
Studies on Pharmacokinetic Drug Interaction Potential of Vinpocetine

Abstract
:Abstract
Background
Methods
Results
Conclusion
1. Introduction

2. Experimental
3. Culture of hMDR1-MDCK-II, MDCK-II and HepG2 Cells
4. Reversible Inhibition (Co-Incubation Assay) and Time Dependent Inhibition (TDI/pre-Incubation Assay) of CYPs
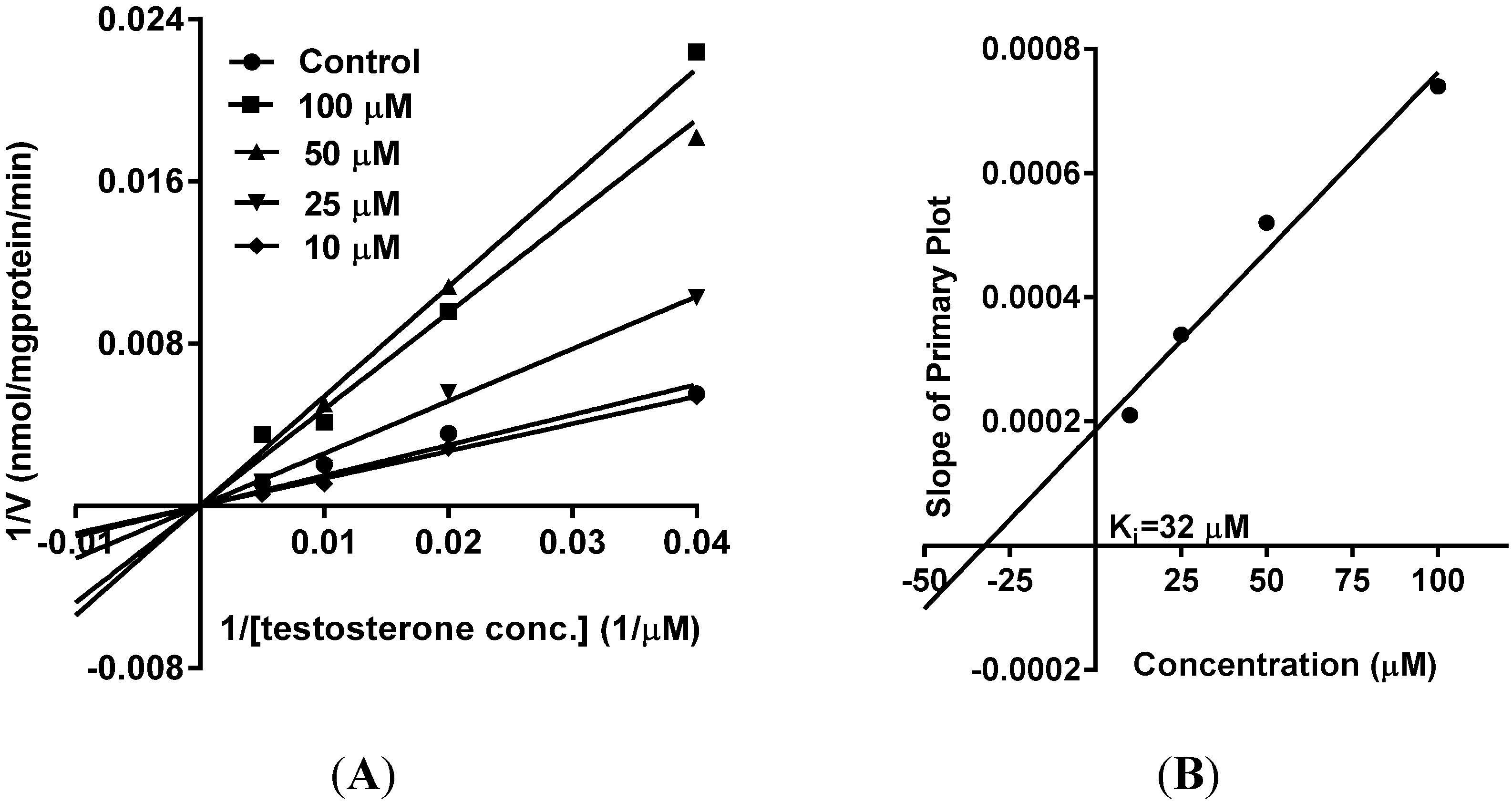
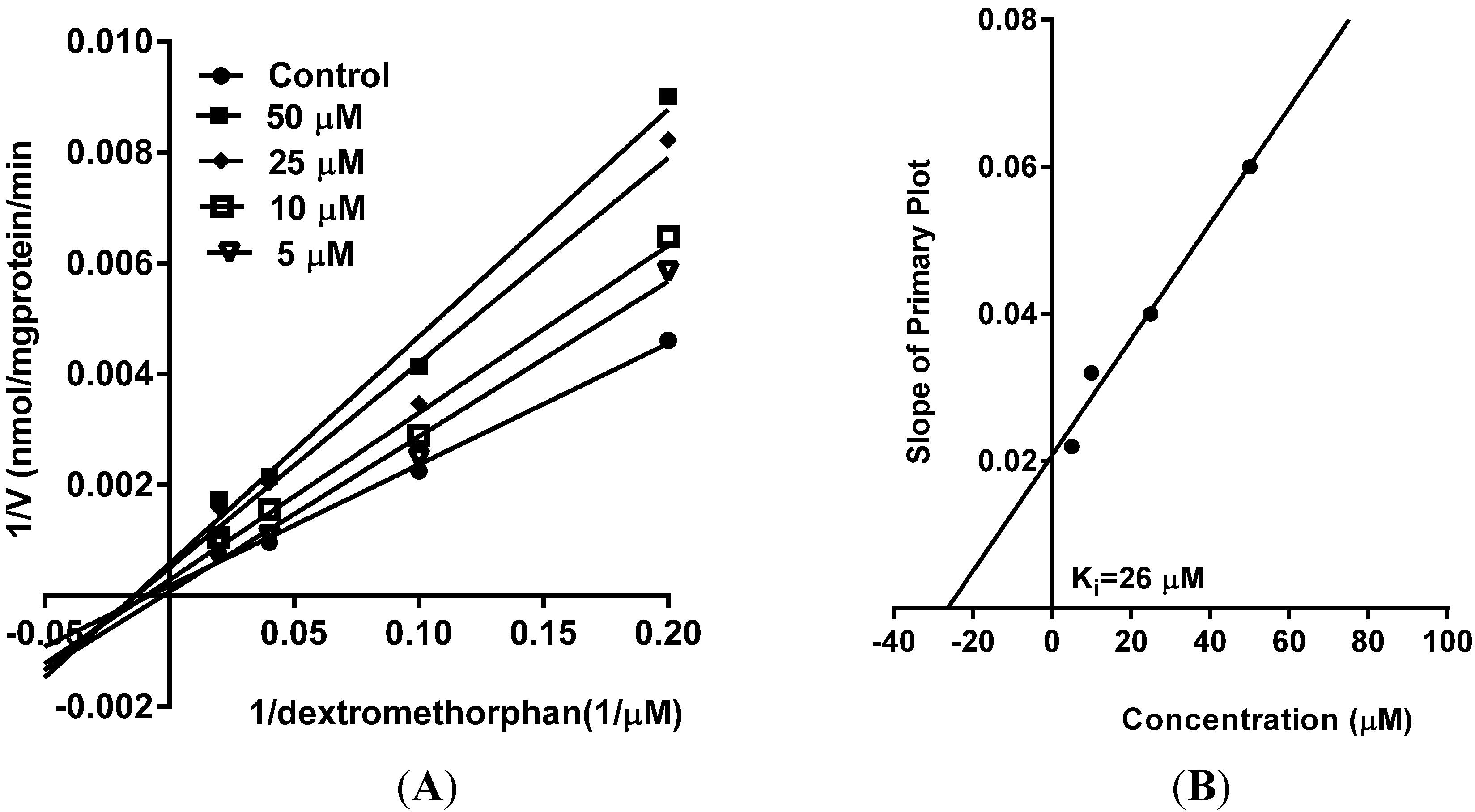
5. Reversible Inhibition and Enzyme Kinetics of CYP3A4 and CYP2D6 in Pooled HLM
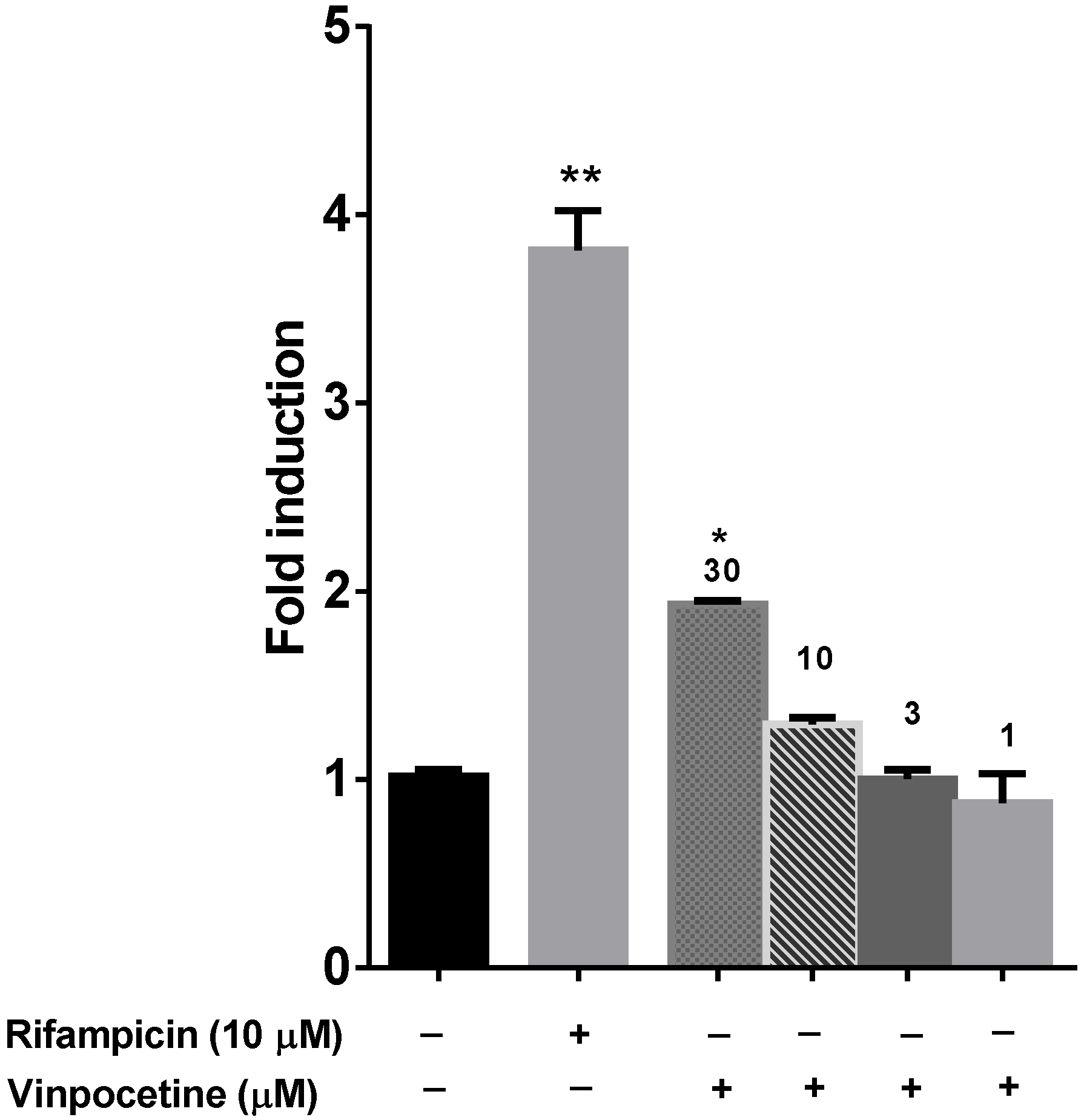
6. PXR Modulation
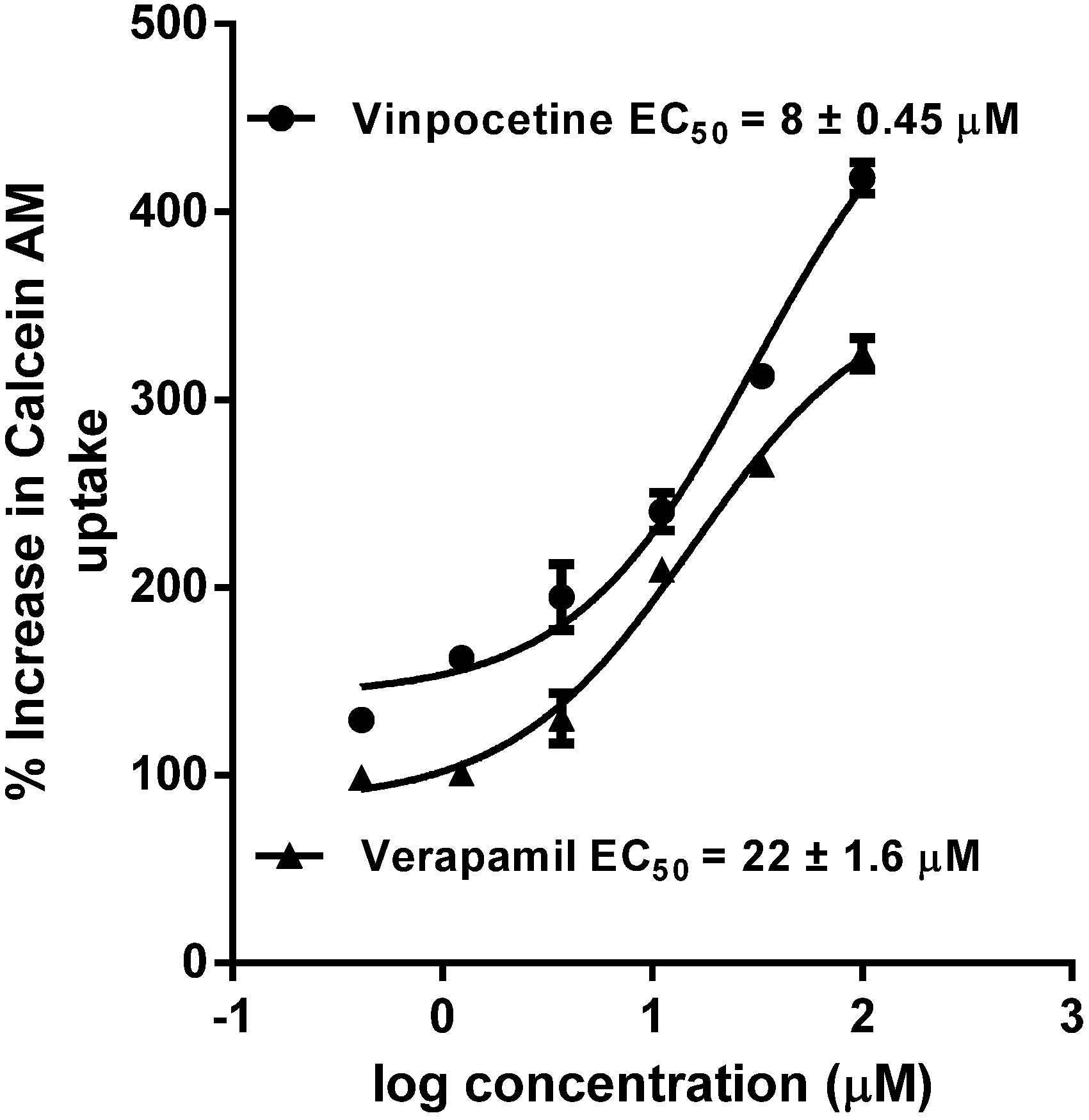
7. P-gp Inhibition
8. Analytical Methods
9. Statistical Methods
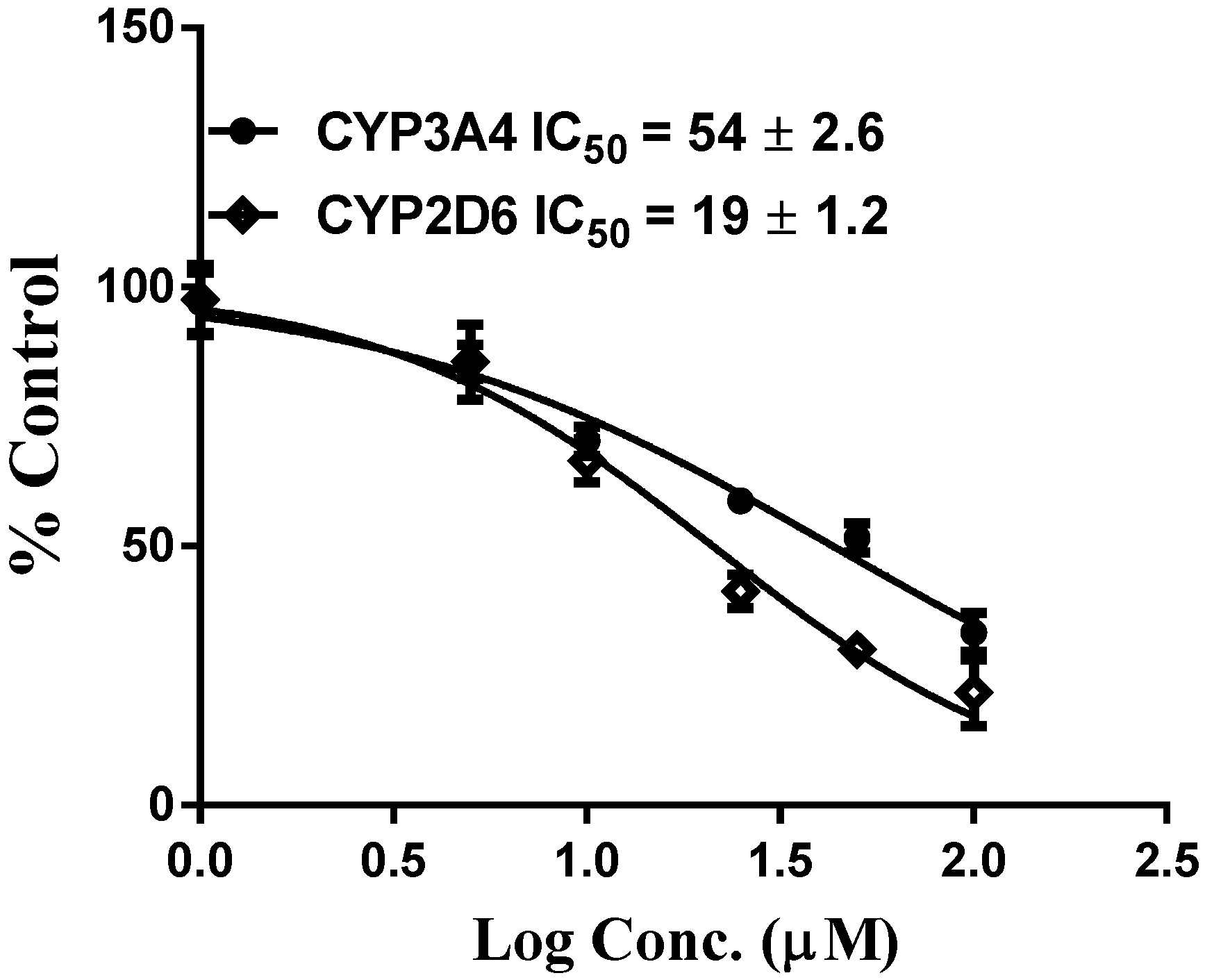
10. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CYP3A4 (recombinant) | ||
|---|---|---|---|
| IC50 (µM) | IC50 (µM) | IC50 | |
| Co-incubation | Pre-incubation | Shift (Fold) | |
| Vinpocetine | 2.8 ± 0.98 | 5.1 ± 0.1 | 0.54 |
| Ketoconazole | 0.04 ± 0.001 | 0.05 ± 0.002 | 0.80 |
| Troleandomycin | 2.5 ± 0.8 | 0.42 ± 0.05 | 5.95 |
| Compound | CYP2D6 (recombinant) | ||
|---|---|---|---|
| IC50 (µM) | IC50 (µM) | IC50 | |
| Co-incubation | Pre-incubation | Shift (Fold) | |
| Vinpocetine | 6.5 ± 1.1 | 37 ± 1.6 | 0.17 |
| Quinidine | 0.05 ± 0.001 | 0.08 ± 0.002 | 0.62 |
| Paroxetine | 3.4 ± 0.9 | 0.62 ± 0.03 | 5.48 |





11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alkuraishy, H.M.; Al-Gareeb, A.I.; Albuhadilly, A.K. Vinpocetine and pyritinol: A new model for blood rheological modulation in cerebrovascular disorders-a randomized controlled clinical study. BioMed. Res. Int. 2014, 2014, 324307. [Google Scholar] [CrossRef] [PubMed]
- Patyar, S.; Prakash, A.; Modi, M.; Medhi, B. Role of vinpocetine in cerebrovascular diseases. Pharmacol. Rep. 2011, 63, 618–628. [Google Scholar] [CrossRef]
- Vohora, D.; Saraogi, P.; Yazdani, M.A.; Bhowmik, M.; Khanam, R.; Pillai, K.K. Recent advances in adjunctive therapy for epilepsy: Focus on sodium channel blockers as third-generation antiepileptic drugs. Drugs Today (Barc.) 2010, 46, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Vinpocetine. Monograph. Altern. Med. Rev. J. Clin. Ther. 2002, 7, 240–243.
- Nagy, Z.; Simon, L. Neuroprotection in ischemic/hypoxic disorders: From the preclinical to the clinical testing. Adv. Exp. Med. Biol. 2004, 541, 39–54. [Google Scholar] [PubMed]
- Lin, C.; Chen, F.; Ye, T.; Zhang, L.; Zhang, W.; Liu, D.; Xiong, W.; Yang, X.; Pan, W. A novel oral delivery system consisting in “drug-in cyclodextrin-in nanostructured lipid carriers” for poorly water-soluble drug: Vinpocetine. Int. J. Pharm. 2014, 465, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Ogunrin, A. Effect of vinpocetine (cognitol) on cognitive performances of a nigerian population. Ann. Med. Health Sci. Res. 2014, 4, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Farina, E.K.; Austin, K.G.; Lieberman, H.R. Concomitant dietary supplement and prescription medication use is prevalent among us adults with doctor-informed medical conditions. J. Acad. Nutr. Diet. 2014, 114, 1784–1790 e1782. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Yoon, I.S. Pharmacokinetic interactions of herbs with cytochrome p450 and p-glycoprotein. Evid.-Based Complement. Alternat. Med. 2015, 2015, 736431. [Google Scholar] [CrossRef] [PubMed]
- Hitzenberger, G.; Sommer, W.; Grandt, R. Influence of vinpocetine on warfarin-induced inhibition of coagulation. Int. J. Clin. Pharmacol. Ther. Toxicol. 1990, 28, 323–328. [Google Scholar] [PubMed]
- Manda, V.K.; Dale, O.R.; Awortwe, C.; Ali, Z.; Khan, I.A.; Walker, L.A.; Khan, S.I. Evaluation of drug interaction potential of labisia pumila (kacip fatimah) and its constituents. Front. Pharmacol. 2014, 5, 178. [Google Scholar] [CrossRef] [PubMed]
- Crespi, C.L.; Miller, V.P.; Penman, B.W. Microtiter plate assays for inhibition of human, drug-metabolizing cytochromes p450. Anal. Biochem. 1997, 248, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.W.; Newton, D.J.; Scheri, T.D.; Lu, A.Y. Human cytochrome p450 3a4-catalyzed testosterone 6 beta-hydroxylation and erythromycin n-demethylation. Competition during catalysis. Drug Metab. Dispos. 1997, 25, 502–507. [Google Scholar] [PubMed]
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor pxr is activated by compounds that regulate cyp3a4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Hodgson, E.; Liddle, C. The orphan human pregnane x receptor mediates the transcriptional activation of cyp3a4 by rifampicin through a distal enhancer module. Mol. Pharmacol. 1999, 56, 1329–1339. [Google Scholar] [PubMed]
- Awortwe, C.; Manda, V.K.; Avonto, C.; Khan, S.I.; Khan, I.A.; Walker, L.A.; Bouic, P.J.; Rosenkranz, B. Echinacea purpurea up-regulates cyp1a2, cyp3a4 and mdr1 gene expression by activation of pregnane x receptor pathway. Xenobiotica 2014, 1–12. [Google Scholar]
- Rautio, J.; Humphreys, J.E.; Webster, L.O.; Balakrishnan, A.; Keogh, J.P.; Kunta, J.R.; Serabjit-Singh, C.J.; Polli, J.W. In vitro p-glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: A recommendation for probe substrates. Drug Metab. Dispos. 2006, 34, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Tee, H.W.; Go, M.L. Functionalized chalcones as selective inhibitors of p-glycoprotein and breast cancer resistance protein. Bioorg. Med. Chem. 2008, 16, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Manda, V.K.; Avula, B.; Ali, Z.; Khan, I.A.; Walker, L.A.; Khan, S.I. Evaluation of in vitro absorption, distribution, metabolism, and excretion (adme) properties of mitragynine, 7-hydroxymitragynine, and mitraphylline. Planta Med. 2014, 80, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Michalets, E.L. Update: Clinically significant cytochrome p-450 drug interactions. Pharmacotherapy 1998, 18, 84–112. [Google Scholar] [PubMed]
- Henderson, L.; Yue, Q.Y.; Bergquist, C.; Gerden, B.; Arlett, P. St john’s wort (hypericum perforatum): Drug interactions and clinical outcomes. Br. J. Clin. Pharmacol. 2002, 54, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Piao, Y.J.; Kang, K.W. Effects of quercetin on the bioavailability of doxorubicin in rats: Role of cyp3a4 and p-gp inhibition by quercetin. Arch. Pharm. Res. 2011, 34, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Strong, J.M.; Zhang, L.; Reynolds, K.S.; Nallani, S.; Temple, R.; Abraham, S.; Habet, S.A.; Baweja, R.K.; Burckart, G.J.; et al. New era in drug interaction evaluation: Us food and drug administration update on cyp enzymes, transporters, and the guidance process. J. Clin. Pharmacol. 2008, 48, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Naritomi, Y.; Teramura, Y.; Terashita, S.; Kagayama, A. Utility of microtiter plate assays for human cytochrome p450 inhibition studies in drug discovery: Application of simple method for detecting quasi-irreversible and irreversible inhibitors. Drug Metab. Pharmacokinet. 2004, 19, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Ekroos, M.; Sjogren, T. Structural basis for ligand promiscuity in cytochrome p450 3a4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.S.; Nilsen, O.G. In vitro cyp3a4 metabolism: Inhibition by echinacea purpurea and choice of substrate for the evaluation of herbal inhibition. Basic Clin. Pharmacol. Toxicol. 2008, 103, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Vlase, L.; Bodiu, B.; Leucuta, S.E. Pharmacokinetics and comparative bioavailability of two vinpocetine tablet formulations in healthy volunteers by using the metabolite apovincaminic acid as pharmacokinetic parameter. Arzneimittel-Forschung 2005, 55, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Adkins, C.E.; Mittapalli, R.K.; Manda, V.K.; Nounou, M.I.; Mohammad, A.S.; Terrell, T.B.; Bohn, K.A.; Yasemin, C.; Grothe, T.R.; Lockman, J.A.; et al. P-glycoprotein mediated efflux limits substrate and drug uptake in a preclinical brain metastases of breast cancer model. Front. Pharmacol. 2013, 4, 136. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, H.M.; Al-Abd, A.M.; El-Dine, R.S.; El-Halawany, A.M. P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: A review. J. Adv. Res. 2015, 6, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Willson, T.M.; Kliewer, S.A. Pxr, car and drug metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manda, V.K.; Avula, B.; Dale, O.R.; Chittiboyina, A.G.; Khan, I.A.; Walker, L.A.; Khan, S.I. Studies on Pharmacokinetic Drug Interaction Potential of Vinpocetine. Medicines 2015, 2, 93-105. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines2020093
Manda VK, Avula B, Dale OR, Chittiboyina AG, Khan IA, Walker LA, Khan SI. Studies on Pharmacokinetic Drug Interaction Potential of Vinpocetine. Medicines. 2015; 2(2):93-105. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines2020093
Chicago/Turabian StyleManda, Vamshi K., Bharathi Avula, Olivia R. Dale, Amar G. Chittiboyina, Ikhlas A. Khan, Larry A. Walker, and Shabana I. Khan. 2015. "Studies on Pharmacokinetic Drug Interaction Potential of Vinpocetine" Medicines 2, no. 2: 93-105. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines2020093