Sleeping Sickness); StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Bottieau, E.; Clerinx, J. Human African Trypanosomiasis: Progress and stagnation. Infect. Dis. Clin. North Am. 2019, 3, 61–77. [Google Scholar] [CrossRef]
- Franco, J.R.; Cecchi, G.; Priotto, G.; Paone, M.; Diarra, A.; Grout, L.; Simarro, P.P.; Zhao, W.; Argaw, D. Monitoring the elimination of human African trypanosomiasis: Update to 2016. PLoS Negl. Trop. Dis. 2018, 12, e0006890. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Control and Surveillance of Human African Trypanosomiasis: Report of a WHO Expert Committee (World Health Organization). 2013. Available online: http://www.who.int/iris/handle/10665/95732 (accessed on 1 December 2019).
- GBD 2015 DALYs and HALE Collaborators. Global, Regional, and National Disability-Adjusted Life-Years (DALYs) for 315 Diseases and Injuries and Healthy Life Expectancy (HALE), 1990–2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1603–1658. [Google Scholar] [CrossRef] [Green Version]
- Checchi, F.; Filipe, J.A.; Barrett, M.P.; Chandramohan, D. The natural progression of Gambiense sleeping sickness: What is the evidence? PLoS Negl. Trop. Dis. 2008, 2, e303. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, P.G. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 2013, 12, 186–194. [Google Scholar] [CrossRef]
- Jamonneau, V.; Ilboudo, H.; Kaboré, J.; Kaba, D.; Koffi, M.; Solano, P.; Garcia, A.; Courtin, D.; Laveissière, C.; Lingue, K.; et al. Untreated human infections by Trypanosoma brucei gambiense are not 100% fatal. PLoS Negl. Trop. Dis. 2012, 6, e1691. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Echeverria, L.E.; Morillo, C.A. American Trypanosomiasis (Chagas Disease). Infect. Dis. Clin. North Am. 2019, 33, 119–134. [Google Scholar] [CrossRef]
- Bern, C. Chagas’ Disease. N. Engl. J. Med. 2015, 373, 456–466. [Google Scholar] [CrossRef]
- Gunter, S.M.; Ronca, S.E.; Sandoval, M.; Coffman, K.; Leining, L.; Gorchakov, R.; Murray, K.O.; Nolan, M.S. Chagas disease infection prevalence and vector exposure in a high-risk population of Texas hunters. Am. J. Trop. Med. Hyg. 2020, 102, 294–297. [Google Scholar] [CrossRef]
- Monge-Maillo, B.; López-Vélez, R. Challenges in the management of Chagas disease in Latin-American migrants in Europe. Clin. Microbiol. Infect. 2017, 23, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Saravia, S.G.; Haberland, A.; Wallukat, G.; Schimke, I. Chronic Chagas’ heart disease: A disease on its way to becoming a worldwide health problem: Epidemiology, etiopathology, treatment, pathogenesis and laboratory medicine. Heart Fail. Rev. 2012, 17, 45–64. [Google Scholar] [CrossRef]
- Jabari, S.; de Oliveira, E.C.; Brehmer, A.; da Silveira, A.B. Chagasic megacolon: Enteric neurons and related structures. Histochem. Cell. Biol. 2014, 142, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Antinori, S.; Corbellino, M. Chagas disease in Europe: A long way to go. Eur. J. Intern. Med. 2018, 48, e29–e30. [Google Scholar] [CrossRef]
- Angheben, A.; Boix, L.; Buonfrate, D.; Gobbi, F.; Bisoffi, Z.; Pupella, S.; Gandini, G.; Aprili, G. Chagas disease and transfusion medicine: A perspective from non-endemic countries. Blood Transfus. 2015, 13, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Huprikar, S.; Bosserman, E.; Patel, G.; Moore, A.; Pinney, S.; Anyanwu, A.; Neofytos, D.; Ketterer, D.; Striker, R.; Silveira, F.; et al. Donor-derived Trypanosoma cruzi infection in solid organ recipients in the United States, 2001–2011. Am. J. Transplant. 2013, 13, 2418–2425. [Google Scholar] [CrossRef] [PubMed]
- Carlier, Y.; Sosa-Estani, S.; Luquetti, A.O.; Buekens, P. Congenital Chagas disease: An update. Mem. Inst. Oswaldo Cruz 2015, 110, 363–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Esmenjaud, J.R.; Arenasa, R. Leishmaniasis: A review. F1000Res 2017, 6, 750. [Google Scholar] [CrossRef] [PubMed]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Colmenares, M.; Kar, S.; Goldsmith-Pestana, K.; McMahon-Pratt, D. Mechanisms of pathogenesis: Differences amongst Leishmania species. Trans. R. Soc. Trop. Med. Hyg. 2002, 96, 3–7. [Google Scholar] [CrossRef]
- Reithinger, R.; Dujardin, J.C.; Louzir, H.; Pirmez, C.; Alexander, B.; Brooker, S. Cutaneous leishmaniasis. Lancet Infect. Dis. 2007, 7, 581–596. [Google Scholar] [CrossRef] [Green Version]
- Handler, M.Z.; Patel, P.A.; Kapila, R.; Al-Qubati, Y.; Schwartz, R.A. Cutaneous and mucocutaneous leishmaniasis: Clinical perspectives. J. Am. Acad. Dermatol. 2015, 73, 897–908. [Google Scholar] [CrossRef]
- Van Griensven, J.; Diro, E. Visceral leishmaniasis. Infect. Dis. Clin. North Am. 2012, 26, 309–322. [Google Scholar] [CrossRef]
- Zijlstra, E.E.; Musa, A.M.; Khalil, E.A.; el-Hassan, I.M.; el-Hassan, A.M. Post-kala-azar dermal leishmaniasis. Lancet Infect. Dis. 2003, 3, 87–98. [Google Scholar] [CrossRef]
- Alvar, J.; Velez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M. WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Leishmaniasis. Geneva: World Health Organization. Available online: http://www.who.int/mediacentre/factsheets/fs375/en/ (accessed on 9 January 2019).
- Singh, O.P.; Hasker, E.; Boelaert, M.; Sundar, S. Elimination of visceral leishmaniasis on the Indian subcontinent. Lancet Infect. Dis. 2016, 16, e304–e309. [Google Scholar] [CrossRef] [Green Version]
- Hendrickx, S.; Guerin, P.J.; Caljon, G.; Croft, S.L.; Maes, L. Evaluating drug resistance in visceral leishmaniasis: The challenges. Parasitology 2018, 145, 453–463. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Malaria Report. 2012. Available online: http://www.who.int/malaria/publications/world_malaria_report_2012/wmr2012_full_report.pdf (accessed on 11 December 2019).
- Feachem, R.G.A.; Chen, I.; Akbari, O.; Bertozzi-Villa, A.; Bhatt, S.; Binka, F.; Boni, M.F.; Buckee, C.; Dieleman, J.; Dondorp, A.; et al. Malaria eradication within a generation: Ambitious, achievable, and necessary. Lancet 2019, 394, 1056–1112. [Google Scholar] [CrossRef]
- African Leaders Malaria Alliance. About ALMA. 2016. Available online: http://alma2030.org/about (accessed on 17 September 2018).
- Mohebali, M.; Nadim, A.; Khamesipour, A. An overview of leishmanization experience: A successful control measure and a tool to evaluate candidate vaccines. Acta Trop. 2019, 200, 105173. [Google Scholar] [CrossRef]
- Reguera, R.M.; Morán, M.; Pérez-Pertejo, Y.; García-Estrada, C.; Balaña-Fouce, R. Current status on prevention and treatment of canine leishmaniasis. Vet. Parasitol. 2016, 227, 98–114. [Google Scholar] [CrossRef]
- Vandoolaeghe, P.; Schuerman, L. The RTS,S/AS01 malaria vaccine in children 5 to 17 months of age at first vaccination. Expert Rev. Vaccin. 2016, 15, 1481–1493. [Google Scholar] [CrossRef] [Green Version]
- Laurens, M.B. RTS,S/AS01 vaccine (Mosquirix™): An overview. Hum. Vaccin. Immunother. 2019, 22, 1–10. [Google Scholar] [CrossRef]
- Goupil, L.S.; McKerrow, J.H. Introduction: Drug discovery and development for neglected diseases. Chem. Rev. 2014, 114, 11131–11137. [Google Scholar] [CrossRef]
- Njoroge, M.; Njuguna, N.M.; Mutai, P.; Ongarora, D.S.; Smith, P.W.; Chibale, K. Recent approaches to chemical discovery and development against malaria and the neglected tropical diseases human African trypanosomiasis and schistosomiasis. Chem. Rev. 2014, 114, 11138–11163. [Google Scholar] [CrossRef]
- Hedley, L.; Fink, D.; Sparkes, D.; Chiodini, P.L. African sleeping sickness. Br. J. Hosp. Med. 2016, 77, C157–C160. [Google Scholar] [CrossRef] [PubMed]
- Doua, F.; Miezan, T.W.; Sanon Singaro, J.R.; Boa Yapo, F.; Baltz, T. The efficacy of pentamidine in the treatment of early-late stage Trypanosoma brucei gambiense trypanosomiasis. Am. J. Trop. Med. Hyg. 1996, 55, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Lejon, V.; Legros, D.; Savignoni, A.; Etchegorry, M.G.; Mbulamberi, D.; Buscher, P. Neuro-inflammatory risk factors for treatment failure in “early second stage” sleeping sickness patients treated with pentamidine. J. Neuroimmunol. 2003, 144, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Choi, G.; No, J.H. Antileishmanial mechanism of diamidines involves targeting kinetoplasts. Antimicrob. Agents Chemother. 2016, 60, 6828–6836. [Google Scholar] [CrossRef] [Green Version]
- Reguera, R.M.; Redondo, C.M.; Pérez-Pertejo, Y.; Balaña-Fouce, R. S-Adenosyl-methionine in protozoan parasites: Functions, synthesis and regulation. Mol. Biochem. Parasitol. 2007, 152, 1–10. [Google Scholar] [CrossRef]
- Song, J.; Baker, N.; Rothert, M.; Henke, B.; Jeacock, L.; Horn, D.; Beitz, E. Pentamidine is not a permeant but a nanomolar inhibitor of the Trypanosoma brucei aquaglyceroporin-2. PLoS Pathog. 2016, 12, e1005436. [Google Scholar] [CrossRef] [Green Version]
- Lutje, V.; Seixas, J.; Kennedy, A. Chemotherapy for second-stage Human African trypanosomiasis. Cochrane Database Syst. Rev. 2013, D006201. [Google Scholar] [CrossRef]
- Barrett, M.P.; Boykin, D.W.; Brun, R.; Tidwell, R.R. Human African trypanosomiasis: Pharmacological re-engagement with a neglected disease. Br. J. Pharmacol. 2007, 152, 1155–1171. [Google Scholar] [CrossRef] [PubMed]
- Burri, C. Chemotherapy against human African trypanosomiasis: Is there a road to success? Parasitology 2010, 137, 1987–1994. [Google Scholar] [CrossRef] [Green Version]
- Collins, J.M.; Klecker, R.W., Jr.; Yarchoan, R.; Lane, H.C.; Fauci, A.S.; Redfield, R.R.; Broder, S.; Myers, C.E. Clinical pharmacokinetics of suramin in patients with HTLV-III/LAV infection. J. Clin. Pharmacol. 1986, 26, 22–26. [Google Scholar]
- Schultzberg, M.; Ambatsis, M.; Samuelsson, E.B.; Kristensson, K.; Meirvenne, N. Spread of Trypanosoma brucei to the nervous system: Early attack on circumventricular organs and sensory ganglia. J. Neurosci. Res. 1988, 21, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Hall, B.S.; Field, M.C. Evidence for a non-LDL-mediated entry route for the trypanocidal drug suramin in Trypanosoma brucei. Mol. Biochem. Parasitol. 2002, 122, 217–221. [Google Scholar] [CrossRef]
- Wierenga, R.K.; Swinkels, B.; Michels, P.A.; Osinga, K.; Misset, O.; Van Beeumen, J.; Gibson, W.C.; Postma, J.P.; Borst, P.; Opperdoes, F.R. Common elements on the surface of glycolytic enzymes from Trypanosoma brucei may serve as topogenic signals for import into glycosomes. EMBO J. 1987, 6, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Burri, C.; Brun, R. Human African trypanosomiasis. In Manson’s Tropical Diseases; Chapter 76; Cook, G., Zumla, A., Eds.; Saunders: London, ON, Canada, 2008; pp. 1307–1325. [Google Scholar]
- Schmid, C.; Richer, M.; Bilenge, C.M.; Josenando, T.; Chappuis, F.; Manthelot, C.R.; Nangouma, A.; Doua, F.; Asumu, P.N.; Simarro, P.P.; et al. Effectiveness of a 10-day melarsoprol schedule for the treatment of late-stage human African trypanosomiasis: Confirmation from a multinational study (IMPAMEL II). J. Infect. Dis. 2005, 191, 1922–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairlamb, A.H.; Horn, D. Melarsoprol resistance in African Trypanosomiasis. Trends Parasitol. 2018, 34, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairlamb, A.H.; Henderson, G.B.; Cerami, A. Trypanothione is the primary target for arsenical drugs against African trypanosomes. Proc. Natl. Acad. Sci. USA 1989, 86, 2607–2611. [Google Scholar] [CrossRef] [Green Version]
- Maser, P.; Sütterlin, C.; Kralli, A.; Kaminsky, R. A nucleoside transporter from Trypanosoma brucei involved in drug resistance. Science 1999, 285, 242–244. [Google Scholar]
- Matovu, E.; Stewart, M.L.; Geiser, F.; Brun, R.; Mäser, P.; Wallace, L.J.; Burchmore, R.J.; Enyaru, J.C.; Barrett, M.P.; Kaminsky, R.; et al. Mechanisms of arsenical and diamidine uptake and resistance in Trypanosoma brucei. Eukaryot. Cell 2003, 2, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Graf, F.E.; Ludin, P.; Wenzler, T.; Kaiser, M.; Brun, R.; Pyana, P.P.; Büscher, P.; de Koning, H.P.; Horn, D.; Mäser, P. Aquaporin 2 mutations in Trypanosoma brucei gambiense field isolates correlate with decreased susceptibility to pentamidine and melarsoprol. PLoS Negl. Trop. Dis. 2013, 7, e2475. [Google Scholar] [CrossRef] [Green Version]
- Graf, F.E.; Baker, N.; Munday, J.C.; de Koning, H.P.; Horn, D.; Mäser, P. Chimerization at the AQP2-AQP3 locus is the genetic basis of melarsoprol-pentamidine cross-resistance in clinical Trypanosoma brucei gambiense isolates. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 65–68. [Google Scholar] [CrossRef] [Green Version]
- Blum, J.; Nkunku, S.; Burri, C. Clinical description of encephalopathic syndromes and risk factors for their occurrence and outcome during melarsoprol treatment of human African trypanosomiasis. Trop. Med. Int. Health 2001, 6, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burri, C.; Brun, R. Eflornithine for the treatment of human African trypanosomiasis. Parasitol. Res. 2003, 90, S49–S52. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, C.J.; Nathan, H.C.; Hutner, S.H.; McCann, P.P.; Sjoerdsma, A. Polyamine metabolism: A potential therapeutic target in trypanosomes. Science 1980, 210, 332–334. [Google Scholar] [CrossRef]
- Bacchi, C.J.; Yarlett, N. Polyamine metabolism as chemotherapeutic target in protozoan parasites. Mini Rev. Med. Chem. 2002, 2, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Haegele, K.D.; Alken, R.G.; Grove, J.; Schechter, P.J.; Koch-Weser, J. Kinetics of alpha-difluoromethylornithine: An irreversible inhibitor of ornithine decarboxylase. Clin. Pharmacol. Ther. 1981, 30, 210–217. [Google Scholar] [CrossRef] [PubMed]
- LoGiudice, N.; Le, L.; Abuan, I.; Leizorek, Y.; Roberts, S.C. Alpha-difluoromethylornithine, an irreversible inhibitor of polyamine biosynthesis, as a therapeutic strategy against hyperproliferative and infectious diseases. Med. Sci. 2018, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, L.; Dogruel, M.; Rodgers, J.; Bradley, B.; Thomas, S.A. The blood–brain barrier significantly limits eflornithine entry into Trypanosoma brucei brucei infected mouse brain. J. Neurochem. 2008, 107, 1136–1146. [Google Scholar]
- Pegg, A.E. Regulation of ornithine decarboxylase. J. Biol. Chem. 2006, 281, 14529–14532. [Google Scholar] [CrossRef] [Green Version]
- Milord, F.; Pépin, J.; Loko, L.; Ethier, L.; Mpia, B. Efficacy and toxicity of eflornithine for treatment of Trypanosoma brucei gambiense sleeping sickness. Lancet 1992, 340, 652–655. [Google Scholar] [CrossRef]
- Baker, C.H.; Welburn, S.C. The long wait for a new drug for Human African Trypanosomiasis. Trends Parasitol. 2018, 34, 818–827. [Google Scholar] [CrossRef]
- Yun, O.; Priotto, G.; Tong, J.; Flevaud, L.; Chappuis, F. NECT is next: Implementing the new drug combination therapy for Trypanosoma brucei gambiense sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4, e720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priotto, G.; Kasparian, S.; Mutombo, W.; Ngouama, D.; Ghorashian, S.; Arnold, U.; Ghabri, S.; Baudin, E.; Buard, V.; Kazadi-Kyanza, S.; et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: A multicentre, randomised, phase III, non-inferiority trial. Lancet 2009, 374, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Deeks, E.D. Fexinidazole: First global approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Pollastri, M.P. Fexinidazole: A new drug for African sleeping sickness on the horizon. Trends Parasitol. 2018, 34, 178–179. [Google Scholar] [CrossRef]
- Mesu, V.K.B.K.; Kalonji, W.M.; Bardonneau, C.; Mordt, O.V.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Kuziena, W.; Lubaki, J.F.; et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: A pivotal multicentre, randomised, non-inferiority trial. Lancet 2018, 391, 144–154. [Google Scholar] [CrossRef]
- Kratz, J.M. Drug discovery for Chagas disease: A viewpoint. Acta Trop. 2019, 198, 105–107. [Google Scholar] [CrossRef]
- Ribeiro, V.; Dias, N.; Paiva, T.; Hagström-Bex, L.; Nitz, N.; Pratesi, R.; Hecht, M. Current trends in the pharmacological management of Chagas disease. Int. J. Parasitol. Drugs Drug. Resist. 2019, 12, 7–17. [Google Scholar] [CrossRef]
- Kratz, J.M.; Garcia Bournissen, F.; Forsyth, C.J.; Sosa-Estani, S. Clinical and pharmacological profile of benznidazole for treatment of Chagas disease. Expert Rev. Clin. Pharmacol. 2018, 11, 943–957. [Google Scholar] [CrossRef] [Green Version]
- Patterson, S.; Wyllie, S. Nitro drugs for the treatment of trypanosomatid diseases: Past, present, and future prospects. Trends Parasitol. 2014, 30, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, S.R. Trypanocidal activity of nitroaromatic prodrugs: Current treatments and future perspectives. Curr. Top. Med. Chem. 2011, 11, 2072–2084. [Google Scholar] [CrossRef] [PubMed]
- Caldas, I.S.; Santos, E.G.; Novaes, R.D. An evaluation of benznidazole as a Chagas disease therapeutic. Expert Opin. Pharmacother. 2019, 20, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Molina, I.; Salvador, F.; Sánchez-Montalvá, A.; Artaza, M.A.; Moreno, R.; Perin, L.; Esquisabel, A.; Pinto, L.; Pedraz, J.L. Pharmacokinetics of benznidazole in healthy volunteers and implications in future clinical trials. Antimicrob. Agents Chemother. 2017, 61, e01912-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altcheh, J.; Moscatelli, G.; Mastrantonio, G.; Moroni, S.; Giglio, N.; Marson, M.E.; Ballering, G.; Bisio, M.; Koren, G.; García-Bournissen, F. Population pharmacokinetic study of benznidazole in pediatric Chagas disease suggests efficacy despite lower plasma concentrations than in adults. PLoS Negl. Trop. Dis. 2014, 8, e2907. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, M.G.; Vigliano, C.; Lococo, B.; Bertocchi, G.; Viotti, R. Prevention of congenital Chagas disease by Benznidazole treatment in reproductive-age women. An observational study. Acta Trop. 2017, 174, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Pinazo, M.J.; Guerrero, L.; Posada, E.; Rodríguez, E.; Soy, D.; Gascon, J. Benznidazole-related adverse drug reactions and their relationship to serum drug concentrations in patients with chronic Chagas disease. Antimicrob. Agents Chemother. 2013, 57, 390–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermudez, J.; Davies, C.; Simonazzia, A.; Real, J.P.; Palma, S. Current drug therapy and pharmaceutical challenges for Chagas disease. Acta Tropica 2016, 156, 1–16. [Google Scholar] [CrossRef]
- Sales, P.A., Jr.; Molina, I.; Fonseca Murta, S.M.; Sánchez-Montalvá, A.; Salvador, F.; Corrêa-Oliveira, R.; Martins Carneiro, C. Experimental and clinical treatment of Chagas disease: A review. Am. J. Trop. Med. Hyg. 2017, 97, 1289–1303. [Google Scholar] [CrossRef]
- Wilkinson, S.R.; Taylor, M.C.; Horn, D.; Kelly, J.M.; Cheeseman, I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 5022–5027. [Google Scholar] [CrossRef] [Green Version]
- González-Martin, G.; Thambo, S.; Paulos, C.; Vásquez, I.; Paredes, J. The pharmacokinetics of nifurtimox in chronic renal failure. Eur. J. Clin. Pharmacol. 1992, 42, 671–673. [Google Scholar] [CrossRef]
- Castro, J.A.; De Mecca, M.M.; Bartel, L.C. Toxic side effects of drugs used to treat CD (American trypanosomiasis). Hum. Exp. Toxicol. 2006, 25, 471–479. [Google Scholar] [CrossRef]
- Kaiser, M.; Bray, M.A.; Cal, M.; Bourdin Trunz, B.; Torreele, E.; Brun, R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.M.; O’Connor, P.D.; Blaser, A.; Yardley, V.; Maes, L.; Gupta, S.; Launay, D.; Martin, D.; Franzblau, S.G.; Wan, B.; et al. Repositioning antitubercular 6-Nitro-2,3-dihydroimidazo[2,1-b][1,3]oxazoles for Neglected Tropical Diseases: Structure-activity studies on a preclinical candidate for visceral leishmaniasis. J. Med. Chem. 2016, 59, 2530–2550. [Google Scholar] [CrossRef] [PubMed]
- Alves, F.; Bilbe, G.; Blesson, S.; Goyal, V.; Monnerat, S.; Mowbray, C.; Muthoni Ouattara, G.; Pécoul, B.; Rijal, S.; Rode, J.; et al. Recent development of visceral leishmaniasis treatments: Successes, pitfalls, and perspectives. Clin. Microbiol. Rev. 2018, 31, e00048-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reguera, R.M.; Gutiérrez-Corbo, C.; Ordóñez, C.; Pérez-Pertejo, M.Y.; Balaña-Fouce, R. Current and promising novel drug candidates against visceral leishmaniasis. Pure Appl. Chem. 2019, 91, 1385–1404. [Google Scholar] [CrossRef]
- Caridha, D.; Vesely, B.; van Bocxlaer, K.; Arana, B.; Mowbray, C.E.; Rafati, S.; Uliana, S.; Reguera, R.; Kreishman-Deitrick, M.; Sciotti, R.; et al. Route map for the discovery and pre-clinical development of new drugs and treatments for cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug. Resist. 2019, 11, 106–117. [Google Scholar] [CrossRef]
- Frézard, F.; Demicheli, C.; Ribeiro, R.R. Pentavalent antimonials: New perspectives for old drugs. Molecules 2009, 14, 2317–2336. [Google Scholar] [CrossRef] [Green Version]
- Salih, M.A.M.; Fakiola, M.; Lyons, P.A.; Younis, B.M.; Musa, A.M.; Elhassan, A.M.; Anderson, D.; Syn, G.; Ibrahim, M.E.; Blackwell, J.M.; et al. Expression profiling of Sudanese visceral leishmaniasis patients pre- and post-treatment with sodium stibogluconate. Parasite Immunol. 2017, 39, e12431. [Google Scholar] [CrossRef] [Green Version]
- Shaked-Mishan, P.; Ulrich, N.; Ephros, M.; Zilberstein, D. Novel Intracellular SbV reducing activity correlates with antimony susceptibility in Leishmania donovani. J. Biol. Chem. 2001, 276, 3971–3976. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.; Chakravarty, J. Leishmaniasis: An update of current pharmacotherapy. Expert Opin. Pharmacother. 2012, 14, 53–63. [Google Scholar] [CrossRef]
- Perry, M.; Wyllie, S.; Prajapati, V.; Menten, J.; Raab, A.; Feldmann, J.; Chakraborti, D.; Sundar, S.; Boelaert, M.; Picado, A.; et al. Arsenic, antimony, and Leishmania: Has arsenic contamination of drinking water in India led to treatment- resistant kala-azar? Lancet 2015, 385, S80. [Google Scholar] [CrossRef]
- Mukhopadhyay, D.; Dalton, J.E.; Kaye, P.M.; Chatterjee, M. Post kala-azar dermal leishmaniasis: An unresolved mystery. Trends Parasitol. 2014, 30, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundar, S.; Chakravarty, J. Antimony toxicity. Int. J. Environ. Res. Public Health 2010, 7, 4267–4277. [Google Scholar] [CrossRef] [PubMed]
- Loo, A.S.; Muhsin, S.A.; Walsh, T.J. Toxicokinetic and mechanistic basis for the safety and tolerability of liposomal amphotericin B. Expert Opin. Drug Saf. 2013, 12, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Brajtburg, J.; Powderly, W.G.; Kobayashi, G.S.; Medoff, G. Amphotericin B: Current understanding of mechanisms of action. Antimicrob. Agents Chemother. 1990, 34, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Stone, N.R.H.; Bicanic, T.; Salim, R.; Hope, W. Liposomal Amphotericin B (AmBisome®): A review of the pharmacokinetics, pharmacodynamics, clinical experience and future directions. Drugs 2016, 76, 485–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundar, S.; Chakravarty, J.; Agarwal, D.; Rai, M.; Murray, H.W. Single-dose liposomal amphotericin B for visceral leishmaniasis in India. N. Engl. J. Med. 2010, 362, 504–512. [Google Scholar] [CrossRef]
- Jensen, G.M. The care and feeding of a commercial liposomal product: Liposomal amphotericin B (AmBisome®). J. Liposome Res. 2017, 27, 173–179. [Google Scholar] [CrossRef]
- Deray, G. Amphotericin B nephrotoxicity. J. Antimicrob. Chemother. 2002, 49, 37–41. [Google Scholar] [CrossRef]
- Jha, T.K.; Sundar, S.; Thakur, C.P.; Bachmann, P.; Karbwang, J.; Fischer, C.; Voss, A.; Berman, J. Oral miltefosine for Indian visceral leishmaniasis. N. Engl. J. Med. 1999, 341, 1795–1800. [Google Scholar] [CrossRef]
- Verma, N.K.; Dey, C.S. Possible mechanism of miltefosine-mediated death of Leishmania donovani. Antimicrob. Agents Chemother. 2004, 48, 3010–3015. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Roy, K.; Roy, S. Immunomodulatory effects of antileishmanial drugs. J. Antimicrob. Chemother. 2013, 68, 2834–2838. [Google Scholar] [CrossRef] [PubMed]
- Dorlo, T.P.; Balasegaram, M.; Beijnen, J.H.; de Vries, P.J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Singh, A. Chemotherapeutics of visceral leishmaniasis: Present and future developments. Parasitology 2018, 145, 481–489. [Google Scholar] [CrossRef]
- Mbui, J.; Olobo, J.; Omollo, R.; Solomos, A.; Kip, A.E.; Kirigi, G.; Sagaki, P.; Kimutai, R.; Were, L.; Omollo, T.; et al. Pharmacokinetics, safety, and efficacy of an allometric miltefosine regimen for the treatment of visceral leishmaniasis in eastern African children: An open-label, phase ii clinical trial. Clin. Infect. Dis. 2019, 68, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Olliaro, P.L. Miltefosine in the treatment of leishmaniasis: Clinical evidence for informed clinical risk management. Ther. Clin. Risk Manag. 2007, 3, 733–740. [Google Scholar]
- Jamil, K.M.; Haque, R.; Rahman, R.; Faiz, M.A.; Bhuiyan, A.T.; Kumar, A.; Hassan, S.M.; Kelly, H.; Dhalaria, P.; Kochhar, S.; et al. Effectiveness study of paromomycin im injection (PMIM) for the treatment of Visceral Leishmaniasis (VL) in Bangladesh. PLoS Negl. Trop. Dis. 2015, 9, e0004118. [Google Scholar] [CrossRef]
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Sinha, P.K.; Bhattacharya, S.K. Injectable paromomycin for Visceral leishmaniasis in India. N. Engl. J. Med. 2007, 356, 2571–2581. [Google Scholar] [CrossRef] [Green Version]
- Diro, E.; Ritmeijer, K.; Boelaert, M.; Alves, F.; Mohammed, R.; Abongomera, C.; Ravinetto, R.; De Crop, M.; Fikre, H.; Adera, C.; et al. Long-term clinical outcomes in visceral leishmaniasis/human immunodeficiency virus-coinfected patients during and after pentamidine secondary prophylaxis in Ethiopia: A single-arm clinical trial. Clin. Infect. Dis. 2018, 66, 444–451. [Google Scholar] [CrossRef] [Green Version]
- Rahman, R.; Goyal, V.; Haque, R.; Jamil, K.; Faiz, A.; Samad, R.; Ellis, S.; Balasegaram, M.; Boer, M.D.; Rijal, S.; et al. Safety and efficacy of short course combination regimens with AmBisome, miltefosine and paromomycin for the treatment of visceral leishmaniasis (VL) in Bangladesh. PLoS Negl. Trop. Dis. 2017, 11, e0005635. [Google Scholar] [CrossRef] [Green Version]
- Melaku, Y.; Collin, S.M.; Keus, K.; Gatluak, F.; Ritmeijer, K.; Davidson, R.N. Treatment of kala-azar in southern Sudan using a 17-day regimen of sodium stibogluconate combined with paromomycin: A retrospective comparison with 30-day sodium stibogluconate monotherapy. Am. J. Trop. Med. Hyg. 2007, 77, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Tse, E.G.; Korsik, M.; Todd, M.H. The past, present and future of anti-malarial medicines. Malar. J. 2019, 18, 93. [Google Scholar] [CrossRef] [Green Version]
- Su, X.-Z.; Miller, L.H. The discovery of artemisinin and the Nobel Prize in Physiology or Medicine. Sci. China Life Sci. 2015, 58, 1175–1179. [Google Scholar] [CrossRef] [Green Version]
- Morris, C.A.; Duparc, S.; Borghini-Fuhrer, I.; Jung, D.; Shin, C.S.; Fleckenstein, L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar. J. 2011, 10, 263. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.V.; Tekwani, B.L.; Singh, R.L.; Chauhan, V.S. Artemisinin, an endoperoxide antimalarial, disrupts the hemoglobin catabolism and heme detoxification systems in malarial parasite. J. Biol. Chem. 1999, 274, 19383–19388. [Google Scholar] [CrossRef] [Green Version]
- Meshnick, S.R. Artemisinin: Mechanisms of action, resistance and toxicity. Int. J. Parasitol. 2002, 32, 1655–1660. [Google Scholar] [CrossRef]
- WHO. Guidelines for the Treatment of Malaria. Third Edition April. 2015. Available online: https://www.who.int/malaria/publications/atoz/9789241549127/en/ (accessed on 10 November 2019).
- International Artemisinin Study Group. Artesunate combinations for treatment of malaria: Meta-analysis. Lancet 2004, 363, 9–17. [Google Scholar] [CrossRef]
- Sinclair, D.; Zani, B.; Donegan, S.; Olliaro, P.; Garner, P. Artemisinin-based combination therapy for treating uncomplicated malaria. Cochrane Database Syst. Rev. 2009, 8, CD007483. [Google Scholar] [CrossRef]
- Medhi, B.; Patyar, S.; Rao, R.S.; Byrav, P.; Prakash, A. Pharmacokinetic and toxicological profile of artemisinin compounds: An update. Pharmacology 2009, 84, 323–332. [Google Scholar] [CrossRef]
- Sullivan, D.J., Jr.; Gluzman, I.Y.; Goldberg, D.E. Plasmodium hemozoin formation mediated by histidine-rich proteins. Science 1996, 271, 219–222. [Google Scholar] [CrossRef]
- Tekwani, B.L.; Walker, L.A. Targeting the hemozoin synthesis pathway for new antimalarial drug discovery: Technologies for in vitro β-hematin formation assay. Comb. Chem. High Throughput Screen. 2005, 8, 63–79. [Google Scholar] [CrossRef]
- Noubiap, J.J.N. Shifting from quinine to artesunate as first-line treatment of severe malaria in children and adults: Saving more lives. J. Infect. Public Health 2014, 7, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Ashley, E.A.; Phyo, A.P. Drugs in development for malaria. Drugs 2018, 78, 861–879. [Google Scholar] [CrossRef] [Green Version]
- Karbwang, J.; White, N.J. Clinical pharmacokinetics of mefloquine. Clin. Pharmacokinet. 1990, 19, 264–279. [Google Scholar] [CrossRef]
- Alkadi, H.O. Antimalarial drug toxicity: A review. Chemotherapy 2007, 53, 385–391. [Google Scholar] [CrossRef]
- Palmer, K.J.M.; Holliday, S.M.; Brogden, R.N. Mefloquine. A review of its antimalarial activity, pharmacokinetic properties and therapeutic efficacy. Drugs 1993, 45, 430–475. [Google Scholar] [CrossRef]
- Olliaro, P.; Nevill, C.; LeBras, J.; Ringwald, P.; Mussano, P.; Garner, P.; Brasseur, P. Systematic review of amodiaquine treatment in uncomplicated malaria. Lancet 1996, 348, 1196–1201. [Google Scholar] [CrossRef]
- Backman, J.T.; Filppula, A.M.; Niemi, M.; Neuvonen, P.J. Role of cytochrome P450 2C8 in drug metabolism and interactions. Pharm. Rev. 2015, 68, 168–241. [Google Scholar] [CrossRef]
- Ebstie, Y.A.; Abay, S.M.; Tadesse, W.T.; Ejigu, D.A. Tafenoquine and its potential in the treatment and relapse prevention of Plasmodium vivax malaria: The evidence to date. Drug Des. Dev. Ther. 2016, 10, 2387–2399. [Google Scholar] [CrossRef] [Green Version]
- Imwong, M.; Snounou, G.; Pukrittayakamee, S.; Tanomsing, N.; Kim, J.R.; Nandy, A.; Guthmann, J.P.; Nosten, F.; Carlton, J.; Looareesuwan, S.; et al. Relapses of Plasmodium vivax infection usually result from activation of heterologous hypnozoites. J. Infect. Dis. 2007, 195, 927–933. [Google Scholar] [CrossRef] [Green Version]
- Baird, J.K. Tafenoquine for travelers’ malaria: Evidence, rationale and recommendations. J. Travel. Med. 2018, 25, 110. [Google Scholar] [CrossRef] [Green Version]
- Rishikesh, K.; Saravu, K. Primaquine treatment and relapse in Plasmodium vivax malaria. Pathog. Glob. Health 2016, 110, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ramos Júnior, W.M.; Sardinha, J.F.; Costa, M.R.; Santana, M.S.; Alecrim, M.G.; Lacerda, M.V. Clinical aspects of hemolysis in patients with Plasmodium vivax malaria treated with primaquine, in the Brazilian Amazon. Braz. J. Infect. Dis. 2010, 14, 410–412. [Google Scholar] [CrossRef] [Green Version]
- Adam, I.; Ibrahim, Y.; Gasim, G.I. Efficacy and safety of artemisinin-based combination therapy for uncomplicated Plasmodium falciparum malaria in Sudan: A systematic review and meta-analysis. Malar. J. 2018, 17, 110. [Google Scholar] [CrossRef]
- Müller, I.B.; Hyde, J.E. Folate metabolism in human malaria parasites—75 years on. Mol. Biochem. Parasitol. 2013, 188, 63–77. [Google Scholar] [CrossRef]
- Peters, P.J.; Thigpen, M.C.; Parise, M.E.; Newman, R.D. Safety and toxicity of sulfadoxine/pyrimethamine: Implications for malaria prevention in pregnancy using intermittent preventive treatment. Drug Saf. 2007, 30, 481–501. [Google Scholar] [CrossRef]
- Xiao, Z.; Morris-Natschke, S.L.; Lee, K.H. Strategies for the optimization of natural leads to anticancer drugs or drug candidates. Med. Res. Rev. 2016, 36, 32–91. [Google Scholar] [CrossRef] [Green Version]
- Henrich, C.; Beutler, J.A. Matching the power of high throughput screening to the chemical diversity of natural products. Nat. Prod. Rep. 2013, 30, 1284–1298. [Google Scholar] [CrossRef] [Green Version]
- Thornburg, C.C.; Britt, J.R.; Evans, J.R.; Akee, R.K.; Whitt, J.A.; Trinh, S.K.; Harris, M.J.; Thompson, J.R.; Ewing, T.L.; Shipley, S.M.; et al. NCI Program for natural product discovery: A publicly-accessible library of natural product fractions for high-throughput screening. ACS Chem. Biol. 2018, 13, 2484–2497. [Google Scholar] [CrossRef]
- Annang, F.; Pérez-Moreno, G.; García-Hernández, R.; Cordon-Obras, C.; Martín, J.; Tormo, J.R.; Rodríguez, L.; de Pedro, N.; Gómez-Pérez, V.; Valente, M.; et al. High-throughput screening platform for natural product-based drug discovery against 3 neglected tropical diseases: Human African trypanosomiasis, leishmaniasis, and Chagas disease. J. Biomol. Screen. 2015, 20, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Reguera, R.M.; Calvo-Álvarez, E.; Alvarez-Velilla, R.; Balaña-Fouce, R. Target-based vs. phenotypic screenings in Leishmania drug discovery: A marriage of convenience or a dialogue of the deaf? Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 355–357. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, I.H. Drug discovery for neglected diseases: Molecular target-based and phenotypic approaches. J. Med. Chem. 2013, 56, 7719–7726. [Google Scholar] [CrossRef]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- Swinney, D.C. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin. Pharmacol. Ther. 2013, 93, 299–301. [Google Scholar] [CrossRef]
- Varela, J.N.; Lammoglia Cobo, M.F.; Pawar, S.V.; Yadav, V.G. Cheminformatic analysis of antimalarial chemical space illuminates therapeutic mechanisms and offers strategies for therapy development. J. Chem. Inf. Model. 2017, 57, 2119–2131. [Google Scholar] [CrossRef]
- Szymański, P.; Markowicz, M.; Mikiciuk-Olasik, E. Adaptation of High-Throughput Screening in drug discovery—Toxicological screening tests. Int. J. Mol. Sci. 2012, 13, 427–452. [Google Scholar] [CrossRef] [Green Version]
- Zulfiqar, B.; Shelper, T.B.; Avery, V.M. Leishmaniasis drug discovery: Recent progress and challenges in assay development. Drug Discov.Today 2017, 22, 1516–1531. [Google Scholar] [CrossRef]
- Frearson, J.A.; Wyatt, P.G.; Gilbert, I.H.; Fairlamb, A.H. Target assessment for antiparasitic drug discovery. Trends Parasitol. 2007, 23, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Roberts, S.; Ullman, B. Parasite polyamines as pharmaceutical targets. Curr. Pharm. Des. 2017, 23, 3325–3341. [Google Scholar] [CrossRef]
- Persson, L.; Jeppsson, A.; Nasizadeh, S. Turnover of trypanosomal ornithine decarboxylases. Biochem. Soc. Trans. 2003, 31, 411–414. [Google Scholar] [CrossRef] [Green Version]
- Lepesheva, G.I.; Villalta, F.; Waterman, M.R. Targeting Trypanosoma cruzi sterol 14α-demethylase (CYP51). Chagas disease, Part, A. Adv. Parasitol. 2011, 75, 65–87. [Google Scholar]
- Bucknera, F.S.; Urbina, J.A. Recent developments in sterol 14-demethylase inhibitors for Chagas disease. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, I.; Gómez i Prat, J.; Salvador, F.; Treviño, B.; Sulleiro, E.; Serre, N.; Pou, D.; Roure, S.; Cabezos, J.; Valerio, L.; et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N. Engl. J. Med. 2014, 370, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Torrico, F.; Gascon, J.; Ortiz, L.; Alonso-Vega, C.; Pinazo, M.J.; Schijman, A.; Almeida, I.C.; Alves, F.; Strub-Wourgaft, N.; Ribeiro, I.; et al. Treatment of adult chronic indeterminate Chagas disease with benznidazole and three E1224 dosing regimens: A proof-of-concept, randomised, placebo-controlled trial. Lancet Infect. Dis. 2018, 18, 419–430. [Google Scholar] [CrossRef]
- Cleghorn, L.A.; Woodland, A.; Collie, I.T.; Torrie, L.S.; Norcross, N.; Luksch, T.; Mpamhanga, C.; Walker, R.G.; Mottram, J.C.; Brenk, R.; et al. Identification of inhibitors of the Leishmania cdc2-related protein kinase CRK3. ChemMedChem 2011, 6, 2214–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyllie, S.; Thomas, M.; Patterson, S.; Crouch, S.; De Rycker, M.; Lowe, R.; Gresham, S.; Urbaniak, M.D.; Otto, T.D.; Stojanovski, L.; et al. Cyclin-dependent kinase 12 is a drug target for visceral leishmaniasis. Nature 2018, 560, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Price, H.P.; Menon, M.R.; Panethymitaki, C.; Goulding, D.; McKean, P.G.; Smith, D.F. Myristoyl-CoA: Protein N-myristoyltransferase, an essential enzyme and potential drug target in kinetoplastid parasites. J. Biol. Chem. 2003, 278, 7206–7214. [Google Scholar] [CrossRef] [Green Version]
- Brand, S.; Cleghorn, L.A.; McElroy, S.P.; Robinson, D.A.; Smith, V.C.; Hallyburton, I.; Harrison, J.R.; Norcross, N.R.; Spinks, D.; Bayliss, T.; et al. Discovery of a novel class of orally active trypanocidal N-myristoyltransferase inhibitors. J. Med. Chem. 2012, 55, 140–152. [Google Scholar] [CrossRef] [Green Version]
- Spinks, D.; Smith, V.; Thompson, S.; Robinson, D.A.; Luksch, T.; Smith, A.; Torrie, L.S.; McElroy, S.; Stojanovski, L.; Norval, S.; et al. Development of small-molecule Trypanosoma brucei N-myristoyltransferase inhibitors: Discovery and optimisation of a novel binding mode. ChemMedChem 2015, 10, 1821–1836. [Google Scholar] [CrossRef] [Green Version]
- Vickers, T.J.; Beverley, S.M. Folate metabolic pathways in Leishmania. Essays Biochem. 2011, 51, 63–80. [Google Scholar]
- Sienkiewicz, N.; Jaroslawski, S.; Wyllie, S.; Fairlamb, A.H. Chemical and genetic validation of dihydrofolate reductase-thymidylate synthase as a drug target in African trypanosomes. Mol. Microbiol. 2008, 69, 520–533. [Google Scholar] [CrossRef] [Green Version]
- Moraes, C.B.; Witt, G.; Kuzikov, M.; Ellinger, B.; Calogeropoulou, T.; Prousis, K.C.; Mangani, S.; Di Pisa, F.; Landi, G.; Iacono, L.D.; et al. Accelerating drug discovery efforts for trypanosomatidic infections using an integrated transnational academic drug discovery platform. SLAS Discov. 2019, 24, 346–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das Neves, G.M.; Kagami, L.P.; Gonçalves, I.L.; Eifler-Lima, V.L. Targeting pteridine reductase 1 and dihydrofolate reductase: The old is a new trend for leishmaniasis drug discovery. Future Med. Chem. 2019, 11, 2107–2130. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, S.; Oza, S.L.; Patterson, S.; Spinks, D.; Thompson, S.; Fairlamb, A.H. Dissecting the essentiality of the bifunctional trypanothione synthetase-amidase in Trypanosoma brucei using chemical and genetic methods. Mol. Microbiol. 2009, 74, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, N.; Tanwar, N.; Munde, M. Molecular insights into trypanothione reductase-inhibitor interaction: A structure-based review. Archiv. Pharm. 2018, 351, 1700373. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, K.; Paulino, M.; Salas, C.O.; Zarate-Ramos, J.J.; Vera, B.; Rivera, G. Trypanothione reductase: A target for the development of anti-Trypanosoma cruzi drugs. Mini Rev. Med. Chem. 2017, 17, 939–946. [Google Scholar] [CrossRef]
- Santos Mendonça, A.A.; Morais Coelho, C.; Paranho Veloso, M.; Santana Caldas, I.; Vilela Gonçalves, R.; Teixeira, A.L.; Silva de Miranda, A.; Dias Novaes, R. Relevance of trypanothione reductase inhibitors on Trypanosoma cruzi infection: A systematic review, meta-analysis, and in silico integrated approach. Oxid. Med. Cell. Longev. 2018, 2018, 8676578. [Google Scholar]
- Balaña-Fouce, R.; Redondo, C.M.; Pérez-Pertejo, Y.; Díaz-González, R.; Reguera, R.M. Targeting atypical trypanosomatid DNA topoisomerase I. Drug Discov. Today 2006, 15-16, 733–740. [Google Scholar]
- Bakshi, R.P.; Shapiro, T.A. RNA interference of Trypanosoma brucei topoisomerase IB: Both subunits are essential. Mol. Biochem. Parasitol. 2004, 136, 249–255. [Google Scholar] [CrossRef]
- Balaña-Fouce, R.; Prada, C.F.; Requena, J.M.; Cushman, M.; Pommier, Y.; Álvarez-Velilla, R.; Escudero-Martínez, J.M.; Calvo-Álvarez, E.; Pérez-Pertejo, Y.; Reguera, R.M. Indotecan (LMP400) and AM13-55: Two novel indenoisoquinolines show potential for treating visceral leishmaniasis. Antimicrob. Agents Chemother. 2012, 56, 5264–5270. [Google Scholar] [CrossRef] [Green Version]
- Chatelain, E. Chagas disease drug discovery: Toward a new era. J. Biomol. Screen. 2015, 20, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Freitas-Junior, L.H.; Chatelain, E.; Kim, H.A.; Siqueira-Neto, J.L. Visceral leishmaniasis treatment: What do we have, what do we need and how to deliver it? Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo-Álvarez, E.; Álvarez-Velilla, R.; Fernández-Prada, C.; Balaña-Fouce, R.; Reguera, R.M. Trypanosomatids see the light: Recent advances in bioimaging research. Drug Discov. Today 2015, 20, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Andreu, N.; Zelmer, A.; Wiles, S. Noninvasive biophotonic imaging for studies of infectious disease. FEMS Microbiol. Rev. 2011, 35, 360–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, T.; Lecoeur, H.; Prina, E. Imaging Leishmania development in their host cells. Trends Parasitol. 2009, 25, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Lee, N.; Ioset, J.R.; No, J.H. Evaluation of parameters impacting drug susceptibility in intracellular Trypanosoma cruzi assay protocols. SLAS Discov. 2017, 22, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dube, A.; Gupta, R.; Sing, N. Reporter genes facilitating discovery of drugs targeting protozoan parasites. Trends Parasitol. 2009, 25, 432–439. [Google Scholar] [CrossRef]
- Peña, I.; Manzano, M.P.; Cantizani, J.; Kessler, A.; Alonso-Padilla, J.; Bardera, A.I.; Alvarez, E.; Colmenarejo, G.; Cotillo, I.; Roquero, I.; et al. New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: An open resource. Sci. Rep. 2015, 5, 8771. [Google Scholar]
- Calvo-Álvarez, E.; Stamatakis, K.; Punzón, C.; Álvarez-Velilla, R.; Tejería, A.; Escudero-Martínez, J.M.; Pérez-Pertejo, Y.; Fresno, M.; Balaña-Fouce, R.; Reguera, R.M. Infrared fluorescent imaging as a potent tool for in vitro, ex vivo and in vivo models of visceral leishmaniasis. PLoS Negl. Trop. Dis. 2015, 9, e0003666. [Google Scholar] [CrossRef]
- Calvo-Alvarez, E.; Cren-Travaillé, C.; Crouzols, A.; Rotureau, B. A new chimeric triple reporter fusion protein as a tool for in vitro and in vivo multimodal imaging to monitor the development of African trypanosomes and Leishmania parasites. Infect. Genet. Evol. 2018, 63, 391–403. [Google Scholar] [CrossRef]
- Roy, G.; Dumas, C.; Sereno, D.; Wu, Y.; Singh, A.K.; Tremblay, M.J.; Ouellette, M.; Olivier, M.; Papadopoulou, B. Episomal and stable expression of the luciferase reporter gene for quantifying Leishmania spp. infections in macrophages and in animal models. Mol. Biochem. Parasitol. 2000, 110, 195–206. [Google Scholar] [CrossRef]
- Miβlitz, A.; Mottram, J.C.; Overath, P.; Aebischer, T. Targeted integration into a rRNA locus results in uniform and high-level expression of transgenes in Leishmania amastigotes. Mol. Biochem. Parasitol. 2000, 107, 251–261. [Google Scholar] [CrossRef]
- Choy, G.; O’Connor, S.; Diehn, F.E.; Costouros, N.; Alexander, H.R.; Choyke, P.; Libutti, S.K. Comparison of noninvasive fluorescent and bioluminescent small animal optical imaging. Biotechniques 2003, 35, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Ashutosh; Gupta, S.; Ramesh; Sundar, S.; Goyal, N. Use of Leishmania donovani field isolates expressing the luciferase reporter gene in in vitro drug screening. Antimicrob. Agents Chemother. 2005, 49, 3776–3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyard, S.; Dutra, P.L.; Deolindo, P.; Autheman, D.; D’Archivio, S.; Minoprio, P. In vivo imaging of trypanosomes for a better assessment of host–parasite relationships and drug efficacy. Parasitol. Int. 2014, 63, 260–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriques, C.; Henriques-Pons, A.; Meuser-Batista, M.; Ribeiro, A.S.; de Souza, W. In vivo imaging of mice infected with bioluminescent Trypanosoma cruzi unveils novel sites of infection. Parasit. Vectors 2014, 7, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Velilla, R.; Gutiérrez-Corbo, M.C.; Punzón, C.; Pérez-Pertejo, M.Y.; Balaña-Fouce, R.; Fresno, M.; Reguera, R.M. A chronic bioluminescent model of experimental visceral leishmaniasis for accelerating drug discovery. PLoS Negl. Trop. Dis. 2019, 13, e0007133. [Google Scholar] [CrossRef]
- Giroud, C.; Ottones, F.; Coustou, V.; Dacheux, D.; Biteau, N.; Miezan, B.; Van Reet, N.; Carrington, M.; Doua, F.; Baltz, T. Murine models for Trypanosoma brucei gambiense disease progression—From silent to chronic infections and early brain tropism. PLoS Negl. Trop. Dis. 2009, 3, e509. [Google Scholar] [CrossRef]
- Kessler, R.L.; Gradia, D.F.; de Cássia Pontello Rampazzo, R.; Lourenço, É.E.; Fidêncio, N.J.; Manhaes, L.; Probst, C.M.; Ávila, A.R.; Fragoso, S.P. Stage-regulated GFP expression in Trypanosoma cruzi: Applications from host-parasite interactions to drug screening. PLoS ONE 2013, 8, e67441. [Google Scholar] [CrossRef] [Green Version]
- Pescher, P.; Blisnick, T.; Bastin, P.; Späth, G.F. Quantitative proteome profiling informs on phenotypic traits that adapt Leishmania donovani for axenic and intracellular proliferation. Cell. Microbiol. 2011, 13, 978–991. [Google Scholar] [CrossRef]
- Singh, N.; Gupta, R.; Jaiswal, A.K.; Sundar, S.; Dube, A. Transgenic Leishmania donovani clinical isolates expressing green fluorescent protein constitutively for rapid and reliable ex vivo drug screening. J. Antimicrob. Chemother. 2009, 64, 370–374. [Google Scholar] [CrossRef] [Green Version]
- Dagley, M.J.; Saunders, E.C.; Simpson, K.J.; McConville, M.J. High-content assay for measuring intracellular growth of Leishmania in human macrophages. Assay Drug Dev. Technol. 2015, 13, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Balaña-Fouce, R.; Pérez Pertejo, M.Y.; Domínguez-Asenjo, B.; Gutiérrez-Corbo, C.; Reguera, R.M. Walking a tightrope: Drug discovery in visceral leishmaniasis. Drug Discov. Today 2019, 24, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Osorio, Y.; Travi, B.L.; Renslo, A.R.; Peniche, A.G.; Melby, P.C. Identification of small molecule lead compounds for visceral leishmaniasis using a novel ex vivo splenic explant model system. PLoS Negl. Trop. Dis. 2011, 5, e962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peniche, A.G.; Osorio, Y.; Renslo, A.R.; Frantz, D.E.; Melby, P.C.; Travi, B.L. Development of an ex vivo lymph node explant model for identification of novel molecules active against Leishmania major. Antimicrob. Agents Chemother. 2014, 58, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Ettinger, N.A.; Wilson, M.E. Macrophage and T-cell gene expression in a model of early infection with the protozoan Leishmania chagasi. PLoS Negl. Trop. Dis. 2008, 2, e252. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; Hooft van Huijsduijnen, R.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef]
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.; Gray, D.W.; Read, K.; De Rycker, M.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Anti-trypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Hovlid, M.L.; Winzeler, E.A. Phenotypic screens in antimalarial drug discovery. Trends Parasitol. 2016, 32, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Cully, M. Trial watch: Next-generation antimalarial from phenotypic screen shows clinical promise. Nat. Rev. Drug Discov. 2014, 13, 717. [Google Scholar] [CrossRef]
- Guiguemde, W.A.; Shelat, A.A.; Garcia-Bustos, J.F.; Diagana, T.T.; Gamo, F.J.; Guy, R.K. Global phenotypic screening for antimalarials. Chem. Biol. 2012, 19, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Linzke, M.; Yan, S.L.R.; Tárnok, A.; Ulrich, H.; Groves, M.R.; Wrenger, C. Live and let dye: Visualizing the cellular compartments of the malaria parasite Plasmodium falciparum. Cytometry A 2019. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.; Pedro, L.; Mak, T.; McCormick, B.; Rowley, J.; Liu, M.; Di Capua, A.; Williams-Noonan, B.; Pham, N.B.; Pouwer, R.; et al. Fragment-Based Screening of a natural product library against 62 potential malaria drug targets employing native mass spectrometry. ACS Infect. Dis. 2018, 4, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Frame, I.J.; Deniskin, R.; Rinderspacher, A.; Katz, F.; Deng, S.X.; Moir, R.D.; Adjalley, S.H.; Coburn-Flynn, O.; Fidock, D.A.; Willis, I.M.; et al. Yeast-based high-throughput screen identifies Plasmodium falciparum equilibrative nucleoside transporter 1 inhibitors that kill malaria parasites. ACS Chem. Biol. 2015, 10, 775–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Lorenzo, M.G.; Rodríguez-Alejandre, A.; Moliner-Cubel, S.; Martínez-Hoyos, M.; Bahamontes-Rosa, N.; González Del Rio, R.; Ródenas, C.; Fuente, J.; Lavandera, J.L.; García-Bustos, J.F.; et al. Functional screening of selective mitochondrial inhibitors of Plasmodium. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 295–303. [Google Scholar] [CrossRef]
- Alam, M.M.; Sanchez-Azqueta, A.; Janha, O.; Flannery, E.L.; Mahindra, A.; Mapesa, K.; Char, A.B.; Sriranganadane, D.; Brancucci, N.M.B.; Antonova-Koch, Y.; et al. Validation of the protein kinase PfCLK3 as a multistage cross-species malarial drug target. Science 2019, 365, eaau1682. [Google Scholar] [CrossRef] [Green Version]
- Laurent, D.; Jullian, V.; Parenty, A.; Knibiehler, M.; Dorin, D.; Schmitt, S.; Lozach, O.; Lebouvier, N.; Frostin, M.; Alby, F.; et al. Antimalarial potential of xestoquinone, a protein kinase inhibitor isolated from a Vanuatu marine sponge Xestospongia sp. Bioorg. Med. Chem. 2006, 14, 4477–4482. [Google Scholar] [CrossRef]
- Xie, S.C.; Gillett, D.L.; Spillman, N.J.; Tsu, C.; Luth, M.R.; Ottilie, S.; Duffy, S.; Gould, A.E.; Hales, P.; Seager, B.A.; et al. Target validation and identification of novel boronate inhibitors of the Plasmodium falciparum proteasome. J. Med. Chem. 2018, 61, 10053–10066. [Google Scholar] [CrossRef] [Green Version]
- Hoelz, L.V.; Calil, F.A.; Nonato, M.C.; Pinheiro, L.; Boechat, N. Plasmodium falciparum dihydroorotate dehydrogenase: A drug target against malaria. Future Med. Chem. 2018, 10, 1853–1874. [Google Scholar] [CrossRef]
- Baldwin, J.; Michnoff, C.H.; Malmquist, N.A.; White, J.; Roth, M.G.; Rathod, P.K.; Phillips, M.A. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 2005, 280, 21847–21853. [Google Scholar] [CrossRef] [Green Version]
- Llanos-Cuentas, A.; Casapia, M.; Chuquiyauri, R.; Hinojosa, J.C.; Kerr, N.; Rosario, M.; Toovey, S.; Arch, R.H.; Phillips, M.A.; Rozenberg, F.D.; et al. Antimalarial activity of single-dose DSM265, a novel plasmodium dihydroorotate dehydrogenase inhibitor, in patients with uncomplicated Plasmodium falciparum or Plasmodium vivax malaria infection: A proof-of-concept, open-label, phase 2a study. Lancet Infect. Dis. 2018, 18, 874–883. [Google Scholar] [CrossRef]
- Hooft van Huijsduijnen, R.; Wells, T.N. The antimalarial pipeline. Curr. Opin. Pharmacol. 2018, 42, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.H.; Ackerman, H.C.; Su, X.Z.; Wellems, T.E. Malaria biology and disease pathogenesis: Insights for new treatments. Nat. Med. 2013, 19, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.; Sarma, P.; Sehgal, R.; Medhi, B. Development in assay methods for in vitro antimalarial drug efficacy testing: A systematic review. Front. Pharmacol. 2017, 8, 754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joanny, F.; Held, J.; Mordmüller, B. In vitro activity of fluorescent dyes against asexual blood stages of Plasmodium falciparum. Antimicrob. Agents Chemother. 2012, 56, 5982–5985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nambati, E.A.; Kiarie, W.C.; Kimani, F.; Kimotho, J.H.; Otinga, M.S.; Too, E.; Kaniaru, S.; Limson, J.; Bulimo, W. Unclear association between levels of Plasmodium falciparum lactate dehydrogenase (PfLDH) in saliva of malaria patients and blood parasitaemia: Diagnostic implications? Malar. J. 2018, 17, 9. [Google Scholar] [CrossRef] [Green Version]
- Hasenkamp, S.; Sidaway, A.; Devine, O.; Roye, R.; Horrocks, P. Evaluation of bioluminescence-based assays of anti-malarial drug activity. Malar. J. 2013, 12, 58. [Google Scholar] [CrossRef] [Green Version]
- Swann, J.; Corey, V.; Scherer, C.A.; Kato, N.; Comer, E.; Maetani, M.; Antonova-Koch, Y.; Reimer, C.; Gagaring, K.; Ibanez, M.; et al. High-throughput luciferase-based assay for the discovery of therapeutics that prevent malaria. ACS Infect. Dis. 2016, 2, 281–293. [Google Scholar] [CrossRef]
- Cui, L.; Miao, J.; Wang, J.; Li, Q.; Cui, L. Plasmodium falciparum: Development of a transgenic line for screening antimalarials using firefly luciferase as the reporter. Exp. Parasitol. 2008, 120, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Caridha, D.; Hickman, M.; Xie, L.; Ngundam, F.; Milner, E.; Schenk, A.; Butler, K.; Nugent, D.; Lee, P.; Roncal, N.; et al. Updating the modified Thompson test by using whole-body bioluminescence imaging to replace traditional efficacy testing in experimental models of murine malaria. Malar. J. 2019, 18, 38. [Google Scholar] [CrossRef]
- Leidenberger, M.; Voigtländer, C.; Simon, N.; Kappes, B. SYBR® Green I-based fluorescence assay to assess cell viability of malaria parasites for routine use in compound screening. Methods Mol. Biol. 2017, 1601, 97–110. [Google Scholar]
- Rason, M.A.; Randriantsoa, T.; Andrianantenaina, H.; Ratsimbasoa, A.; Menard, D. Performance and reliability of the SYBR Green I based assay for the routine monitoring of susceptibility of Plasmodium falciparum clinical isolates. Trans. R. Soc. Trop. Med. Hyg. 2008, 102, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Cheruiyot, A.C.; Auschwitz, J.M.; Lee, P.J.; Yeda, R.A.; Okello, C.O.; Leed, S.E.; Talwar, M.; Murthy, T.; Gaona, H.W.; Hickman, M.R.; et al. Assessment of the worldwide antimalarial resistance network standardized procedure for in vitro malaria drug sensitivity testing using SYBR green assay for field samples with various initial parasitemia levels. Antimicrob. Agents Chemother. 2016, 60, 2417–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Co, E.M.; Dennull, R.A.; Reinbold, D.D.; Waters, N.C.; Johnson, J.D. Assessment of malaria in vitro drug combination screening and mixed-strain infections using the malaria SYBR green I-based fluorescence assay. Antimicrob. Agents Chemother. 2009, 53, 2557–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karl, S.; Wong, R.P.; St Pierre, T.G.; Davis, T.M. A comparative study of a flow-cytometry-based assessment of in vitro Plasmodium falciparum drug sensitivity. Malar. J. 2009, 8, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, M.H.; Deharo, E.; Valentin, A.; Garavito, G. Adaptation and optimization of a fluorescence-based assay for in vivo antimalarial drug screening. Parasitol. Res. 2017, 116, 1955–1962. [Google Scholar] [CrossRef]
- Somsak, V.; Srichairatanakool, S.; Yuthavong, Y.; Kamchonwongpaisan, S.; Uthaipibull, C. Flow cytometric enumeration of Plasmodium berghei-infected red blood cells stained with SYBR green I. Acta Trop. 2012, 122, 113–118. [Google Scholar] [CrossRef]
- Bennett, T.N.; Paguio, M.; Gligorijevic, B.; Seudieu, C.; Kosar, A.D.; Davidson, E.; Roepe, P.D. Novel, rapid, and inexpensive cell-based quantification of antimalarial drug efficacy. Antimicrob. Agents Chemother. 2004, 48, 1807–1810. [Google Scholar] [CrossRef] [Green Version]
- Che, P.; Cui, L.; Kutsch, O.; Cui, L.; Li, Q. Validating a firefly luciferase-based high-throughput screening assay for antimalarial drug discovery. Assay Drug Dev. Technol. 2012, 10, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Lucumi, E.; Darling, C.; Jo, H.; Napper, A.D.; Chandramohanadas, R.; Fisher, N.; Shone, A.E.; Jing, H.; Ward, S.A.; Biagini, G.A.; et al. Discovery of potent small-molecule inhibitors of multidrug-resistant Plasmodium falciparum using a novel miniaturized high-throughput luciferase-based assay. Antimicrob. Agents Chemother. 2010, 54, 3597–3604. [Google Scholar] [CrossRef] [Green Version]
- Cervantes, S.; Prudhomme, J.; Carter, D.; Gopi, K.G.; Li, Q.; Chang, Y.T.; Le Roch, K.G. High-content live cell imaging with RNA probes: Advancements in high-throughput antimalarial drug discovery. BMC Cell Biol. 2009, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Cervantes, S.; Stout, P.E.; Prudhomme, J.; Engel, S.; Bruton, M.; Cervantes, M.; Carter, D.; Tae-Chang, Y.; Hay, M.E.; Aalbersberg, W.; et al. High content live cell imaging for the discovery of new antimalarial marine natural products. BMC Infect. Dis. 2012, 12, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, N.; March, S.; Bhatia, S.N.; Marti, M. Phenotypic screening of small molecules with antimalarial activity for three different parasitic life stages. Methods Mol. Biol. 2018, 1787, 41–52. [Google Scholar] [PubMed]
- Poonam;Gupta, Y.; Gupta, N.; Singh, S.; Wu, L.; Chhikara, B.S.; Rawat, M.; Rathi, B. Multistage inhibitors of the malaria parasite: Emerging hope for chemoprotection and malaria eradication. Med. Res. Rev. 2018, 38, 1511–1535. [Google Scholar] [CrossRef] [PubMed]
- Lucantoni, L.; Fidock, D.A.; Avery, V.M. Luciferase-based, high-throughput assay for screening and profiling transmission-blocking compounds against Plasmodium falciparum gametocytes. Antimicrob. Agents Chemother. 2016, 60, 2097–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delves, M.J.; Angrisano, F.; Blagborough, A.M. Antimalarial transmission-blocking interventions: Past, present, and future. Trends Parasitol. 2018, 34, 735–746. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, S.; Silvestrini, F.; Dechering, K.; Corbett, Y.; Parapini, S.; Timmerman, M.; Galastri, L.; Basilico, N.; Sauerwein, R.; Alano, P.; et al. A Plasmodium falciparum screening assay for anti-gametocyte drugs based on parasite lactate dehydrogenase detection. J. Antimicrob. Chemother. 2013, 68, 2048–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.Q.; Dehdashti, S.J.; Nguyen, D.T.; McKew, J.C.; Zheng, W.; Williamson, K.C. A quantitative high throughput assay for identifying gametocytocidal compounds. Mol. Biochem. Parasitol. 2013, 188, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Spicer, T.P.; Gardiner, D.L.; Schoenen, F.J.; Roy, S.; Griffin, P.R.; Chase, P.; Scampavia, L.; Hodder, P.; Trenholme, K.R. Identification of antimalarial inhibitors using late-stage gametocytes in a phenotypic live/dead assay. SLAS Discov. 2019, 24, 38–46. [Google Scholar] [CrossRef]
- Delves, M.J.; Miguel-Blanco, C.; Matthews, H.; Molina, I.; Ruecker, A.; Yahiya, S.; Straschil, U.; Abraham, M.; León, M.L.; Fischer, O.J.; et al. A high throughput screen for next-generation leads targeting malaria parasite transmission. Nat. Commun. 2018, 9, 3805. [Google Scholar] [CrossRef] [Green Version]
- Lucantoni, L.; Silvestrini, F.; Signore, M.; Siciliano, G.; Eldering, M.; Dechering, K.J.; Avery, V.M.; Alano, P. A simple and predictive phenotypic High Content Imaging assay for Plasmodium falciparum mature gametocytes to identify malaria transmission blocking compounds. Sci. Rep. 2015, 5, 16414. [Google Scholar] [CrossRef] [Green Version]
- Lucantoni, L.; Duffy, S.; Adjalley, S.H.; Fidock, D.A.; Avery, V.M. Identification of MMV malaria box inhibitors of Plasmodium falciparum early-stage gametocytes using a luciferase-based high-throughput assay. Antimicrob. Agents Chemother. 2013, 57, 6050–6062. [Google Scholar] [CrossRef] [Green Version]
- Plouffe, D.M.; Wree, M.; Du, A.Y.; Meister, S.; Li, F.; Patra, K.; Lubar, A.; Okitsu, S.L.; Flannery, E.L.; Kato, N.; et al. High-throughput assay and discovery of small molecules that interrupt malaria transmission. Cell Host Microbe 2016, 19, 114–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alessandro, S.; Camarda, G.; Corbett, Y.; Siciliano, G.; Parapini, S.; Cevenini, L.; Michelini, E.; Roda, A.; Leroy, D.; Taramelli, D.; et al. A chemical susceptibility profile of the Plasmodium falciparum transmission stages by complementary cell-based gametocyte assays. J. Antimicrob. Chemother. 2016, 71, 1148–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchholz, K.; Burke, T.A.; Williamson, K.C.; Wiegand, R.C.; Wirth, D.F.; Marti, M. A high-throughput screen targeting malaria transmission stages opens new avenues for drug development. J. Infect. Dis. 2011, 203, 1445–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucantoni, L.; Avery, V. Whole-cell in vitro screening for gametocytocidal compounds. Future Med. Chem. 2012, 4, 2337–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguel-Blanco, C.; Lelièvre, J.; Delves, M.J.; Bardera, A.I.; Presa, J.L.; López-Barragán, M.J.; Ruecker, A.; Marques, S.; Sinden, R.E.; Herreros, E. Imaging-based high-throughput screening assay to identify new molecules with transmission-blocking potential against Plasmodium falciparum female gamete formation. Antimicrob. Agents Chemother 2015, 59, 3298–3305. [Google Scholar] [CrossRef] [Green Version]
- Sanders, N.G.; Sullivan, D.J.; Mlambo, G.; Dimopoulos, G.; Tripathi, A.K. Gametocytocidal screen identifies novel chemical classes with Plasmodium falciparum transmission blocking activity. PLoS ONE 2014, 9, e105817. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Huang, X.; Li, H.; Tawa, G.; Fisher, E.; Tanaka, T.Q.; Shinn, P.; Huang, W.; Williamson, K.C.; Zheng, W. Novel lead structures with both Plasmodium falciparum gametocytocidal and asexual blood stage activity identified from high throughput compound screening. Malar. J. 2017, 16, 147. [Google Scholar] [CrossRef]
- Da Cruz, F.P.; Martin, C.; Buchholz, K.; Lafuente-Monasterio, M.J.; Rodrigues, T.; Sönnichsen, B.; Moreira, R.; Gamo, F.J.; Marti, M.; Mota, M.M.; et al. Drug screen targeted at Plasmodium liver stages identifies a potent multistage antimalarial drug. J. Infect. Dis. 2012, 205, 1278–1286. [Google Scholar] [CrossRef]
- Kåhrström, C.T. Techniques & applications: A new tool for liver-stage malaria. Nat. Rev. Microbiol. 2013, 11, 596. [Google Scholar]
- Roth, A.; Maher, S.P.; Conway, A.J.; Ubalee, R.; Chaumeau, V.; Andolina, C.; Kaba, S.A.; Vantaux, A.; Bakowski, M.A.; Thomson-Luque, R.; et al. A comprehensive model for assessment of liver stage therapies targeting Plasmodium vivax and Plasmodium falciparum. Nat. Commun. 2018, 9, 1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campo, B.; Vandal, O.; Wesche, D.L.; Burrows, J.N. Killing the hypnozoite—Drug discovery approaches to prevent relapse in Plasmodium vivax. Pathog. Glob. Health 2015, 109, 107–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gego, A.; Silvie, O.; Franetich, J.F.; Farhati, K.; Hannoun, L.; Luty, A.J.; Sauerwein, R.W.; Boucheix, C.; Rubinstein, E.; Mazier, D. New approach for high-throughput screening of drug activity on Plasmodium liver stages. Antimicrob. Agents Chemother. 2006, 50, 1586–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- March, S.; Ng, S.; Velmurugan, S.; Galstian, A.; Shan, J.; Logan, D.J.; Carpenter, A.E.; Thomas, D.; Sim, B.K.; Mota, M.M.; et al. A microscale human liver platform that supports the hepatic stages of Plasmodium falciparum and vivax. Cell Host Microbe 2013, 14, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, R.; Velmurugan, S.; Chakiath, C.; Andrews Donkor, L.; Milhous, W.; Barnwell, J.W.; Collins, W.E.; Hoffman, S.L. Establishment of an in vitro assay for assessing the effects of drugs on the liver stages of Plasmodium vivax malaria. PLoS ONE 2010, 5, e14275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonova-Koch, Y.; Meister, S.; Abraham, M.; Luth, M.R.; Ottilie, S.; Lukens, A.K.; Sakata-Kato, T.; Vanaerschot, M.; Owen, E.; Jado, J.C.; et al. Open-source discovery of chemical leads for next-generation chemoprotective antimalarials. Science 2018, 362, eaat9446. [Google Scholar] [CrossRef] [Green Version]
- Lage, O.M.; Ramos, M.C.; Calisto, R.; Almeida, E.; Vasconcelos, V.; Vicente, F. Current screening methodologies in drug discovery for selected human diseases. Mar. Drugs 2018, 16, 279. [Google Scholar] [CrossRef] [Green Version]
- Torres, F.A.E.; Passalacqua, T.G.; Velásquez, A.M.A.; de Souza, R.A.; Colepicolo, P.; Graminha, M.A.S. New drugs with antiprotozoal activity from marine algae: A review. Rev. Bras. Farmacol. 2014, 24, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Tchokouaha Yamthe, L.R.; Appiah-Opong, R.; Tsouh Fokou, P.V.; Tsabang, N.; Fekam Boyom, F.; Nyarko, A.K.; Wilson, M.D. Marine algae as source of novel antileishmanial drugs: A review. Mar. Drugs 2017, 15, 323. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, A.O.; Britta, E.A.; Bianco, E.M.; Ueda-Nakamura, T.; Filho, B.P.; Pereira, R.C.; Nakamura, C.V. 4-Acetoxydolastane diterpene from the Brazilian brown alga Canistrocarpus cervicornis as antileishmanial agent. Mar. Drugs 2011, 9, 2369–2383. [Google Scholar] [CrossRef] [Green Version]
- Soares, D.C.; Calegari-Silva, T.C.; Lopes, U.G.; Teixeira, V.L.; de Palmer Paixão, I.C.; Cirne-Santos, C.; Bou-Habib, D.C.; Saraiva, E.M. Dolabelladienetriol, a compound from Dictyota pfaffii algae, inhibits the infection by Leishmania amazonensis. PLoS Negl. Trop. Dis. 2012, 6, e1787. [Google Scholar] [CrossRef] [PubMed]
- Chiboub, O.; Sifaoui, I.; Lorenzo-Morales, J.; Abderrabba, M.; Mejri, M.; Fernández, J.J.; Piñero, J.E.; Díaz-Marrero, A.R. Spiralyde A, an antikinetoplastid dolabellane from the brown alga Dictyota spiralis. Mar. Drugs 2019, 17, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallé, J.B.; Attioua, B.; Kaiser, M.; Rusig, A.M.; Lobstein, A.; Vonthron-Sénécheau, C. Eleganolone, a diterpene from the French marine alga Bifurcaria bifurcata inhibits growth of the human pathogens Trypanosoma brucei and Plasmodium falciparum. Mar. Drugs 2013, 11, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Bruno de Sousa, C.; Gangadhar, K.N.; Morais, T.R.; Conserva, G.A.; Vizetto-Duarte, C.; Pereira, H.; Laurenti, M.D.; Campino, L.; Levy, D.; Uemi, M.; et al. Antileishmanial activity of meroditerpenoids from the macroalgae Cystoseira baccata. Exp. Parasitol. 2017, 174, 1–9. [Google Scholar] [CrossRef]
- Soares, D.C.; Szlachta, M.M.; Teixeira, V.L.; Soares, A.R.; Saraiva, E.M. The brown alga Stypopodium zonale (Dictyotaceae): A potential source of anti-leishmania drugs. Mar. Drugs 2016, 14, 163. [Google Scholar] [CrossRef] [PubMed]
- Becerra, M.; Boutefnouchet, S.; Córdoba, O.; Pinto Vitorino, G.; Brehu, L.; Lamour, I.; Laimay, F.; Efstathiou, A.; Smirlis, D.; Michel, S.; et al. Antileishmanial activity of fucosterol recovered from Lessonia vadosa Searles (Lessoniaceae) by SFE, PSE and CPC. Phytochem. Lett. 2015, 11, 418–423. [Google Scholar] [CrossRef]
- Díaz-Marrero, A.R.; López-Arencibia, A.; Bethencout-Estrella, C.J.; Cen-Pacheco, F.; Sifaoui, I.; Hernández Creus, A.; Duque-Ramírez, M.C.; Souto, M.L.; Hernández Daranas, A.; Lorenzo-Morales, J.; et al. Antiprotozoal activities of marine polyether triterpenoids. Bioorg. Chem. 2019, 92, 103276. [Google Scholar] [CrossRef]
- Dos Santos, A.O.; Veiga-Santos, P.; Ueda-Nakamura, T.; Filho, B.P.; Sudatti, D.B.; Bianco, E.M.; Pereira, R.C.; Nakamura, C.V. Effect of elatol, isolated from red seaweed Laurencia dendroidea, on Leishmania amazonensis. Mar. Drugs. 2010, 8, 2733–2743. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Machado, F.L.; Pacienza-Lima, W.; Rossi-Bergmann, B.; de Souza Gestinari, L.M.; Fujii, M.T.; Campos de Paula, J.; Costa, S.S.; Lopes, N.P.; Kaiser, C.R.; Soares, A.R. Antileishmanial sesquiterpenes from the Brazilian red alga Laurencia dendroidea. Planta Med. 2011, 77, 733–735. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Sharma, G.; Das, P.K. Fucoidan cures infection with both antimony-susceptible and -resistant strains of Leishmania donovani through Th1 response and macrophage-derived oxidants. J. Antimicrob. Chemother. 2011, 66, 618–625. [Google Scholar] [CrossRef]
- Anjum, K.; Abbas, S.Q.; Shah, S.A.; Akhter, N.; Batool, S.; Hassan, S.S. Marine sponges as a drug treasure. Biomol. Ther. 2016, 24, 347–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; MacIntyre, L.; Abdelmohsen, U.R.; Horn, H.; Polymenakou, P.N.; Edrada-Ebel, R.; Hentschel, U. Biodiversity, anti-trypanosomal activity screening, and metabolomic profiling of actinomycetes isolated from Mediterranean sponges. PLoS ONE 2015, 10, e0138528. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Tatsuo, H.; Jefford, C.W.; Bernardinelli, G. Manzamine A, a novel antitumor alkaloid from a sponge. J. Am. Chem. Soc. 1986, 108, 6404–6405. [Google Scholar] [CrossRef]
- Ang, K.K.; Holmes, M.J.; Higa, T.; Hamann, M.T.; Kara, U.A. In vivo antimalarial activity of the beta-carboline alkaloid manzamine A. Antimicrob. Agents Chemother. 2000, 44, 1645–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousaf, M.; Hammond, N.L.; Peng, J.; Wahyuono, S.; McIntosh, K.A.; Charman, W.N.; Mayer, A.M.; Hamann, M.T. New manzamine alkaloids from an Indo-Pacific sponge. Pharmacokinetics, oral availability, and the significant activity of several manzamines against HIV-I, AIDS opportunistic infections, and inflammatory diseases. J. Med. Chem. 2004, 47, 3512–3517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilabin, A.G.; Kasanah, N.; Tekwani, B.L.; Hamann, M.T. Kinetic studies and bioactivity of potential manzamine prodrugs. J. Nat. Prod. 2008, 71, 1218–1221. [Google Scholar] [CrossRef] [Green Version]
- Ashok, P.; Ganguly, S.; Murugesan, S. Manzamine alkaloids: Isolation, cytotoxicity, antimalarial activity and SAR studies. Drug Discov. Today 2014, 19, 1781–1791. [Google Scholar] [CrossRef]
- Ashok, P.; Lathiya, H.; Murugesan, S. Manzamine alkaloids as antileishmanial agents: A review. Eur. J. Med. Chem. 2015, 97, 928–936. [Google Scholar] [CrossRef]
- Shady, N.H.; Fouad, M.A.; Kamel, M.S.; Schirmeister, T.; Abdelmohsen, U.R. Natural product repertoire of the Genus Amphimedon. Mar. Drugs 2019, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Skropeta, D.; Pastro, N.; Zivanovic, A. Kinase Inhibitors from marine sponges. Mar. Drugs 2011, 9, 2131–2154. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Peng, W.; Chen, H.; Ma, Y.; Liu, X.; Hou, X.; Guan, H.; Xu, A. DNA binding properties of 9-substituted harmine derivatives. Biochem. Biophys. Res. Commun. 2005, 338, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Fattorusso, E.; Taglialatela-Scafati, O. Marine antimalarials. Mar. Drugs 2009, 7, 130–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.D.; McCluskey, A.; Robertson, M.J.; MacGregor, K.A.; Gordon, C.P.; Guenther, J. Anti-malarial, anti-algal, anti-tubercular, anti-bacterial, anti-photosynthetic, and anti-fouling activity of diterpene and diterpene isonitriles from the tropical marine sponge Cymbastela hooperi. Org. Biomol. Chem. 2011, 9, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.D.; Wang, H.; Gurrath, M.; König, G.M.; Kocak, G.; Neumann, G.; Loria, P.; Foley, M.; Tilley, L. Inhibition of heme detoxification processes underlies the antimalarial activity of terpene isonitrile compounds from marine sponges. J. Med. Chem. 2001, 44, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Witowski, C.G.; Maschek, A.; Vesely, B.; Kyle, D.E.; McClintock, J.; Amsler, C.D.; Baker, B.J. Characterization of membranolides B-H from Dendrilla membranosa and their activity against leishmaniasis. Planta Med. 2014, 80, PB8. [Google Scholar] [CrossRef]
- Orhan, I.; Sener, B.; Kaiser, M.; Brun, R.; Tasdemir, D. Inhibitory activity of marine sponge-derived natural products against parasitic protozoa. Mar. Drugs 2010, 8, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Regalado, E.L.; Tasdemir, D.; Kaiser, M.; Cachet, N.; Amade, P.; Thomas, O.P. Antiprotozoal steroidal saponins from the marine sponge Pandaros acanthifolium. J. Nat. Prod. 2010, 73, 1404–1410. [Google Scholar] [CrossRef]
- Cunningham, L.V.; Kazan, B.H.; Kuwahara, S.S. Effect of long-chain fatty acids on some trypanosomatid flagellates. J. Gen. Microbiol. 1972, 70, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Kumaratilake, L.M.; Robinson, B.S.; Ferrante, A.; Poulos, A. Antimalarial properties of n-3 and n-6 polyunsaturated fatty acids: In vitro effects on Plasmodium falciparum and in vivo effects on P. berghei. J. Clin. Invest. 1992, 89, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Carballeira, N.M.; Cartagena, M.M.; Prada, C.F.; Rubio, C.F.; Balaña-Fouce, R. Total synthesis and antileishmanial activity of the natural occurring acetylenic fatty acids 6-heptadecynoic acid and 6-icosynoic acid. Lipids 2009, 44, 953–961. [Google Scholar] [CrossRef] [Green Version]
- Balaña-Fouce, R.; Alvarez-Velilla, R.; Fernández-Prada, C.; García-Estrada, C.; Reguera, R.M. Trypanosomatids topoisomerase re-visited. New structural findings and role in drug discovery. Int. J. Parasitol. Drugs Drug. Resist. 2014, 4, 326–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carballeira, N.M.; Montano, N.; Cintrón, G.A.; Márquez, C.; Rubio, C.F.; Prada, C.F.; Balaña-Fouce, R.R. First total synthesis and antileishmanial activity of (Z)-16-methyl-11-heptadecenoic acid, a new marine fatty acid from the sponge Dragmaxia undata. Chem. Phys. Lipids 2011, 164, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carballeira, N.M.; Montano, N.; Reguera, R.M.; Balaña-Fouce, R. The first total synthesis of the (±)-17-methyl-trans-4,5-methyleneoctadecanoic acid and related analogs with antileishmanial activity. Tetrahedron Lett. 2010, 51, 6153–6155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carballeira, N.M.; Cartagena, M.; Sanabria, D.; Tasdemir, D.; Prada, C.F.; Reguera, R.M.; Balaña-Fouce, R. 2-Alkynoic fatty acids inhibit topoisomerase IB from Leishmania donovani. Bioorg. Med. Chem. Lett. 2012, 22, 6185–6189. [Google Scholar] [CrossRef] [Green Version]
- Carballeira, N.M. Recent developments in the antiprotozoal and anticancer activities of the 2-alkynoic fatty acids. Chem. Phys. Lipids 2013, 172, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Carballeira, N.M.; Montano, N.; Amador, L.A.; Rodríguez, A.D.; Golovko, M.Y.; Golovko, S.A.; Reguera, R.M.; Álvarez-Velilla, R.; Balaña-Fouce, R. Novel very long-chain α-methoxylated α5,9 fatty acids from the sponge Asteropus niger are effective inhibitors of topoisomerases IB. Lipids 2016, 51, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Carballeira, N.M.; Montano, N.; Alvarez-Velilla, R.; Prada, C.F.; Reguera, R.M.; Balaña-Fouce, R. Synthesis of marine α-methoxylated fatty acid analogs that effectively inhibit the topoisomerase IB from Leishmania donovani with a mechanism different from that of camptothecin. Mar. Drugs 2013, 11, 3661–3675. [Google Scholar] [CrossRef]
- Tasdemir, D.; Sanabria, D.; Lauinger, I.L.; Tarun, A.; Herman, R.; Perozzo, R.; Zloh, M.; Kappe, S.H.; Brun, R.; Carballeira, N.M. 2-Hexadecynoic acid inhibits plasmodial FAS-II enzymes and arrests erythrocytic and liver stage Plasmodium infections. Bioorg. Med. Chem. 2010, 18, 7475–7485. [Google Scholar] [CrossRef] [Green Version]
- Carballeira, N.M.; Bwalya, A.G.; Itoe, M.A.; Andricopulo, A.D.; Cordero-Maldonado, M.L.; Kaiser, M.; Mota, M.M.; Crawford, A.D.; Guido, R.V.; Tasdemir, D. 2-Octadecynoic acid as a dual life stage inhibitor of Plasmodium infections and plasmodial FAS-II enzymes. Bioorg. Med. Chem. Lett. 2014, 24, 4151–4157. [Google Scholar] [CrossRef]
- Mayer, A.M.S.; Guerrero, A.J.; Rodríguez, A.D.; Taglialatela-Scafati, O.; Nakamura, F.; Fusetani, N. Marine pharmacology in 2014–2015: Marine compounds with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis, antiviral, and anthelmintic activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Mar. Drugs 2019, 18, 273. [Google Scholar]
- Thao, N.P.; No, J.H.; Luyen, B.T.; Yang, G.; Byun, S.Y.; Goo, J.; Kim, K.T.; Cuong, N.X.; Nam, N.H.; Van Minh, C.; et al. Secondary metabolites from Vietnamese marine invertebrates with activity against Trypanosoma brucei and T. cruzi. Molecules 2014, 19, 7869–7880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thao, N.P.; Luyen, B.T.; Brun, R.; Kaiser, M.; Van Kiem, P.; Van Minh, C.; Schmidt, T.J.; Kang, J.S.; Kim, Y.H. Anti-protozoal activities of cembrane-type diterpenes from Vietnamese soft corals. Molecules 2015, 20, 12459–12468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimão, J.Q.; Migotto, A.E.; Kossuga, M.H.; Berlinck, R.G.; Tempone, A.G. Antiprotozoan activity of Brazilian marine cnidarian extracts and of a modified steroid from the octocoral Carijoa riisei. Parasitol. Res. 2008, 103, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
















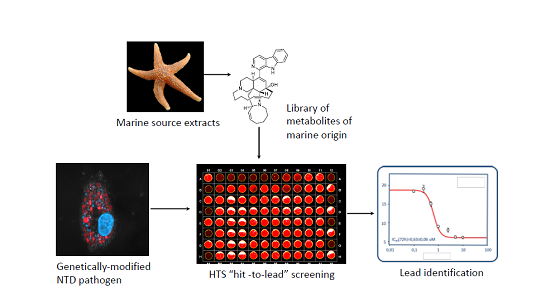{kind=link}
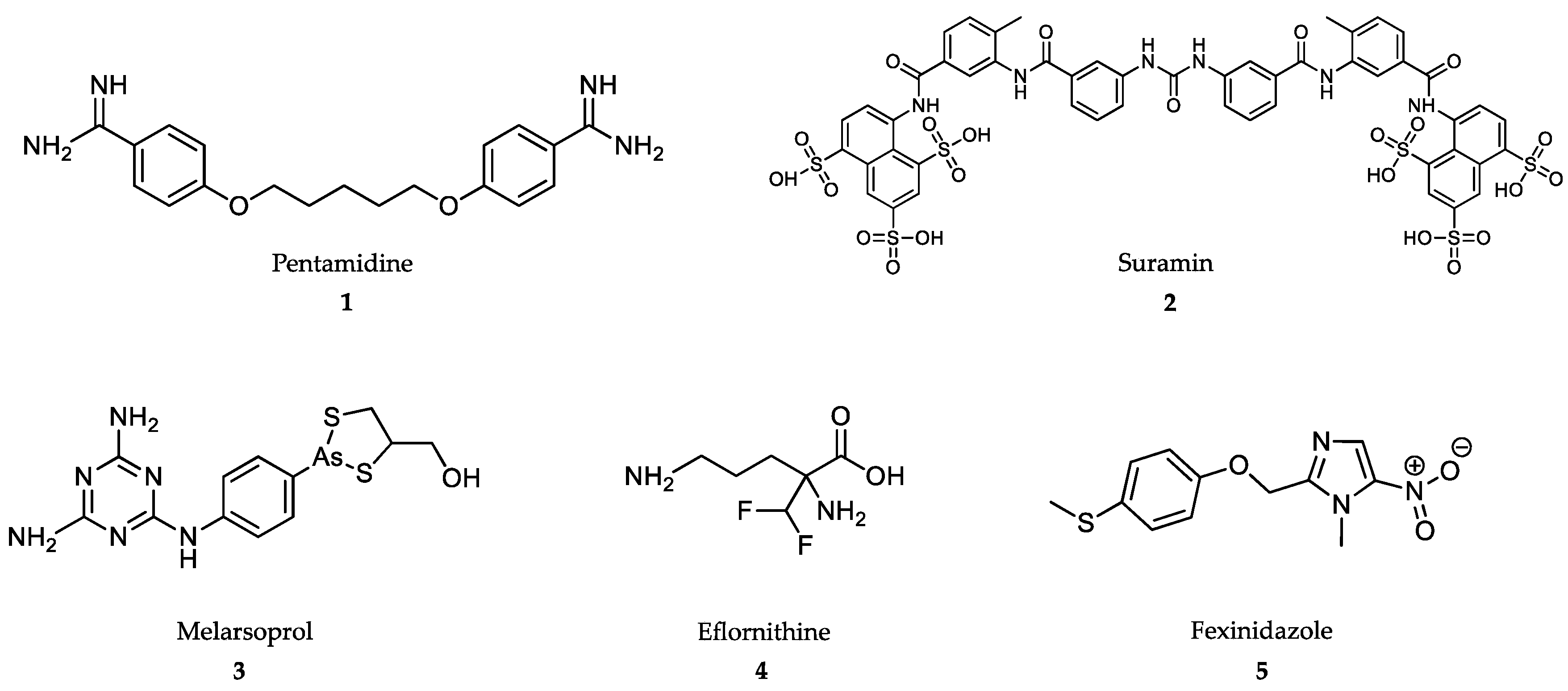{kind=link}
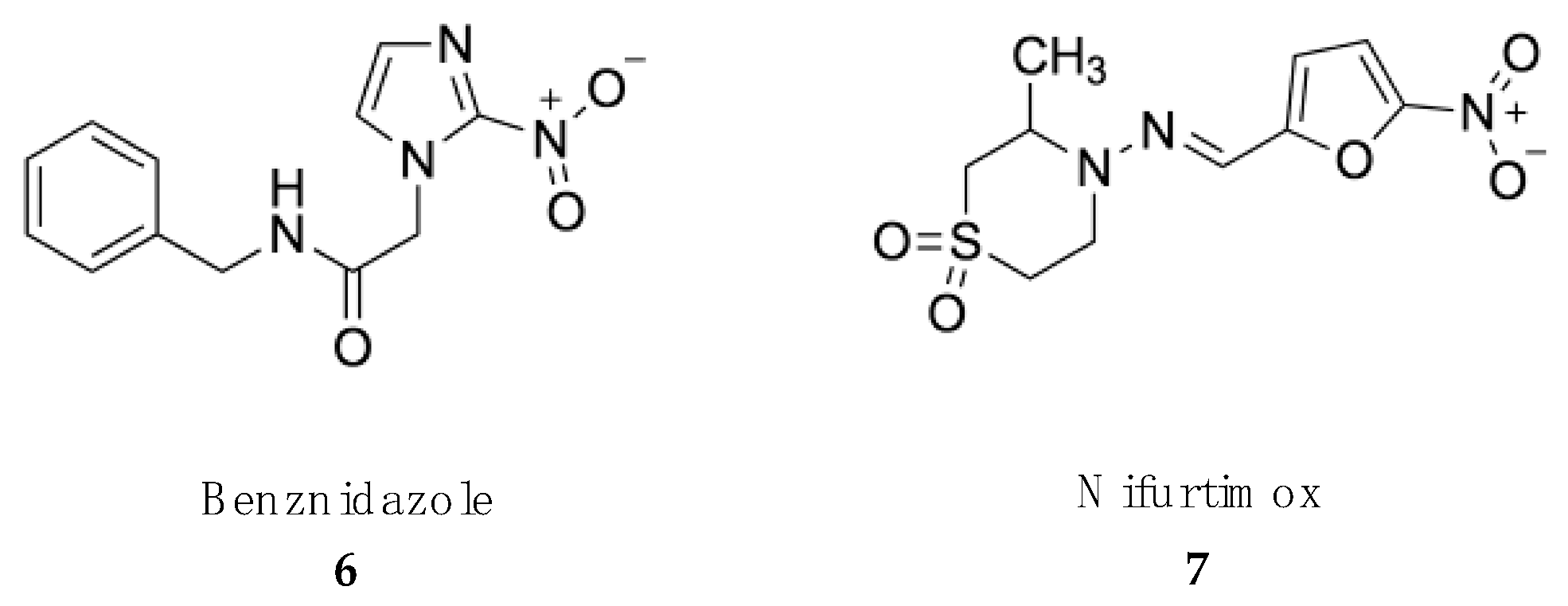{kind=link}
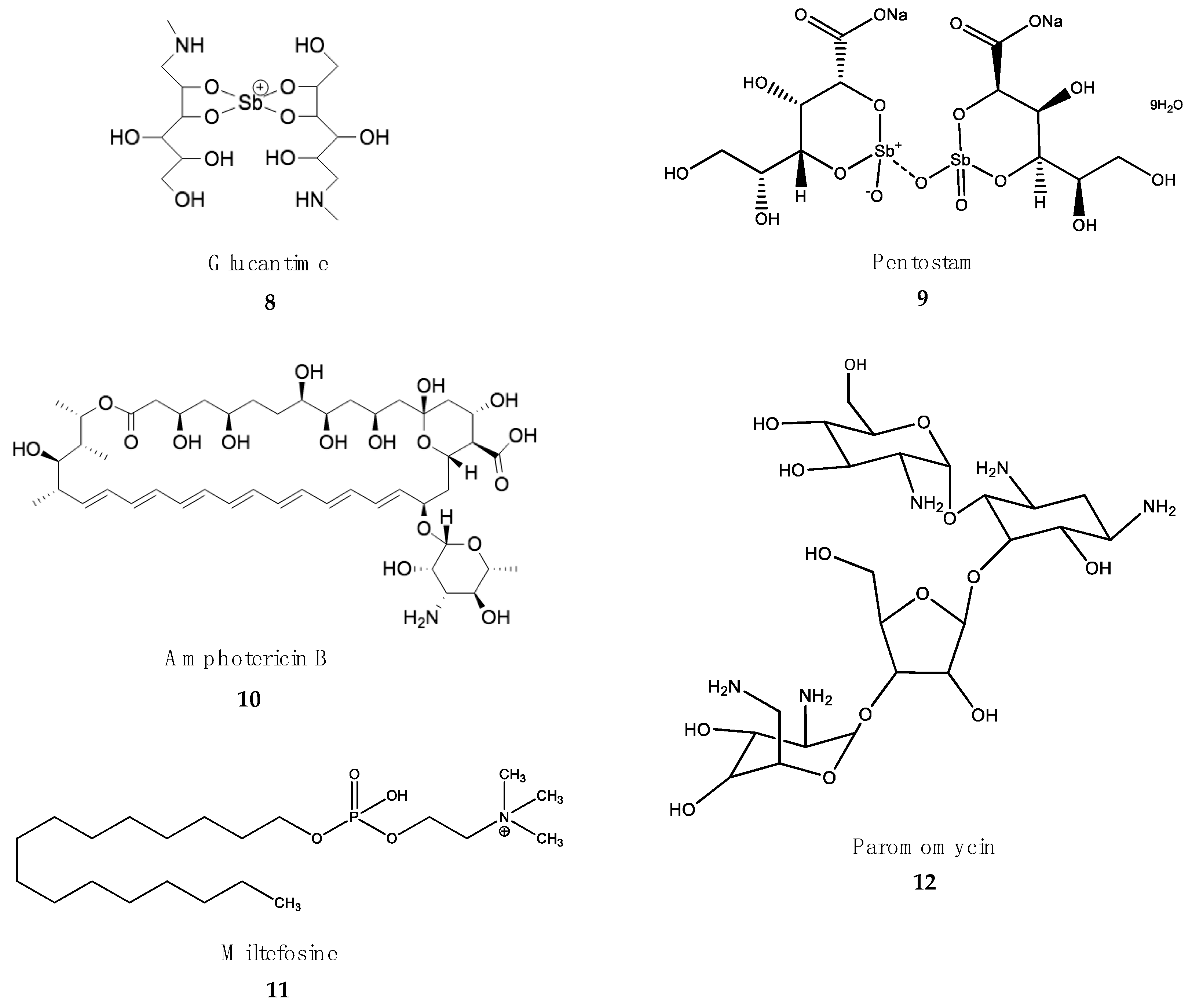{kind=link}
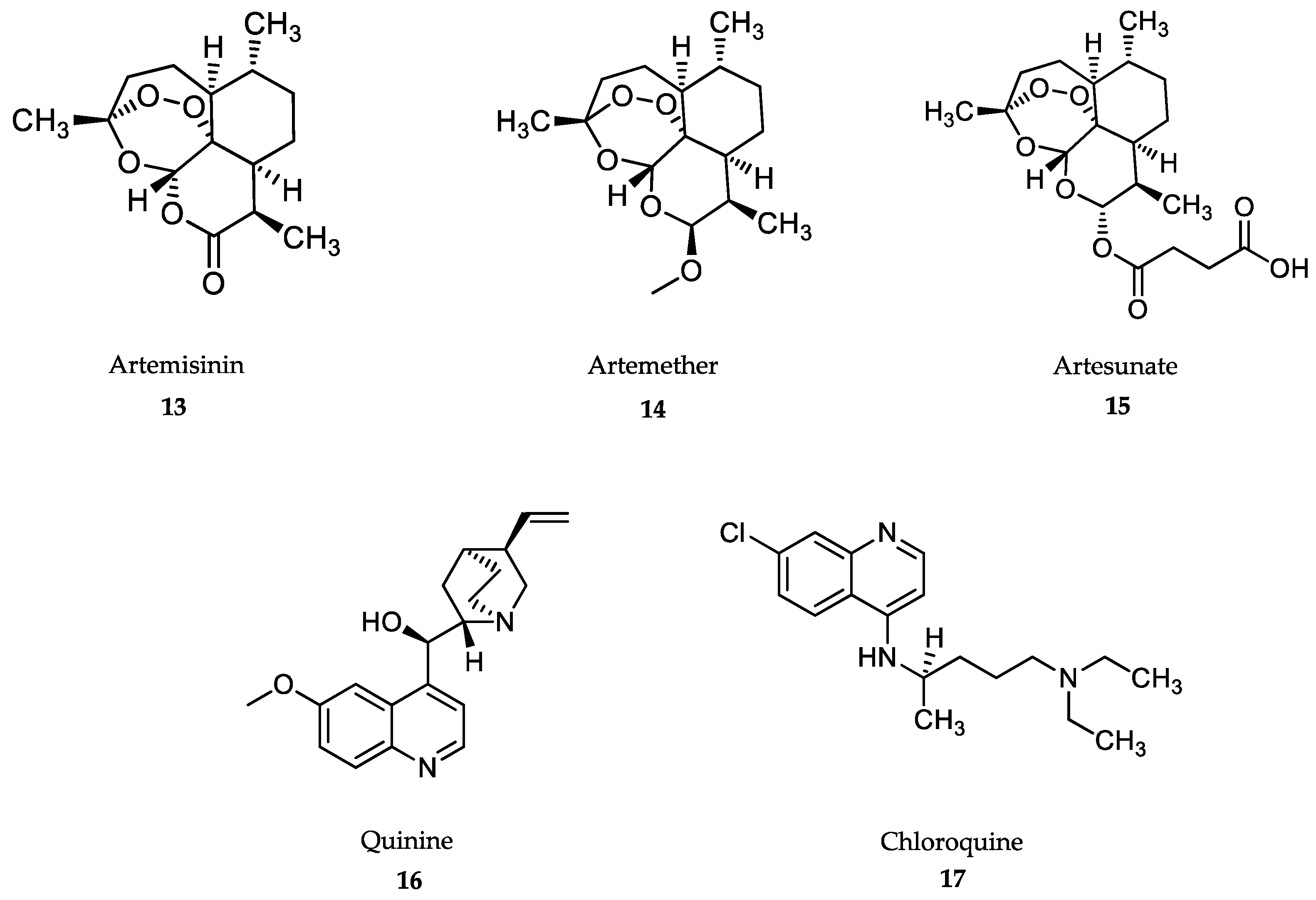{kind=link}
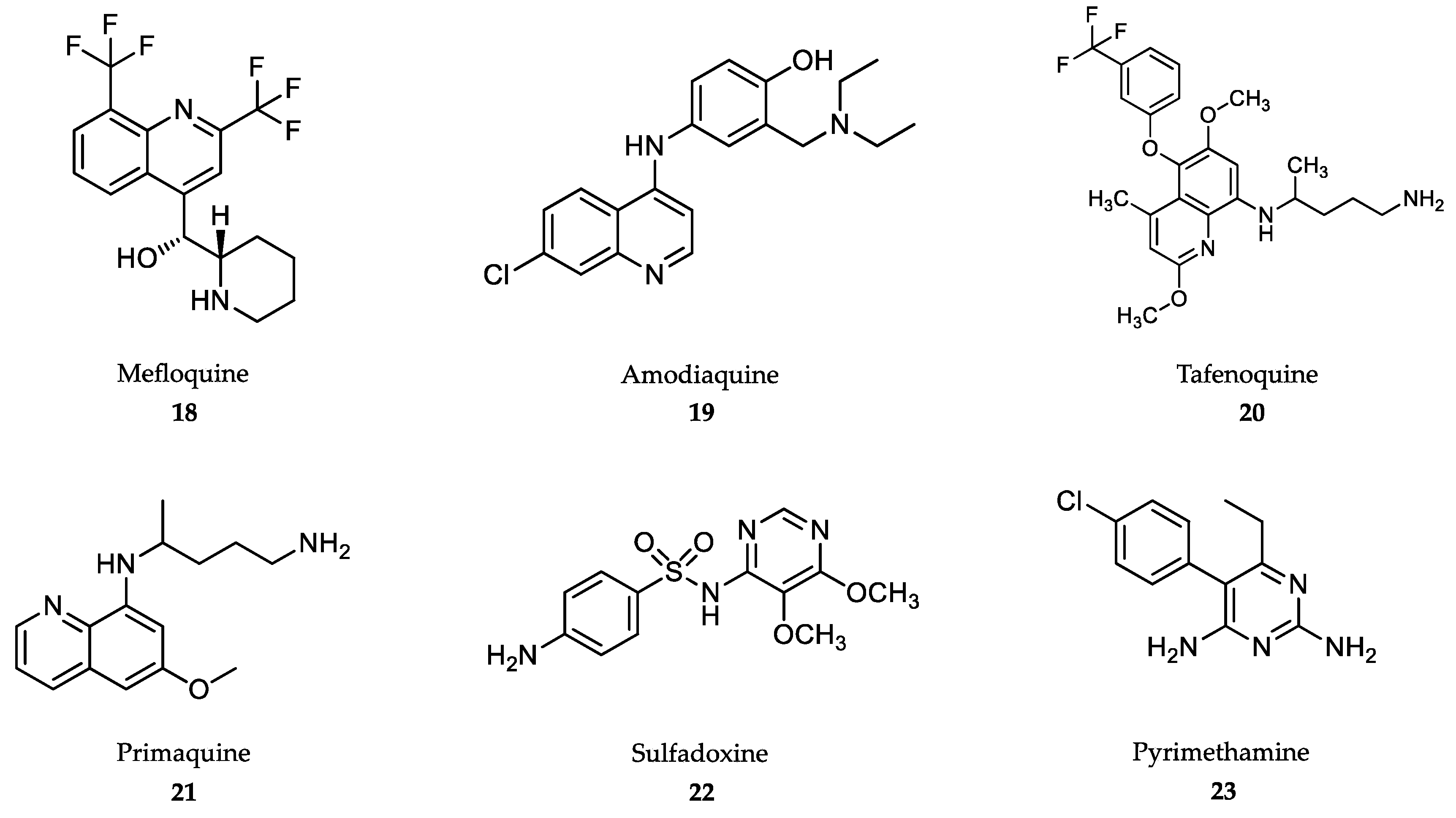{kind=link}
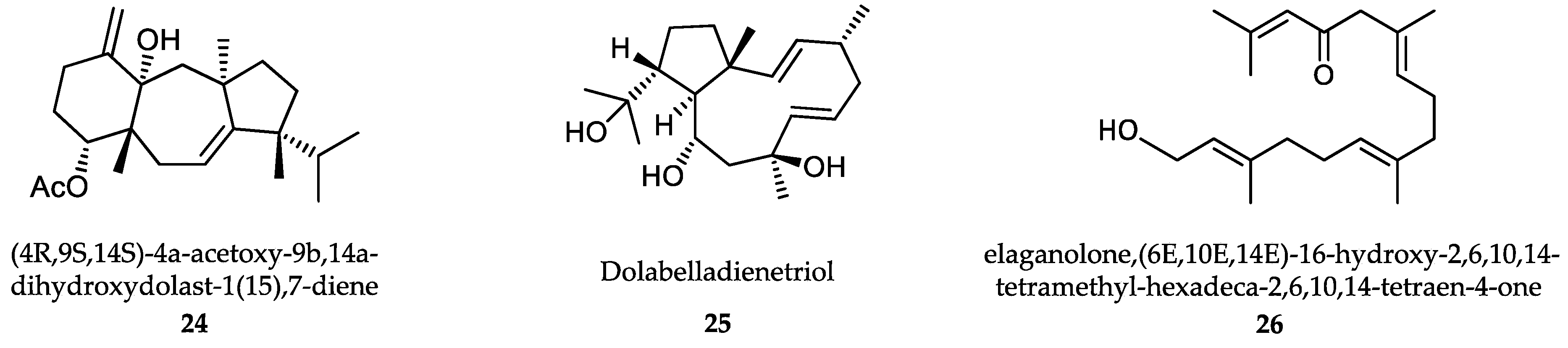{kind=link}
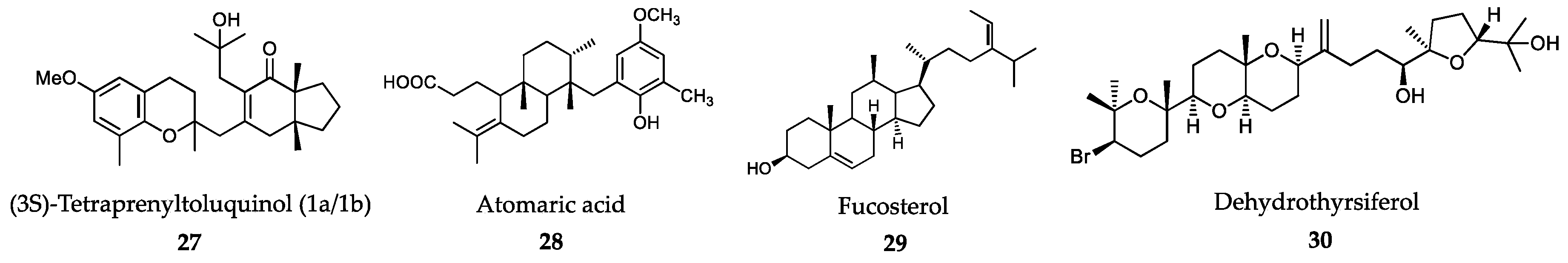{kind=link}
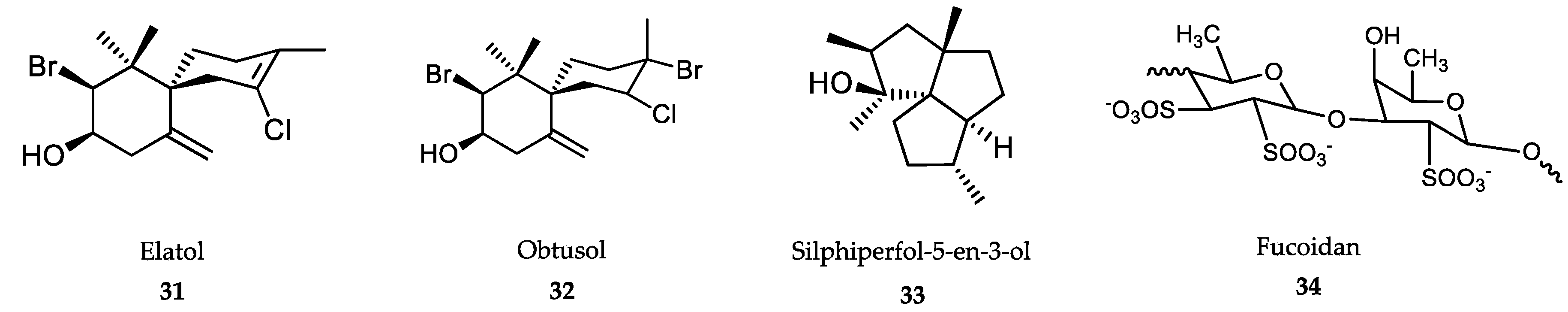{kind=link}
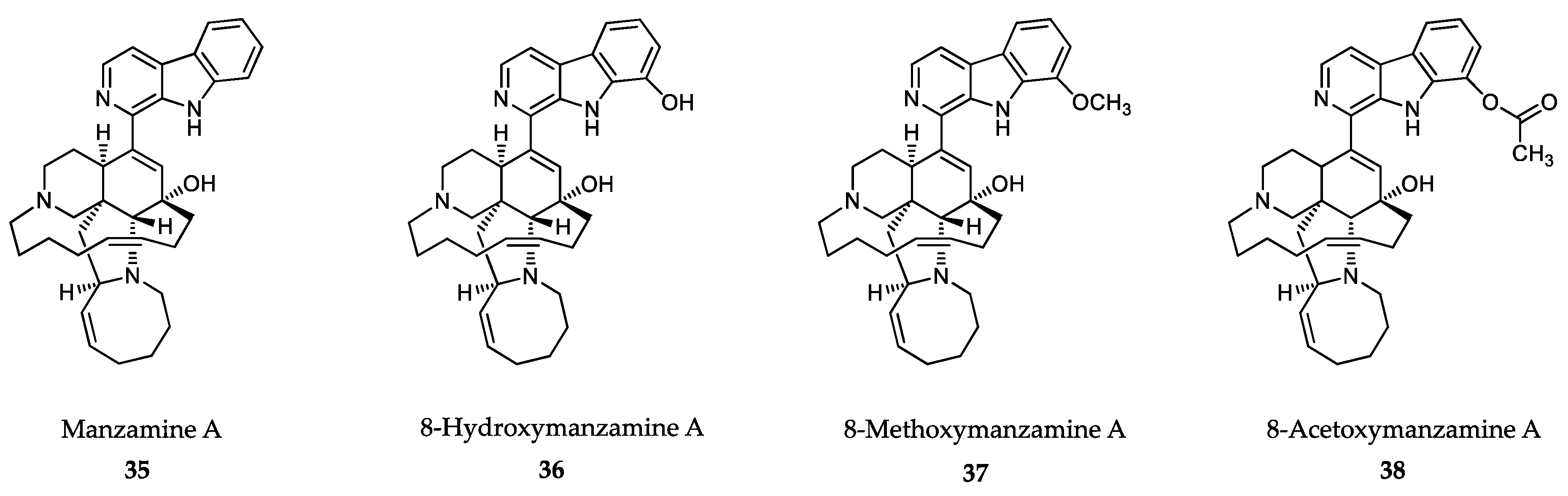{kind=link}
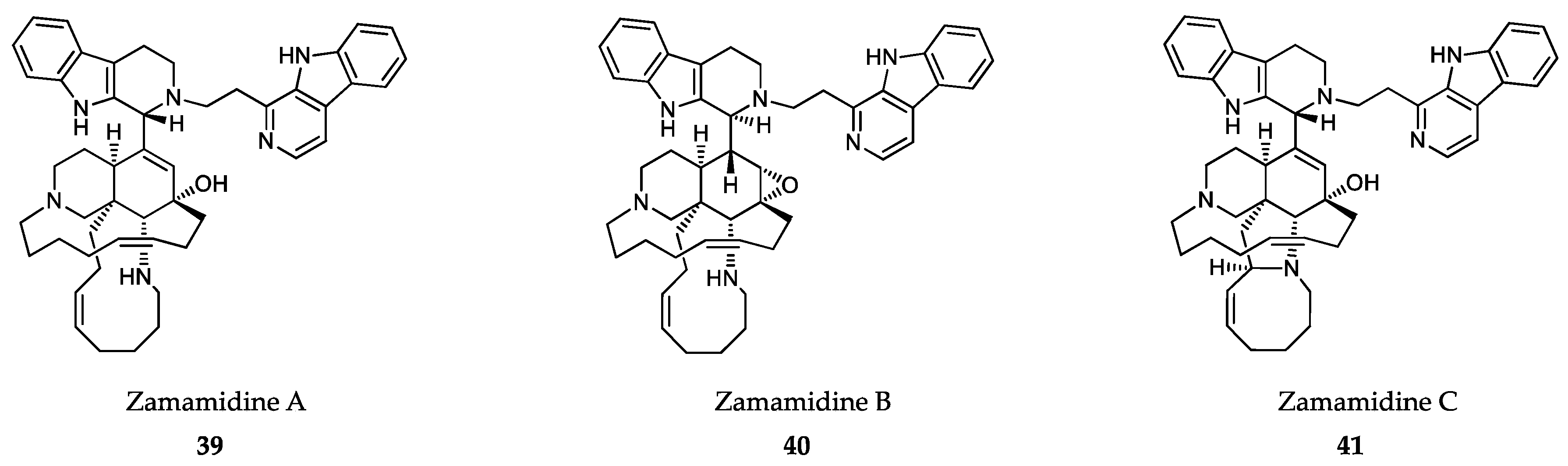{kind=link}
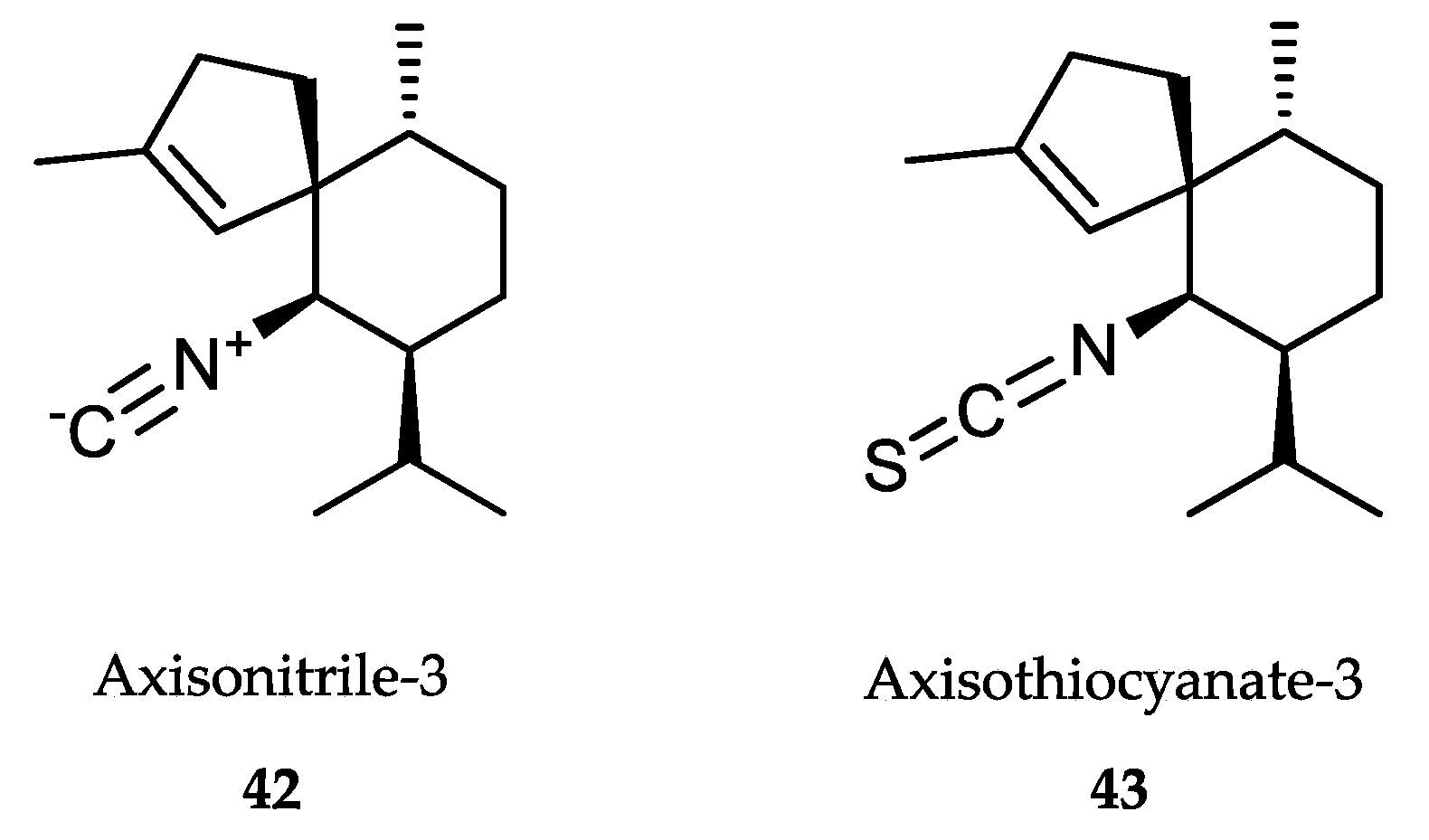{kind=link}
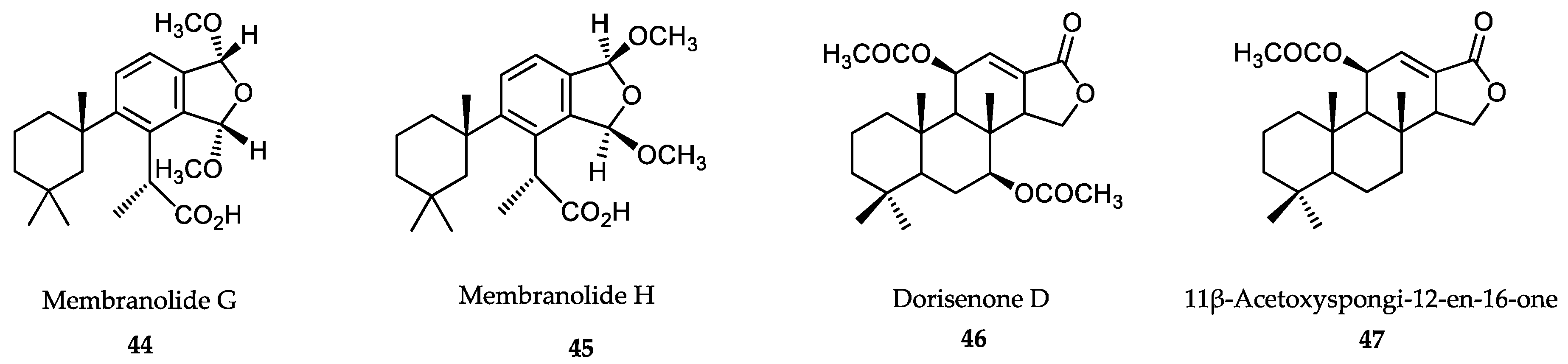{kind=link}
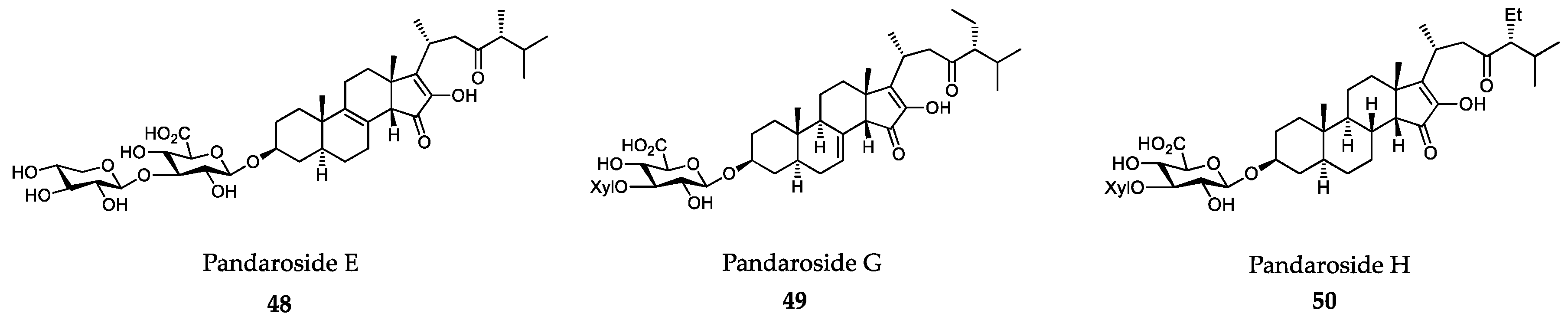{kind=link}
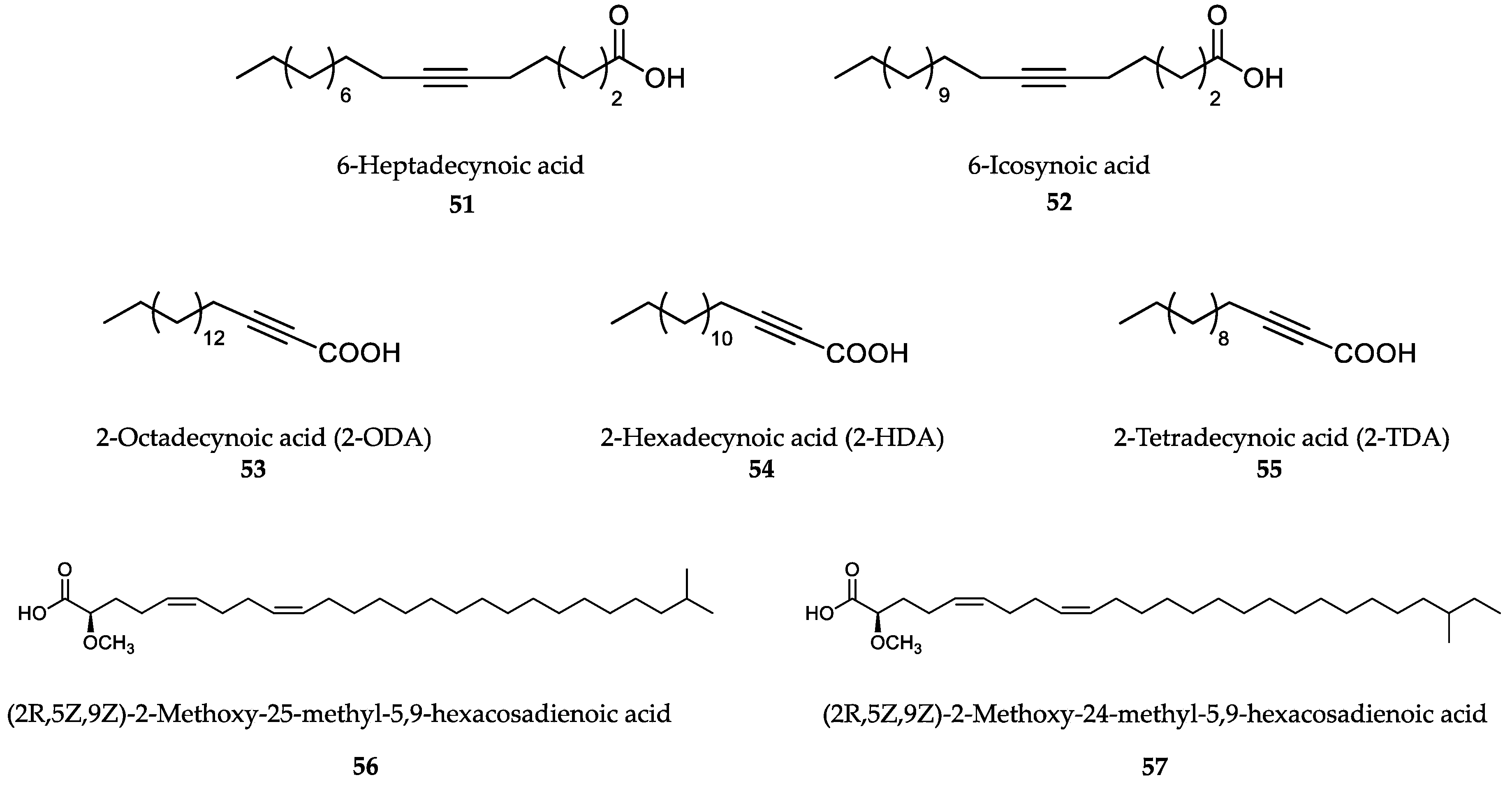{kind=link}
{kind=link}
| Disease/species | Drug | Chemical Class/Origin | Dose | Reference | |
|---|---|---|---|---|---|
| Human African Trypanosomiasis (HAT) | Pentamidine | Aromatic diamidine | Deep intramuscular injection 4 mg/kg body weight daily for 7 days | [57,58,63] | |
| Suramin | Polysulphonated naphthylurea | Slow intravenous infusion Day 1: 5 mg/kg body weight Day 3: 10 mg/kg body weight Days 5, 11, 17, 23, 30: 20 mg/kg body weight | [57,64,65] | ||
| Melarsoprol | Organic arsenical | Slow intravenous infusion 2.2 mg/kg daily for 10 days | [57,71] | ||
| Eflornithine | Amino acid analogue | Intravenous infusion 100 mg/kg body weight four times daily for 14 days | [57,79,80,81] | ||
| American trypanosomiasis | Benznidazole | Nitroheterocyclic compound | Oral 5-7 mg/kg body weight for 60 days | [95,96,97] | |
| Nifurtimox | Nitroheterocyclic compound | Oral 8–10 mg/kg body weight for 90 days | [103,104,105] | ||
| Leishmaniasis | Pentavalent antimony compounds | Antimony derivative | Intramuscular injection (Pentostam): 20 mg/kg body weight/day for 30 days | [113,114,116] | |
| Amphotericin B | Polyene antibiotic | 1 mg/kg body weight per day 15 - 20 intravenous infusion of deoxycholate amphotericin Intravenous infusions of liposomal amphotericin: single dose 10 mg/kg body weight | [116,120,123] | ||
| Miltefosine | Alkyl-phospholipid | Patients with 30-44 kg: one 50 mg capsule twice daily for 28 consecutive days Patients with ≥45 kg: one 50 mg capsule three times daily for 28 consecutive days | [126,129,130,131] | ||
| Paromomycin | Aminoglycoside antibiotic | Intramuscular injection 11 mg/kg body weight for 21 days | [133] | ||
| Malaria | Artemisinin | Artesunate | Sesquiterpene lactone derivative | 4 mg/kg body weight daily, with a daily dose range of 2-10 mg/kg body weight. | [15,139,143] |
| Artemether | Dihydroartemisinin | 3.2 mg/kg body weight by immediate intramuscular injection, followed by 1.6 mg/kg daily | [15,140,143] | ||
| Quinoline derivatives | Mefloquine | Quinoline | 25 mg/kg body weight/day for 3 days | [143,149] | |
| Amodiaquine | 4- aminoquinoline | 10 mg base body weight daily for 3 days | [143,154] | ||
| Tafenoquine | 8-aminoquinoline | Single oral dose of 300 mg | [15,143,156,158] | ||
| Primaquine | 8-aminoquinoline | 0.25 mg/kg body weight daily for 2 weeks | [15,143,156,159] | ||
| Sulfadoxine/pyrimethamine | Sulfadoxine: a synthetic analog of para-aminobenzoic acid (PABA) Pyrimethamine: a synthetic derivative of ethyl-pyrimidine | 25 mg/kg body weight sulfadoxine + 1.25 mg/kg body weight pyrimethamine | [15,19,143,161,162] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Bardón, M.; Pérez-Pertejo, Y.; Ordóñez, C.; Sepúlveda-Crespo, D.; Carballeira, N.M.; Tekwani, B.L.; Murugesan, S.; Martinez-Valladares, M.; García-Estrada, C.; Reguera, R.M.; et al. Screening Marine Natural Products for New Drug Leads against Trypanosomatids and Malaria. Mar. Drugs 2020, 18, 187. https://0-doi-org.brum.beds.ac.uk/10.3390/md18040187
Álvarez-Bardón M, Pérez-Pertejo Y, Ordóñez C, Sepúlveda-Crespo D, Carballeira NM, Tekwani BL, Murugesan S, Martinez-Valladares M, García-Estrada C, Reguera RM, et al. Screening Marine Natural Products for New Drug Leads against Trypanosomatids and Malaria. Marine Drugs. 2020; 18(4):187. https://0-doi-org.brum.beds.ac.uk/10.3390/md18040187
Chicago/Turabian StyleÁlvarez-Bardón, María, Yolanda Pérez-Pertejo, César Ordóñez, Daniel Sepúlveda-Crespo, Nestor M. Carballeira, Babu L. Tekwani, Sankaranarayanan Murugesan, Maria Martinez-Valladares, Carlos García-Estrada, Rosa M. Reguera, and et al. 2020. "Screening Marine Natural Products for New Drug Leads against Trypanosomatids and Malaria" Marine Drugs 18, no. 4: 187. https://0-doi-org.brum.beds.ac.uk/10.3390/md18040187