New Cytotoxic Cerebrosides from the Red Sea Cucumber Holothuria spinifera Supported by In-Silico Studies

 , , , , ,
, , , , ,
Abstract
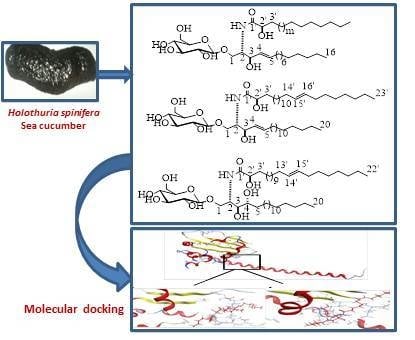
:
1. Introduction
2. Results and Discussion
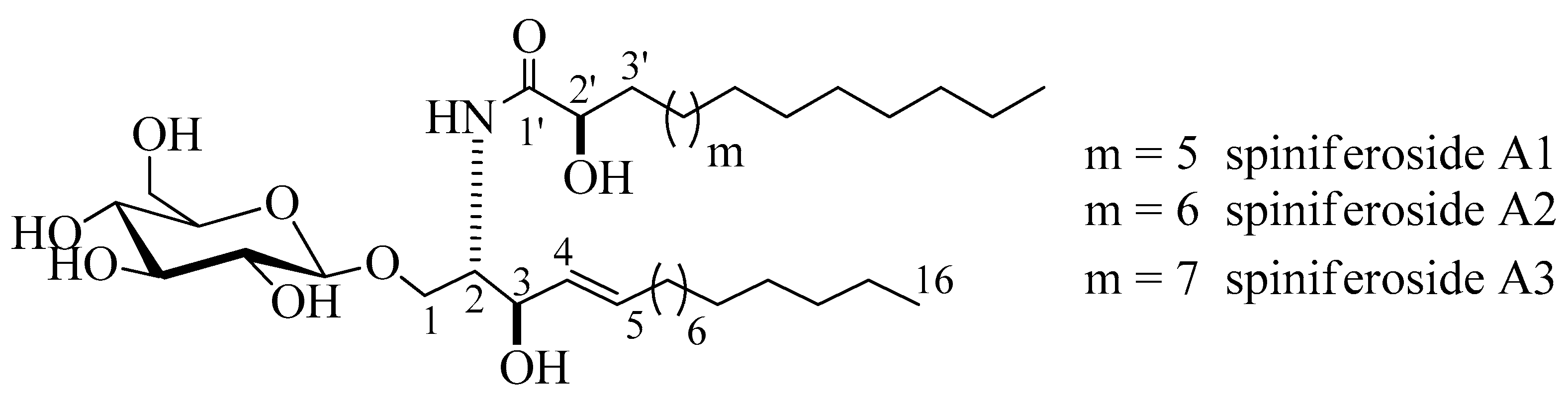
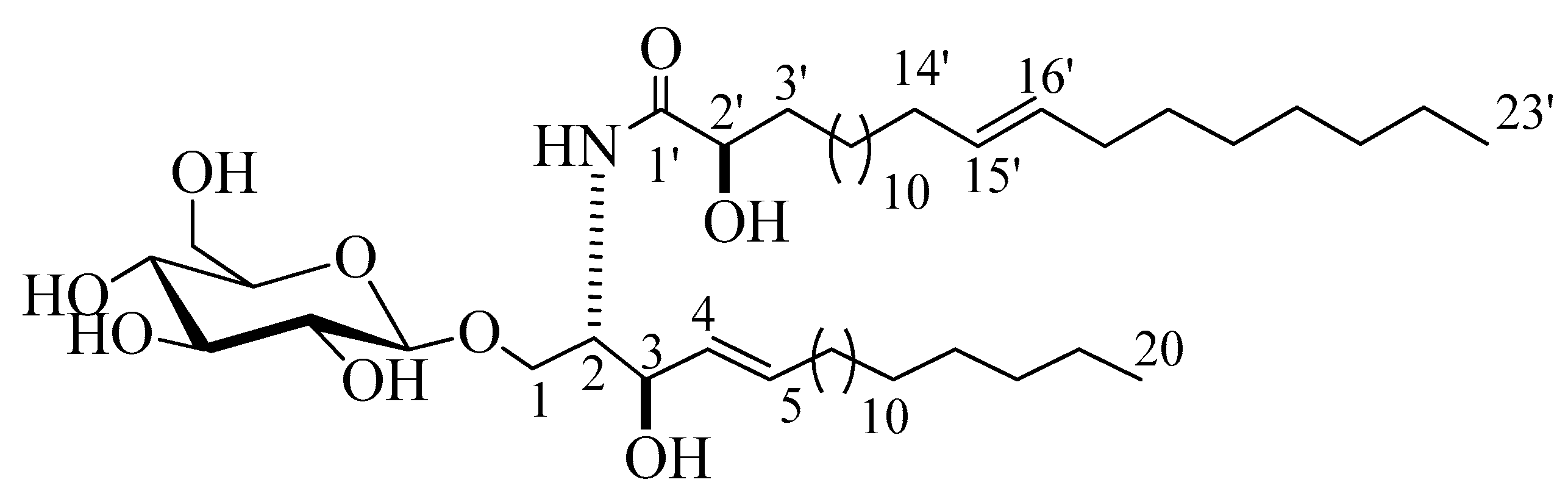
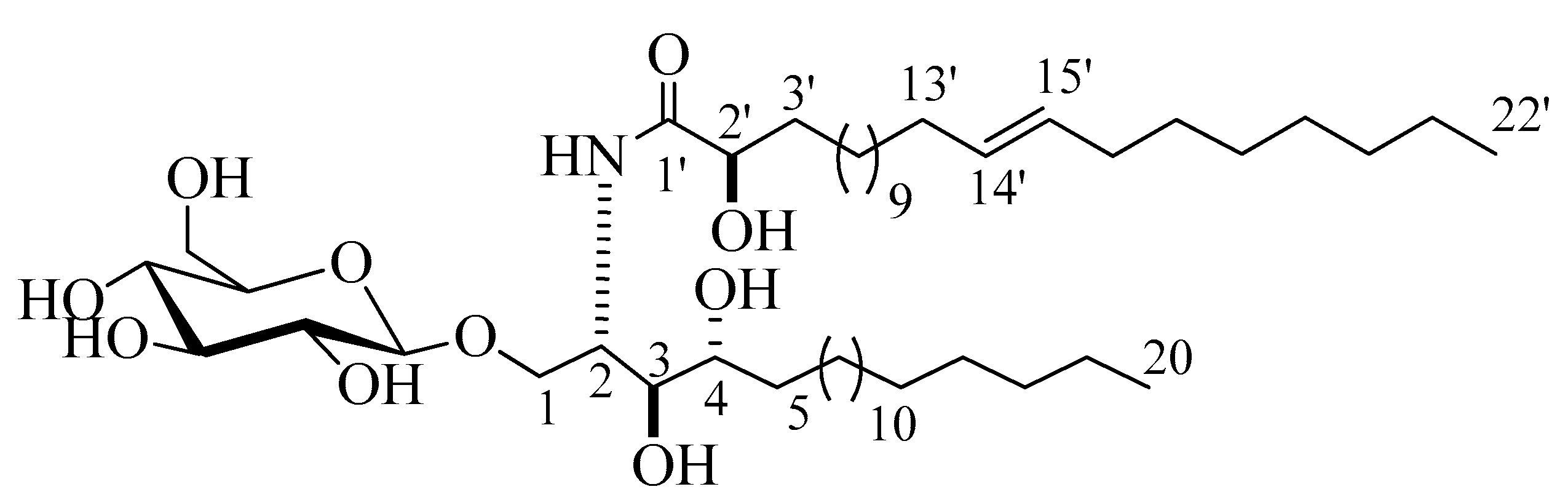
2.1. Structure Elucidation of the Isolated Compounds
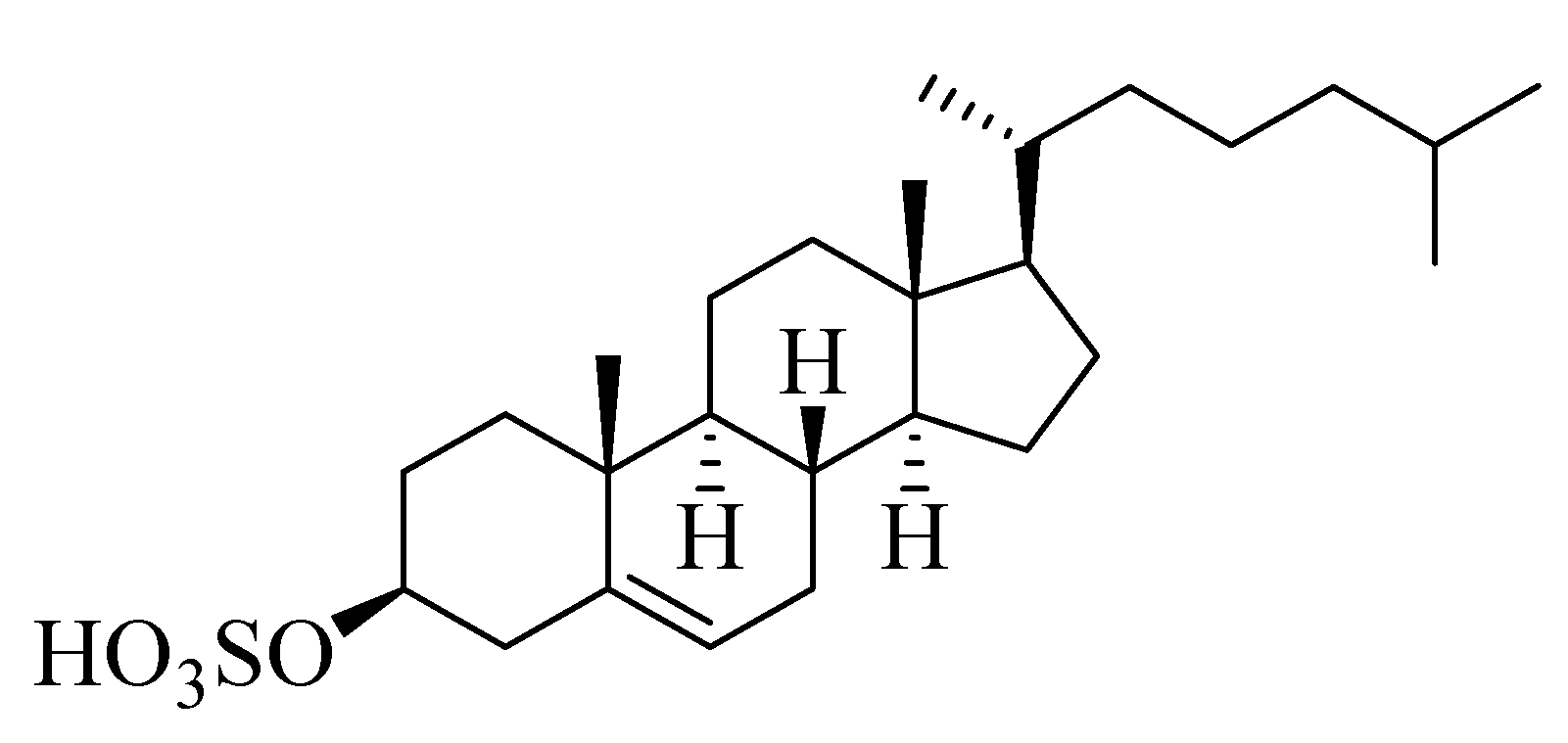
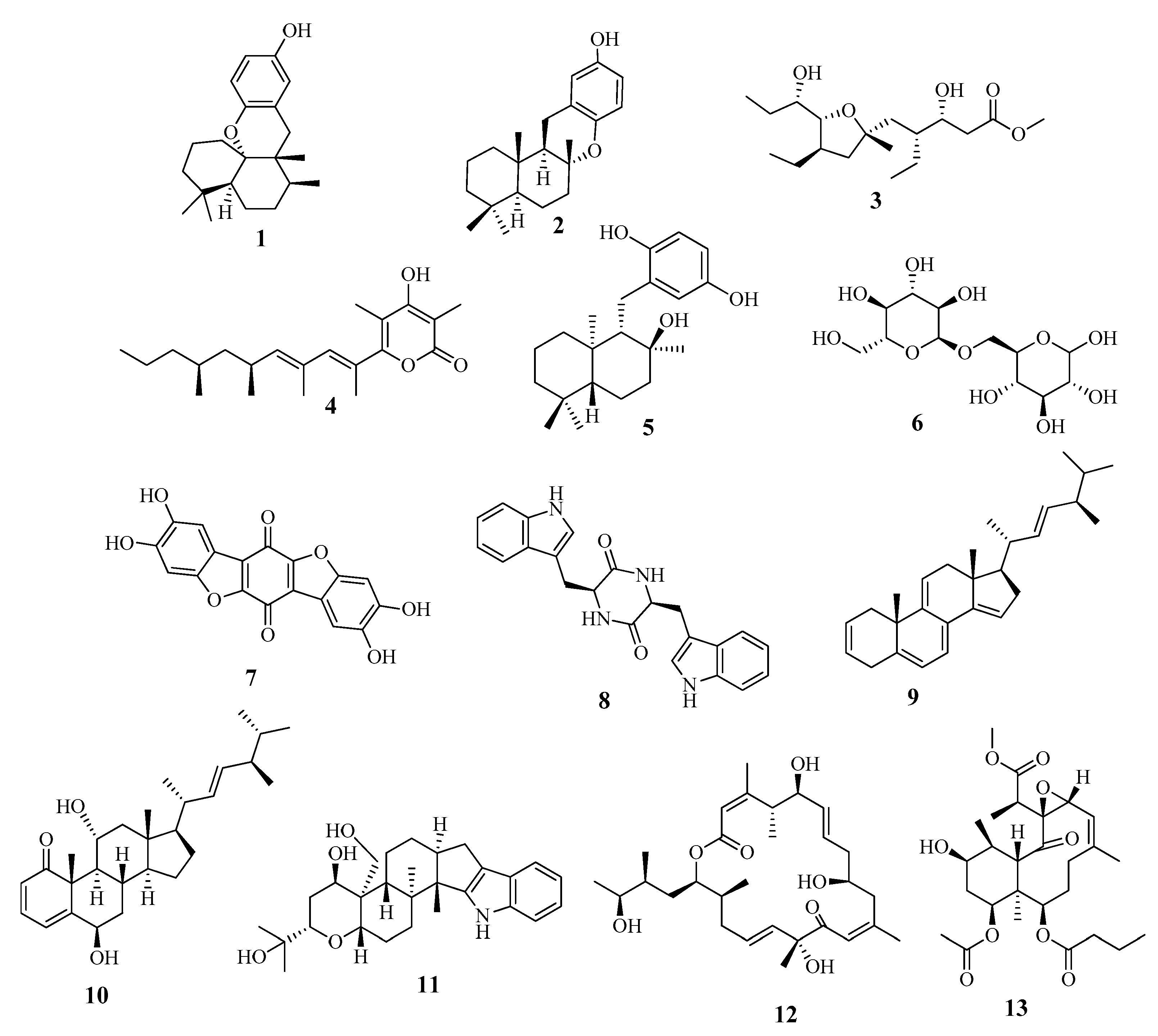
2.2. Metabolic Profiling
2.3. In Vitro Evaluation of the Antitumor Activities of the Isolated Compounds
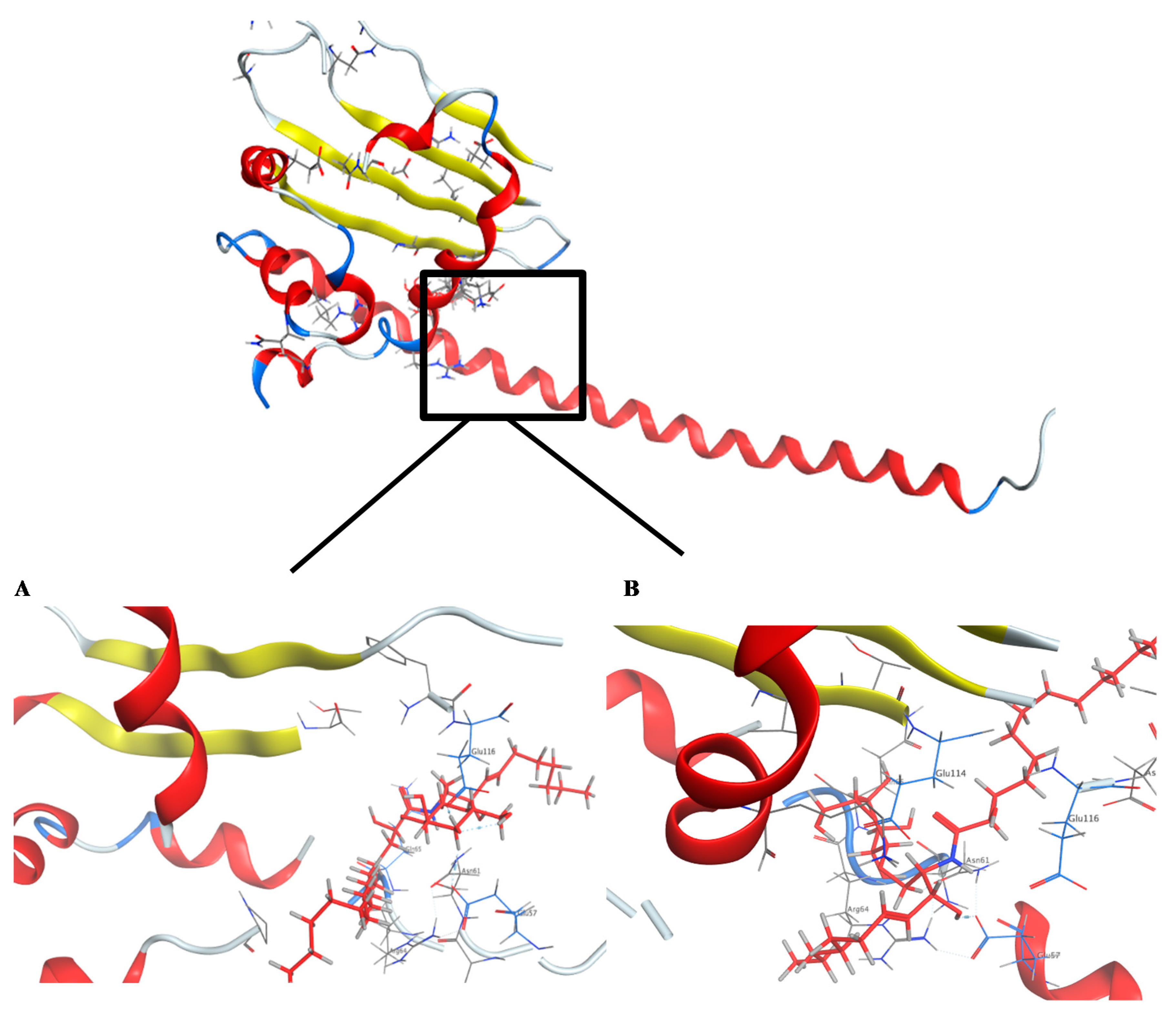
2.4. Molecular Docking Studies
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Sea Cucumber Material
3.3. Extraction and Isolation
3.4. Cerebroside Hydrolysis
3.5. Identification of the Sugar Moiety in Compounds 1, 2, and 3
3.6. Determination of the Configuration of the Sugar Moiety in 1, 2, and 3
3.7. Metabolomic Profiling
3.8. Spectroscopic Data
3.9. Cytotoxicity Assays
3.10. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eltahawy, N.A.; Ibrahim, A.K.; Radwan, M.M.; Zaitone, S.A.; Gomaa, M.; ElSohly, M.A.; Hassanean, H.A.; Ahmed, S.A. Mechanism of action of antiepileptic ceramide from Red Sea soft coral Sarcophyton auritum. Bioorg. Med. Chem. Lett. 2015, 25, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; El-Hossary, E.M.; Oelschlaeger, T.A.; Donia, M.S.; Quinn, R.J.; Abdelmohsen, U.R. Potential of marine natural products against drug-resistant bacterial infections. Lancet Infect. Dis. 2019, 19, 237–245. [Google Scholar] [CrossRef]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.E.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs. 2019, 17, 491. [Google Scholar] [CrossRef] [Green Version]
- Castellano, I.; Seebeck, F.P. On ovothiol biosynthesis and biological roles: From life in the ocean to therapeutic potential. Nat. Prod. Rep. 2018, 35, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Gerdol, M.; Sollitto, M.; Pallavicini, A.; Castellano, I. The complex evolutionary history of sulfoxide synthase in ovothiol biosynthesis. Proc. Biol. Sci. 2019, 286, 20191812. [Google Scholar] [CrossRef]
- Mokhlesi, A.; Saeidnia, S.; Gohari, A.R.; Shahverdi, A.R.; Nasrolahi, A.; Farahani, F.; Khoshnood, R.; Es’haghi, N. Biological activities of the Sea Cucumber Holothuria leucospilota. Asian J. Anim. Vet. Adv. 2012, 7, 243–249. [Google Scholar] [CrossRef]
- Malve, H. Exploring the ocean for new drug developments: Marine pharmacology. J. Pharm. Bioallied Sci. 2016, 8, 83–91. [Google Scholar] [CrossRef]
- Ebada, S.S.; Lin, W.H.; Proksch, P. Bioactive Sesquiterpenes and Triterpenes from Marine Sponges: Occurrence and Pharmacological Significance. Mar. Drugs 2010, 8, 313–346. [Google Scholar] [CrossRef] [Green Version]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef]
- Ahmed, M.I.; Aamer, M.A.; Lawrence, A.J. Identification of the Holothurian species of the Red Sea and Gulf of Aqaba using DNA barcoding technique. Egypt. J. Aquat. Biol. Fish. 2016, 20, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Harada, Y.; Nagaregawa, Y.; Miyamoto, T.; Isobe, R.; Higuchi, R. Isolation and structure of biologically active gangliosides from the sea cucumber Holothuria pervicax. Org. Chem. 1998, 1, 2519–2525. [Google Scholar] [CrossRef]
- Yamada, K.; Sasaki, K.; Harada, Y.; Isobe, R.; Higuchi, R. Isolation and structure of glucocerebrosides from the sea cucumber Holothuria pervicax. Chem. Pharm. Bull. 2002, 50, 1467–1470. [Google Scholar] [CrossRef] [Green Version]
- Abdelmohsen, U.; Cheng, C.; Viegelmann, C.; Zhang, T.; Grkovic, T.; Ahmed, S.; Quinn, R.J.; Hentschel, U.; Edrada-Ebel, R. Dereplication Strategies for Targeted Isolation of New Antitrypanosomal Actinosporins A and B from a Marine Sponge Associated-Actinokineospora sp. EG49. Mar. Drugs. 2014, 12, 1220–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.Y.; Huang, T.T.; Chen, Y.T.; Chen, J.L.; Chu, P.Y.; Huang, C.T.; Wang, W.L.; Lau, K.Y.; Dai, M.S.; Shiau, C.W.; et al. Targeting SET to restore PP2A activity disrupts an oncogenic CIP2Afeedforward loop and impairs triple negative breast cancer progression. EBioMedicine 2019, 40, 263–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruckhäberle, E.; Rody, A.; Engels, K.; Gaetje, R.; Minckwitz, G.V.; Schiffmann, S.; Grösch, S.; Geisslinger, G.; Holtrich, U.; Karn, T.; et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res. Treat. 2008, 112, 41–52. [Google Scholar] [CrossRef]
- Holthuis, J.C.M.; Pomorski, T.; Raggers, R.J.; Sprong, H.; Meer, G.V. The organizing potential of sphingolipids in intracellular membrane transport. Physiol. Rev. 2001, 81, 1689–1723. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J. Spectrometric Identification of Organic Compounds, 3rd ed.; John Wiley & Sons: New York, NY, USA,, 1974. [Google Scholar]
- Thomford, A.K.; Abdelhameed, R.F.A.; Yamada, K. Chemical studies on the parasitic plant Thonningia sanguinea Vahl. RSC Adv. 2018, 8, 21002–21011. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Xu, Y.; Liu, K.; Hua, H.; Zhu, H.; Pei, Y. Gracilarioside and Gracilamides from the Red Alga Gracilaria asiatica. J. Nat. Prod. 2006, 69, 1488–1491. [Google Scholar] [CrossRef]
- Yamada, K.; Matsubara, R.; Kaneko, M.; Miyamoto, T.; Higuchi, R. Isolation and structure of a biologically active ganglioside molecular species from the sea cucumber Holothuria leucospilota. Chem. Pharm. Bull. 2001, 49, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Abdelhameed, R.F.; Ibrahim, A.K.; Yamada, K.; Ahmed, S.A. Cytotoxic and anti-inflammatory compounds from Red Sea grass Thalassodendron ciliatum. Med. Chem. Res. 2018, 27, 1238–1244. [Google Scholar] [CrossRef]
- Goad, L.J.; Akihisa, T. Analysis of Sterols; Blackie Academic and Professional: London, UK, 1997; 1H NMR spectroscopy of sterols; pp. 197–234. [Google Scholar]
- Gallo, C.; D’Ippolito, G.; Nuzzo, G.; Sardo, A.; Fontana, A. Autoinhibitory sterol sulfates mediate programmed cell death in a bloom-forming marine diatom. Nat. Commun. 2017, 8, 1292. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahn, J.M.; Vistica, D.; Warren, J.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Nat. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Vijayarathna, S.; Sasidharan, S. Cytotoxicity of methanol extracts of Elaeis guineensis on MCF-7 and Vero cell lines. Asian Pac. J. Trop. Biomed. 2012, 2, 826–829. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.C.; Liaw, C.C.; Ho, J.R.; Khalil, A.T.; Kuo, Y.H. Isolation of aureol from Smenospongia sp. and cytotoxic activity of some aureol derivatives. Nat. Prod. Res. 2006, 20, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, M. Cytotoxic Terpene Quinones from Marine Sponges. Mar. Drugs 2010, 8, 2849–2870. [Google Scholar] [CrossRef] [Green Version]
- Campagnuolo, C.; Fattorusso, E.; Taglialatela-Scafati, O.; Ianaro, A.; Pisano, B. Plakortethers A−G: A New Class of Cytotoxic Plakortin-Derived Metabolites. Eur. J. Org. Chem. 2002, 1, 61–69. [Google Scholar] [CrossRef]
- Duh, C.Y.; Lo, I.W.; Wang, S.K.; Dai, C.F. New cytotoxic steroids from the soft coral Clavularia viridis. Steroids 2007, 72, 573–579. [Google Scholar] [CrossRef]
- Nagasawa, I.; Kaneko, A.; Suzuki, T.; Nishio, K.; Kinoshita, K.; Shiro, M.; Koyama, k. Potential Anti-angiogenesis Effects of p-Terphenyl Compounds from Polyozellus multiplex. J. Nat. Prod. 2014, 77, 963–968. [Google Scholar] [CrossRef]
- Tomoda, H.; Tabata, N.; Yang, D.-J.; Takayanagi, H.; Omura, S. Terpendoles, novel ACAT inhibitors produced by Albophoma yamanashiensis. J. Antibiot. 1995, 48, 793–804. [Google Scholar] [CrossRef]
- Longeon, A.; Copp, B.R.; Quévrain, E.; Roué, M.; Kientz, B.; Cresteil, T.; Petek, S.; Debitus, C.; Bourguet-Kondracki, M.L. Bioactive indole derivatives from the South Pacific marine sponges Rhopaloeides odorabile and Hyrtios sp. Mar. Drugs 2011, 9, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, M.; Nishikawa, K.; Matsuura, H.; Umezawa, T.; Matsuda, F.; Okino, T. Antioxidants from the Brown Alga Dictyopteris undulata. Molecules 2018, 23, 1214. [Google Scholar] [CrossRef] [Green Version]
- Hochlowski, J.E.; Faulkner, D.J. Antibiotics from the marine pulmonate Siphonaria diemenensis. Tetrahedron Lett. 1983, 24, 1917–1920. [Google Scholar] [CrossRef]
- Ishibashi, F.; Sato, S.; Sakai, K.; Hirao, S.; Kuwano, K. Algicidal sesquiterpene hydroquinones from the brown alga Dictyopteris undulate. Biosci. Biotechnol. Biochem. 2013, 77, 1120–1122. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Vining, L.C. Nutrient utilization in actinomycetes. Induction of α-glucosidases in Streptomyces venezuelae. Can. J. Microbiol. 1981, 27, 639–645. [Google Scholar]
- Wang, X.; Li, Y.; Zhang, X.; Lai, D.; Zhou, L. Structural diversity and biological activities of the cyclodipeptides from fungi. Molecules 2017, 22, 2026. [Google Scholar] [CrossRef] [Green Version]
- Ivonne, J.N.; AVILA, C.; Iris, M. Determination of fatty acids and triterpenoid compounds from the fruiting body of Suillus luteus. Rev. Colomb. Quím. 2008, 37, 297–304. [Google Scholar]
- Tsuda, M.; Oguchi, K.; Iwamoto, R.; Okamoto, Y.; Fukushi, E.; Kawabata, J.; Ozawa, T.; Masuda, A. Iriomoteolides-1b and -1c, 20-Membered Macrolides from a Marine Dinoflagellate Amphidinium Species. J. Nat. Prod. 2007, 70, 1661–1663. [Google Scholar] [CrossRef]
- Mootoo, B.S.; Ramsewak, R.; Sharma, R.; Tinto, W.F.; Lough, A.J.; Mclean, S.; Reynolds, W.F.; Yang, P.J.; Yu, M. Further briareolate esters and briareolides from the Caribbean Gorgonian octocoral Briareum asbestinum. Tetrahedron 1996, 52, 9953–9962. [Google Scholar] [CrossRef]
- Eltamany, E.E.; Ibrahim, A.K.; Radwan, M.M.; ElSohly, M.A.; Hassanean, H.A.; Ahmed, S.A. Cytotoxic ceramides from the Red Sea sponge Spheciospongia vagabunda. Med. Chem. Res. 2015, 24, 3467–3473. [Google Scholar] [CrossRef]
- Raslan, A.E.; Radwan, M.M.; Ahmed, S.A.; Nafady, A.M.; Zaki, M.A.; Wanas, A.S.; Abou-Karam, M.; Shier, T.W.; Hassanean, H.A.; ElSohly, M.A. Monanchoramides A–D, ceramides from the marine sponge Monanchora clathrata with cytotoxic activity. Phytochem. Lett. 2018, 23, 83–89. [Google Scholar] [CrossRef]
- Abdelhameed, R.F.A.; Habib, E.S.; Eltahawy, N.A.; Hassanean, H.A.; Ibrahim, A.K.; Mohammed, A.F.; Fayez, S.; Hayallah, A.M.; Yamada, K.; Behery, F.A.; et al. New Cytotoxic Natural Products from the Red Sea Sponge Stylissa carteri. Mar. Drugs 2020, 18, 241. [Google Scholar] [CrossRef]
- Kaneko, M.; Kisa, F.; Yamada, K.; Miyamoto, T.; Higuchi, R. Structure of a New Neuritogenic-Active Ganglioside from the Sea Cucumber Stichopus japonicus. Eur. J. Org. Chem. 2003, 1004–1008. [Google Scholar] [CrossRef]
- Jia, Z.; Song, Y.; Tao, S.; Cong, P.; Wang, X.; Xue, C.; Xu, J. Structure of Sphingolipids From Sea Cucumber Cucumaria frondosa and Structure-Specific Cytotoxicity Against Human HepG2 Cells. Lipids 2016, 51, 321–334. [Google Scholar] [CrossRef]
- Farokhi, F.; Wielgosz-Collin, G.; Clement, M.; Kornprobst, J.M.; Barnathan, G. Cytotoxicity on human cancer cells of ophidiacerebrosides isolated from the African starfish Narcissia canariensis. Mar. Drug 2010, 8, 2988–2998. [Google Scholar] [CrossRef] [Green Version]
- Umegaki, K.; Taki, Y.; Endoh, K.; Taku, K.; Tanabe, H.; Shinozuka, K.; Sugiyama, T. Bilobalide in Ginkgo biloba Extract Is a Major Substance Inducing Hepatic CYPs. J. Pharm. Pharmacol. 2007, 59, 871–877. [Google Scholar] [CrossRef]
- Souza, P.; Bianchi, S.; Figueiró, F.; Heimfarth, L.; Moresco, K.; Gonçalves, R.; Hoppe, J.; Klein, C.; Salbego, C.; Gelain, D.; et al. Anticancer activity of flavonoids isolated from Achyrocline satureioides in gliomas cell lines. Toxicol. Vitro 2018, 51, 23–33. [Google Scholar] [CrossRef]
- Huang, W.-C.; Chen, C.-L.; Lin, Y.-S.; Lin, C.-F. Apoptotic sphingolipid ceramide in cancer therapy. J. Lipids 2011, 2011, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Oku, H.; Wongtangtintharn, S.; Iwasaki, H.; Inafuku, M.; Shimatani, M.; Toda, T. Tumor specific cytotoxicity of glucosylceramide. Cancer Chemother. Pharm. 2007, 60, 767–775. [Google Scholar] [CrossRef]
- Wei, T.; Xiaojun, X.; Peilong, C. Magnoflorine improves sensitivity to doxorubicin (DOX) of breast cancer cells via inducing apoptosis and autophagy through AKT/mTOR and p38 signaling pathways. Biomed. Pharmacother. 2020, 121, 109139. [Google Scholar] [CrossRef]
- Senchenkov, A.; Litvak, D.A.; Cabot, M.C. Targeting ceramide metabolism--a strategy for overcoming drug resistance. J. Natl. Cancer Inst. 2001, 93, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D. Drug Resistance and the role of combination chemotherapy in improving patient outcomes. Int. J. Breast Cancer 2013, 137414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Switzer, C.; Cheng, R.; Vitek, T.; Christensen, D.J.; Wink, D.A.; Vitek, M.P. Targeting SET/I2PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene 2011, 30, 2504–2513. [Google Scholar] [CrossRef] [Green Version]
- Furuya, H.; Shimizu, Y.; Kawamori, T. Sphingolipids in cancer. Cancer Metastasis Rev. 2011, 30, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Saddoughi, S.A.; Song, P.; Sultan, I.; Ponnusamy, S.; Senkal, C.E.; Snook, C.F.; Arnold, H.K.; Sears, R.C.; Hannun, Y.A.; et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEBJ 2009, 23, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Bayarkhangai, B.; Noureldin, S.; Yu, L.; Zhao, N.; Gu, Y.; Xu, H.; Guo, C. A comprehensive and perspective view of oncoprotein SET in cancer. Cancer Med. 2018, 7, 3084–3094. [Google Scholar] [CrossRef] [PubMed]
- Janghorban, M.; Farrell, A.S.; Allen-Petersen, B.L.; Pelz, C.; Daniel, C.J.; Oddo, J.; Langer, E.M.; Christensen, D.J.; Sears, R.C. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 9157–9162. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Cai, J.; Chen, S.; Zheng, X.; Hu, S.; Dong, W.; Lu, J.; Xing, J.; Dong, Y. Paclitaxel resistance in MCF-7/PTX cells is reversed by paeonol through suppression of the SET/phosphatidylinositol 3-kinase/Akt pathway. Mol. Med. Rep. 2015, 12, 1506–1514. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Hikichi, M.; Yukitake, J.; Harada, N.; Utsumi, T. Estradiol suppresses phosphorylation of ERα serine 167 through upregulation of PP2A in breast cancer cells. Oncol. Lett. 2017, 14, 8060–8065. [Google Scholar] [CrossRef] [Green Version]
- Cristóbal, I.; Torrejón, B.; Martínez-Useros, J.; Madoz-Gurpide, J.; Rojo, F.; García-Foncillas, J. PP2A regulates signaling through hormonal receptors in breast cancer with important therapeutic implications. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 435–438. [Google Scholar] [CrossRef]
- Zhao, H.; Li, D.; Zhang, B.; Qi, Y.; Diao, Y.; Zhen, Y.; Shu, X. PP2A as the Main Node of Therapeutic Strategies and Resistance Reversal in Triple-Negative Breast Cancer. Molecules 2017, 22, 2277. [Google Scholar] [CrossRef] [Green Version]
- Keen, J.C.; Zhou, Q.; Park, B.H.; Pettit, C.; Mack, K.M.; Blair, B.; Brenner, K.; Davidson, N.E. Protein phosphatase 2A regulates estrogen receptor alpha (ER) expression through modulation of ER mRNA stability. J. Biol. Chem. 2005, 280, 29519–29524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsayed, Y.; Refaat, J.; Abdelmohsen, U.R.; Othman, E.M.; Stopper, H.; Fouad, M.A. Metabolomic profiling and biological investigation of the marine sponge-derived bacterium Rhodococcus sp. UA13. Phytochem. Anal. 2018, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Senda, M.; Akai, Y.; Sato, L.; Suzuki, T.; Nagai, R.; Senda, T.; Horikoshi, M. Relationship between the structure of SET/TAF-Ibeta/INHAT and its histone chaperone activity. Proc. Natl. Acad. Sci. USA 2007, 104, 4285–4290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular Operating Environment (MOE), 2019.0101 of Chemical Computing Group. Inc. Available online: http://www. chemcomp.com (accessed on 15 April 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | ||||||
|---|---|---|---|---|---|---|---|---|
| Position | δH (mult., JHz) | δC | Position | δH (mult., JHz) | δC | Position | δH (mult., JHz) | δC |
| 1 | 4.26, m 4.76, dd (4, 12) | 70.5 | 1 | 4.21, m 4.75, m | 70.0 | 1 | 4.34, m 4.61, m | 70.2 |
| 2 | 4.82, m | 54.9 | 2 | 4.79, m | 54.6 | 2 | 5.30, br,m | 51.4 |
| 3 | 4.82, m | 72.6 | 3 | 4.79, m | 72.5 | 3 | 4.35, m | 74.9 |
| 4 | 5.48, m | 132.2 | 4 | 5.96, m | 131.9 | 4 | 4.20, m | 72.1 |
| 5 | 5.96, m | 132.6 | 5 | 5.96, m | 131.1 | 5 | 1.70, m | 33.8 |
| 6 | 2.04, m | 32.6 | 6 | 2.02, m | 32.8 | 6 | 1.26 | 29.4 |
| 7 | 1.25 | 32.0 | 7 | 1.69, m | 31.9 | 7 | 1.26 | 30.1 |
| 8–15 | 1.25 | 29.6 | 8 | 1.25 | 31.9 | 8 | 1.26 | 30.1 |
| 16 | 0.85, t (6.8) | 14.1 | 9–19 | 1.25 | 29.7 | 9–19 | 1.26 | 29.7 |
| 1’ | - | 173.4 | 20 | 0.85, t (8.0) | 14.1 | 20 | 0.87, t (6.8) | 14.0 |
| 2’ | 4.79, m | 72.6 | 1’ | - | 175.7 | 1’ | - | 175.4 |
| 3’ | 2.04, m | 32.0 | 2’ | 4.56, m | 72.4 | 2’ | 4.74, t (8.0) | 72.2 |
| 4’ | 1.25 | 28.0 | 3’ | 2.18, m | 32.94 | 3’ | 1.70, m | 33.8 |
| 5’ | 1.25 | 27.6 | 4’–13’ | 1.25 | 29.7 | 4’–12’ | 1.26 | 29.7 |
| 6’–16’ | 1.25 | 29.6 | 14’ | 2.02, m | 31.9 | 13’ | 2.12, m | 31.9 |
| 17’ | 0.85, t (6.8) | 14.1 | 15’ | 5.25, m | 129.9 | 14’ | 5.30, m | 129.9 |
| NH | 8.37, d (8.0) | - | 16’ | 5.49, m | 132.3 | 15’ | 5.50, m | 130.0 |
| 1’’ | 4.53, d (8.0) | 105.9 | 17’ | 2.02, m | 31.9 | 16’ | 2.12, m | 31.9 |
| 2’’ | 4.06, t (8.0) | 75.2 | 18’–22’ | 1.25 | 29.7 | 17’–21’ | 1.26 | 29.7 |
| 3’’ | 4.22, m | 78.5 | 23’ | 0.85, t (6.8) | 14.1 | 22’ | 0.87, t (6.8) | 14.0 |
| 4’’ | 4.22, m | 71.5 | NH | 8.37, d (8.0) | - | NH | 8.60, d (8.0) | - |
| 5’’ | 3.94, m | 78.5 | 1’’ | 4.98, d (8.0) | 105.6 | 1’’ | 4.97, d (8.0) | 105.1 |
| 6’’ | 4.37, m 4.75, dd (4.0, 12.0) | 62.6 | 2’’ | 4.02, t (8.0) | 75.0 | 2’’ | 4.02, m | 75.5 |
| 3’’ | 4.20, m | 78.4 | 3’’ | 4.55, m | 78.1 | |||
| 4’’ | 4.20, m | 71.4 | 4’’ | 4.74, m | 71.1 | |||
| 5’’ | 3.90, m | 78.5 | 5’’ | 3.88, br,m | 78.3 | |||
| 6’’ | 4.36, dd (12.0, 4.0) 4.56, m | 62.5 | 6’’ | 4.35, dd (4.0, 8.0) 4.53, m | 62.3 | |||
| RT (min) | MZmine ID | Molecular Weight | Name | Source | Reference | |
|---|---|---|---|---|---|---|
| 1 | 12.29 | 171 | 314.2249 | Aureol | Porifera Hyrtios sp. | [33] |
| 2 | 10.87 | 122 | 314.2252 | Epichromazonarol | Dictyopteris undulata | [34] |
| 3 | 5.63 | 96 | 330.2398 | Plakortether E | Porifera Plakortis simplex | [29] |
| 4 | 11.23 | 109 | 332.2338 | Diemenensin A | Siphonaria diemenensis | [35] |
| 5 | 10.91 | 154 | 332.2353 | Yahazunol | Algae Dictyopteris undulata | [36] |
| 6 | 0.68 | 168 | 342.1149 | Isomaltose | Bacillus polymxa and Streptomyces spp. | [37] |
| 7 | 4.85 | 131 | 352.0208 | Thelephoric acid | Basidiomycete Polyozellus multiflex | [31] |
| 8 | 12.63 | 144 | 372.1592 | Fellutanine | Penicillium fellutanum | [38] |
| 9 | 11.78 | 75 | 374.2976 | (22E)-Ergosta-2,5,7,9(11),14,22-hexaene | Suillus luteus | [39] |
| 10 | 11.58 | 115 | 426.3135 | Stoloniferone O | Clavularia viridis | [30] |
| 11 | 9.25 | 24 | 453.2860 | Terpendole F | Albophoma yamanashiensis | [32] |
| 12 | 10.29 | 20 | 506.3224 | Iriomoteolide-1b | Marine Amphidinium species | [40] |
| 13 | 6.72 | 155 | 508.2678 | Briareolate ester D | Cnidaria Briareum asbestinum | [41] |
| Compound No. | IC50 (µM) |
|---|---|
| 1 | 13.83 ± 0.06 * µM |
| 2 | 8.13 ± 0.01 µM |
| 3 | 8.27 ± 0.03 µM |
| 4 | 35.56 ± 0.12 µM |
| Doxorubicin | 8.64 ± 0.02 µM |
| Compound No. | Binding Energy Score | Average Number of Poses per Run |
|---|---|---|
| 1a | −12.493 | 30 |
| 1b | −10.518 | 30 |
| 1c | −12.586 | 30 |
| 2 | −11.482 | 30 |
| 3 | −9.854 | 30 |
| 4 | −7.238 | 30 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelhameed, R.F.A.; Eltamany, E.E.; Hal, D.M.; Ibrahim, A.K.; AboulMagd, A.M.; Al-Warhi, T.; Youssif, K.A.; Abd El-kader, A.M.; Hassanean, H.A.; Fayez, S.; et al. New Cytotoxic Cerebrosides from the Red Sea Cucumber Holothuria spinifera Supported by In-Silico Studies. Mar. Drugs 2020, 18, 405. https://0-doi-org.brum.beds.ac.uk/10.3390/md18080405
Abdelhameed RFA, Eltamany EE, Hal DM, Ibrahim AK, AboulMagd AM, Al-Warhi T, Youssif KA, Abd El-kader AM, Hassanean HA, Fayez S, et al. New Cytotoxic Cerebrosides from the Red Sea Cucumber Holothuria spinifera Supported by In-Silico Studies. Marine Drugs. 2020; 18(8):405. https://0-doi-org.brum.beds.ac.uk/10.3390/md18080405
Chicago/Turabian StyleAbdelhameed, Reda F. A., Enas E. Eltamany, Dina M. Hal, Amany K. Ibrahim, Asmaa M. AboulMagd, Tarfah Al-Warhi, Khayrya A. Youssif, Adel M. Abd El-kader, Hashim A. Hassanean, Shaimaa Fayez, and et al. 2020. "New Cytotoxic Cerebrosides from the Red Sea Cucumber Holothuria spinifera Supported by In-Silico Studies" Marine Drugs 18, no. 8: 405. https://0-doi-org.brum.beds.ac.uk/10.3390/md18080405