
Trauma-Induced Coagulopathy: Overview of an Emerging Medical Problem from Pathophysiology to Outcomes

 ,
, 
Abstract
:1. Introduction
2. Definition
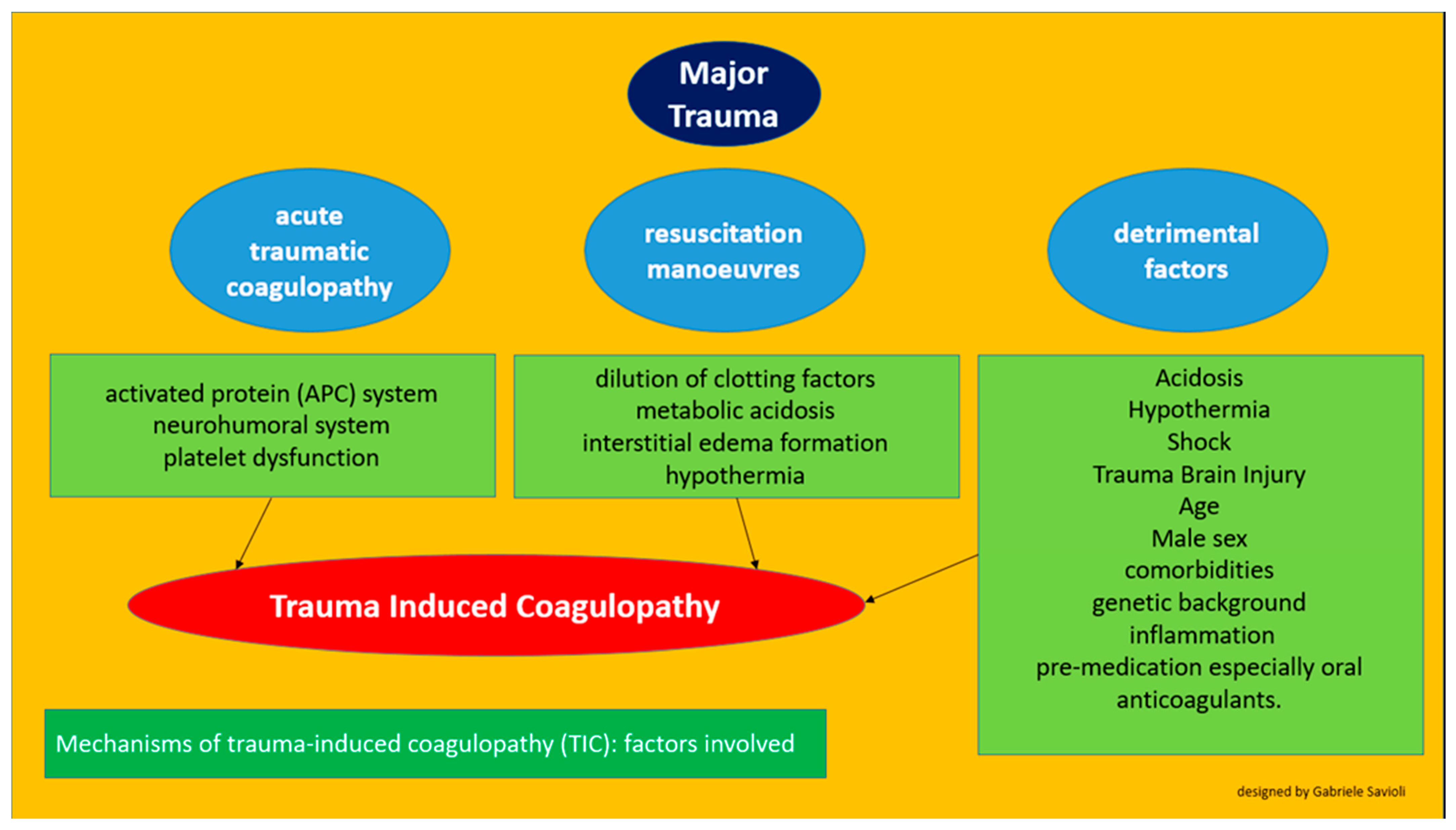
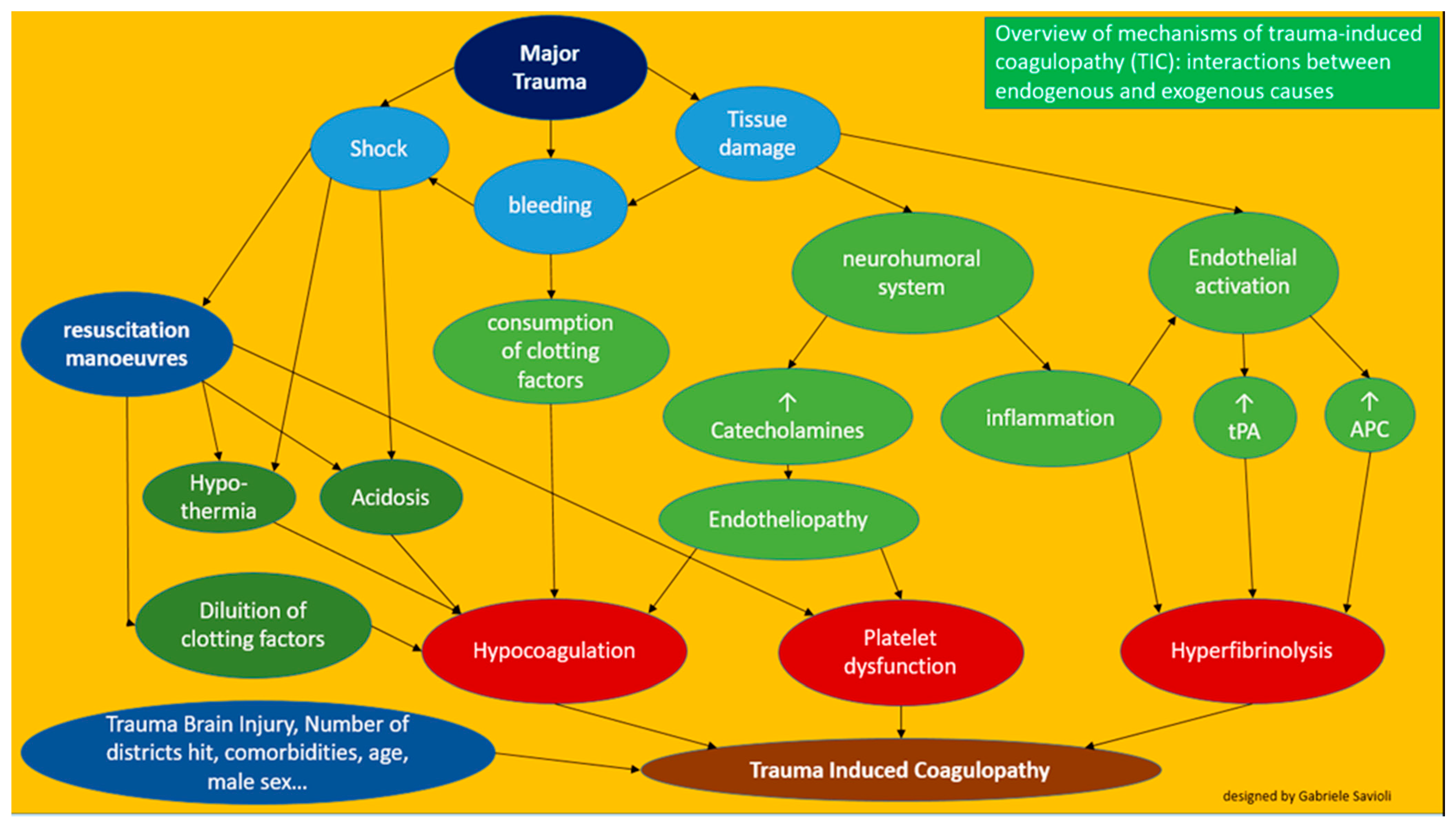
3. Pathophysiology
3.1. Acute Traumatic Hypercoagulability
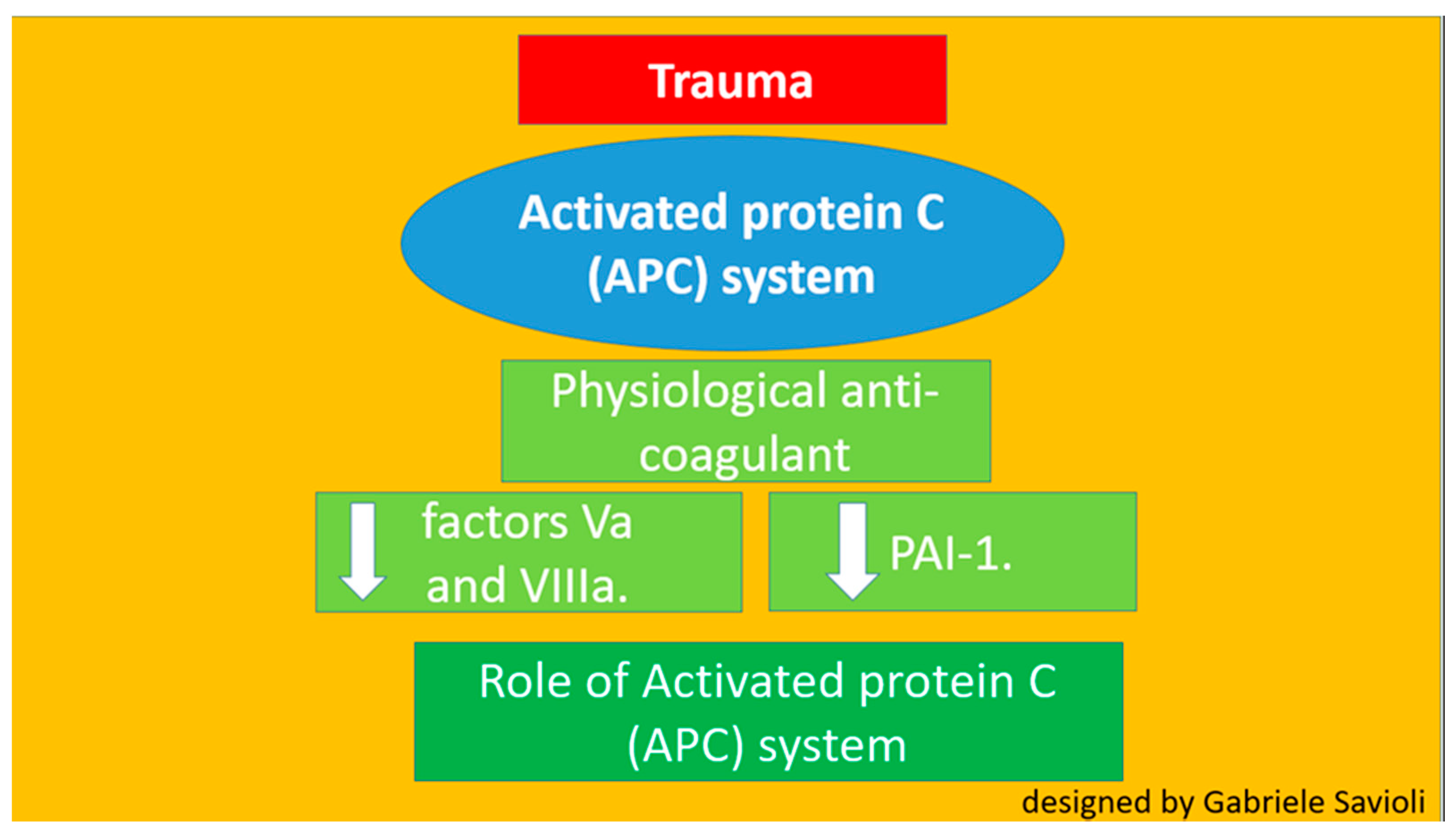
3.1.1. Role of the C Protein
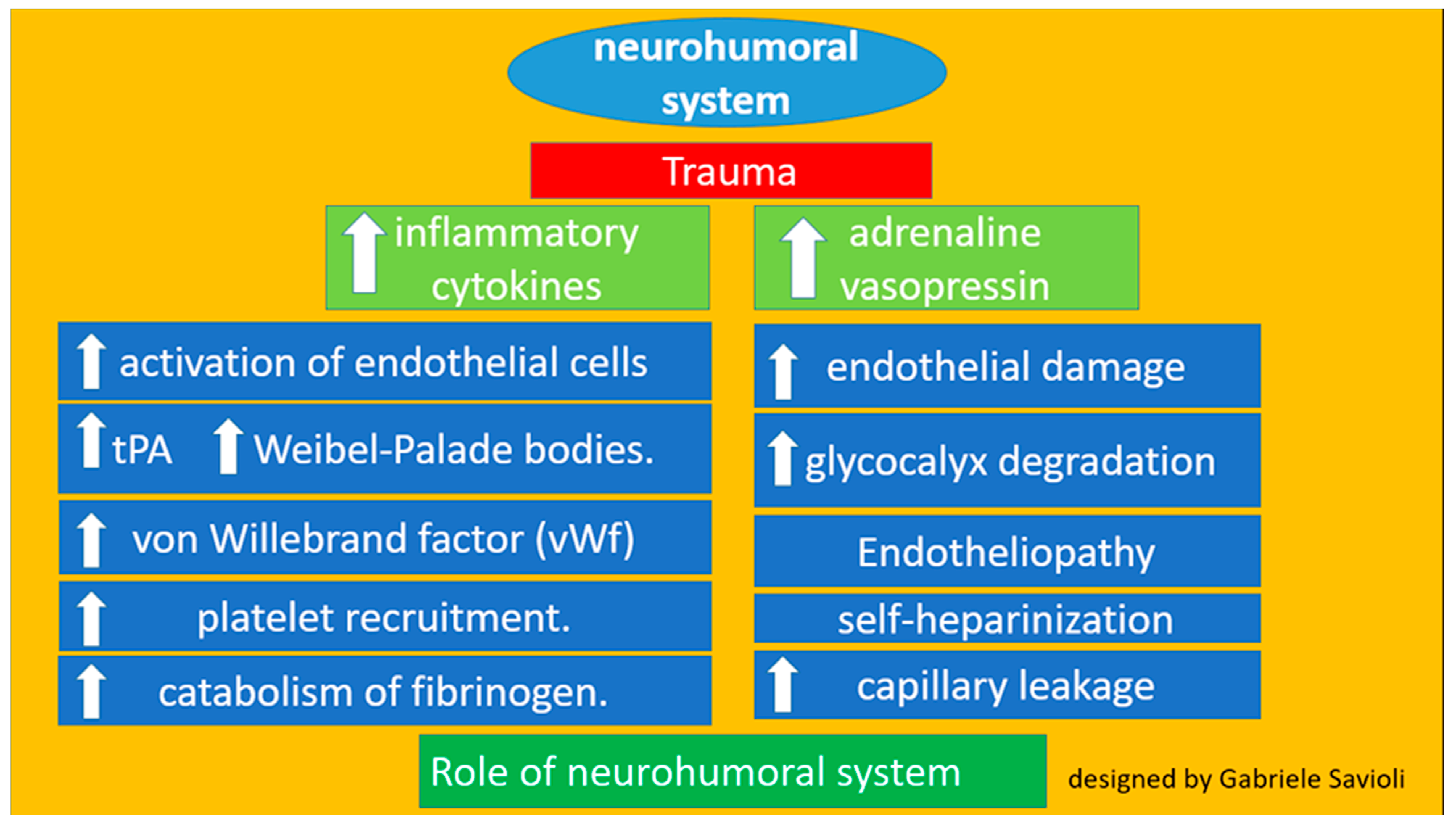
3.1.2. Role of the Neurohumoral System
3.1.3. Role of Platelets
3.2. Coagulopathy Associated with Resuscitation Maneuvers
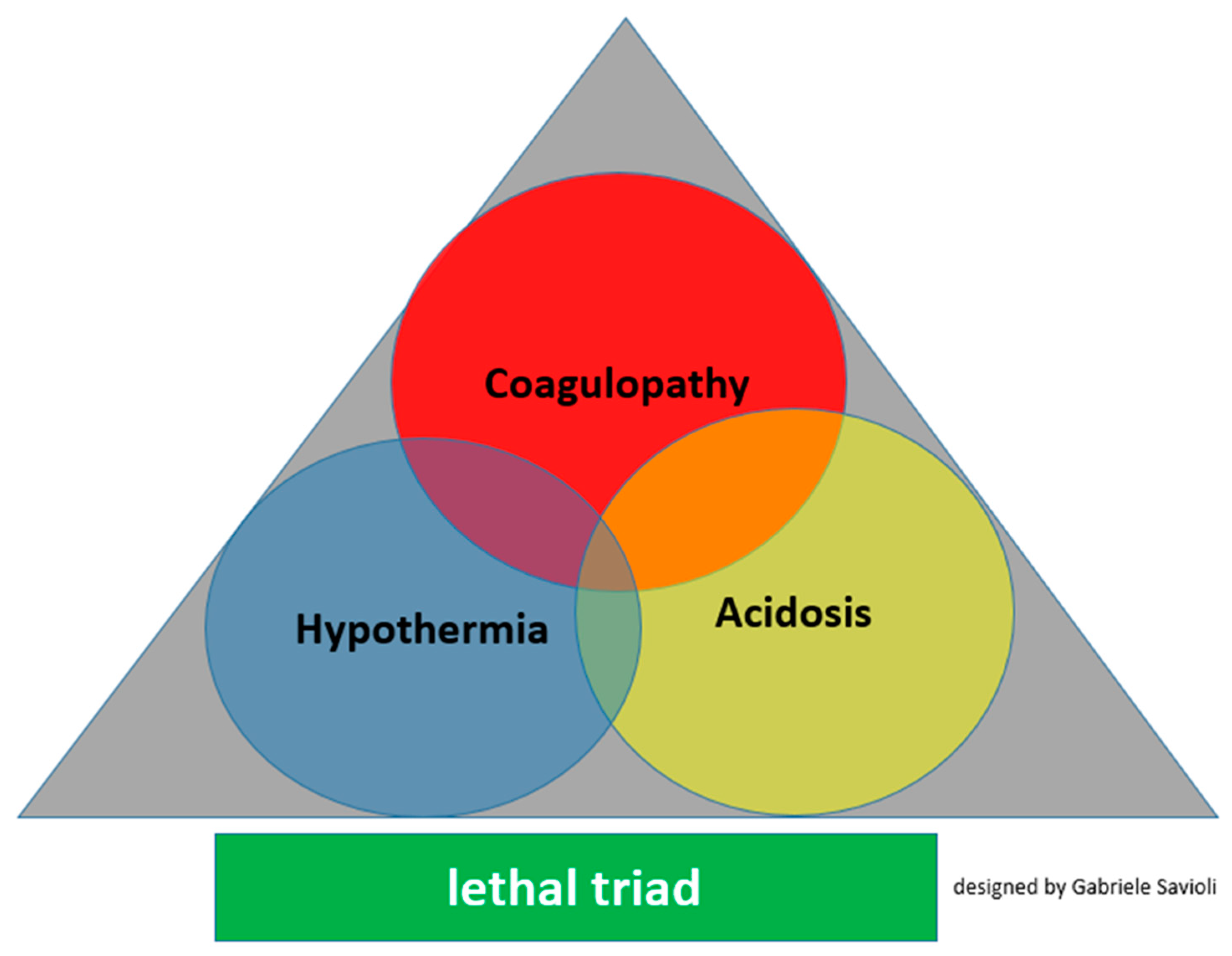
3.3. Detrimental Factors Exacerbating Trauma Coagulopathy
3.3.1. Acidosis
- Changes of platelet shapes and structure;
- Reductions of clotting factor activity;
- Compromised thrombin production;
- Reductions of the fibrinogen concentration;
- Increased fibrinogen degradation (caused by increased fibrinolysis and increased factor XIII levels) without effects on fibrinogen production;
- Increased pro-inflammatory responses by platelet-mediated neutrophils;
3.3.2. Hypothermia
- Negatively affects platelet function;
- Reduces the enzyme activity of clotting factors;
- Induces the activation of fibrinolysis;
3.3.3. Shock
3.3.4. TBI
3.3.5. Age, Male Sex and Comorbidities
3.3.6. Other Factors
4. Specials Clinical Forms of TIC
4.1. Early Primary Hyperfibrinolysis
4.2. Late Hypercoagulability
5. Diagnosis
5.1. Clinical Features
5.2. Laboratory Tests
6. Outcomes
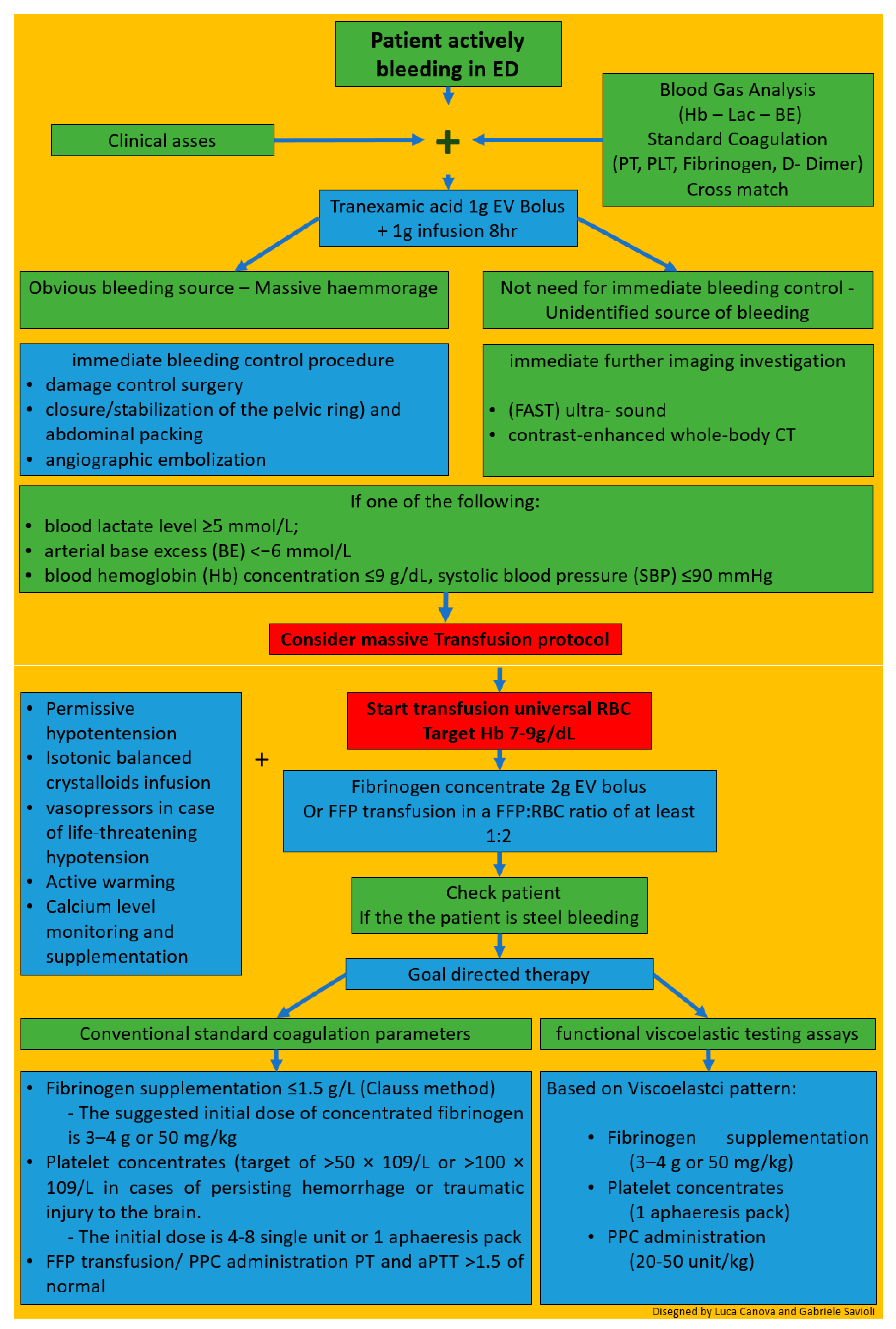
7. Hints for Therapy
8. Management of Patients with Severe Trauma in the ED
9. Conclusions
- TIC is a dynamic sequence coagulation disorder from hyperfibrinolysis, hypercoagulation to its final stage hypocoagulation. The early hypocoagulable state is not related to dilution nor to iatrogenic or hypothermic causes.
- TIC is present in approximately one-third of patients who report MT
- The physiopathology of TIC is complex and features several contributing causes. The role of protein C has been less emphasized
- The diagnosis and management of TIC often encompasses standard coagulation test and functional viscoelastic assays.
- Early initiation of antifibrinolytic therapy and balanced resuscitation of coagulation disorder is the mainstay of TIC
- TIC is related to worse outcomes, among which increased rates of transfusion, infection, thromboembolism, acute lung injury, multi-organ failure, and death.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Department of Violence and Injury Prevention and Disability; World Health Organization. Injuries and Violence: The Facts. Available online: http://whqlibdoc.who.int/publications/2010/9789241599375_eng.pdf (accessed on 22 February 2020).
- GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age–sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 385, 117–171. [Google Scholar] [CrossRef]
- Soreide, K. Epidemiology of major trauma. Br. J. Surg. 2009, 96, 697–698. [Google Scholar] [CrossRef]
- Frith, D.; Goslings, J.C.; Gaarder, C.; Maegele, M.; Cohen, M.J.; Allard, S.; Johansson, P.I.; Stanworth, S.; Thiemermann, C.; Brohi, K. Definition and drivers of acute traumatic coagulopathy: Clinical and experimental investigations. J. Thromb. Haemost. 2010, 8, 1919–1925. [Google Scholar] [CrossRef]
- Maegele, M.; Lefering, R.; Yucel, N.; Tjardes, T.; Rixen, D.; Paffrath, T.; Simanski, C.; Neugebauer, E.; Bouillon, B. Early coagulopathy in multiple injury: An analysis from the German Trauma Registry on 8724 patients. Injury 2007, 38, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Brohi, K.; Singh, J.; Heron, M.; Coats, T. Acute traumatic coagulopathy. J. Trauma 2003, 54, 1127–1130. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, J.B.; Lynn, M.; McKenney, M.G.; Cohn, S.M.; Murtha, M. Early coagulopathy predicts mortality in trauma. J. Trauma 2003, 55, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Schöchl, H.; Nienaber, U.; Maegele, M.; Hochleitner, G.; Primavesi, F.; Steitz, B.; Arndt, C.; Hanke, A.; Voelckel, W.; Solomon, C. Transfusion in trauma: Thromboelastometry-guided coagulation factor concentrate-based therapy versus standard fresh frozen plasma-based therapy. Crit. Care 2011, 15, R83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöchl, H.; Frietsch, T.; Pavelka, M.; Jambor, C. Hyperfibrinolysis after major trauma: Differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J. Trauma 2009, 67, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Maegele, M.; Schochl, H.; Cohen, M.J. An update on the coagulopathy of trauma. Shock 2014, 41, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Davenport, R.; Raza, I.; Glasgow, S.; De’Ath, H.D.; Johansson, P.I.; Curry, N.; Stanworth, S.; Gaarder, C.; Brohi, K. Damage control resuscitation using blood component therapy in standard doses has a limited effect on coagulopathy during trauma hemorrhage. Intensive Care Med. 2015, 41, 239–247. [Google Scholar] [CrossRef]
- Hagemo, J.S.; Christiaans, S.C.; Stanworth, S.J.; Brohi, K.; Johansson, P.I.; Goslings, J.C.; Naess, P.A.; Gaarder, C. Detection of acute traumatic coagulopathy and massive transfusion requirements by means of rotational thromboelastometry: An international prospective validation study. Crit. Care 2015, 19, 97. [Google Scholar] [CrossRef] [Green Version]
- Savioli, G.; Ceresa, I.F.; Macedonio, S.; Gerosa, S.; Belliato, M.; Iotti, G.A.; Luzzi, S.; Del Maestro, M.; Mezzini, G.; Giotta Lucifero, A.; et al. Trauma Coagulopathy and Its Outcomes. Medicina 2020, 56, 205. [Google Scholar] [CrossRef] [Green Version]
- Hagemo, J.S.; Stanworth, S.; Juffermans, N.P.; Brohi, K.; Cohen, M.; Johansson, P.I.; Roislien, J.; Eken, T.; Naess, P.A.; Gaarder, C. Prevalence, predictors and outcome of hypofibrinogenaemia in trauma: A multicentre observational study. Crit. Care 2014, 18, R52. [Google Scholar] [CrossRef] [Green Version]
- Hess, J.R.; Brohi, K.; Dutton, R.P.; Hauser, C.J.; Holcomb, J.B.; Kluger, Y.; Mackway-Jones, K.; Parr, M.J.; Rizoli, S.B.; Yukioka, T.; et al. The coagulopathy of trauma: A review of mechanisms. J. Trauma 2008, 65, 748–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frith, D.; Davenport, R.; Brohi, K. Acute traumatic coagulopathy. Curr. Opin. Anaesthesiol. 2012, 25, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Spivey, M.; Parr, M.J. Therapeutic approaches in trauma-induced coagulopathy. Minerva Anestesiol. 2005, 71, 281–289. [Google Scholar]
- Engels, P.T.; Rezende-Neto, J.B.; Al Mahroos, M.; Scarpelini, S.; Rizoli, S.B.; Tien, H.C. The natural history of trauma-related coagulopathy: Implications for treatment. J. Trauma 2011, 71 (Suppl. 1), S448–S55. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.; Monroe, D.M.I.I.I. A cell-based model of hemostasis. Thromb. Haemost. 2001, 85, 958–965. [Google Scholar]
- Kushimoto, S.; Kudo, D.; Kawazoe, Y. Acute traumatic coagulopathy and trauma-induced coagulopathy: An overview. J. Intensive Care 2017, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Levi, M.; van der Poll, T. The role of natural anticoagulants in the pathogenesis and management of systemic activation of coagulation and inflammation in critically ill patients. Semin. Thromb. Hemost. 2008, 34, 459–468. [Google Scholar] [CrossRef]
- Cohen, M.J.; Kutcher, M.; Redick, B.; Nelson, M.; Call, M.; Knudson, M.M.; Schreiber, M.A.; Bulger, E.M.; Muskat, P.; Alarcon, L.H.; et al. Clinical and mechanistic drivers of acute traumatic coagulopathy. J. Trauma Acute Care Surg. 2013, 75, S40–S47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesebro, B.B.; Rahn, P.; Carles, M.; Esmon, C.T.; Xu, J.; Brohi, K.; Frith, D.; Pittet, J.F.; Cohen, M.J. Increase inactivated protein C mediates acute traumatic coagulopathy in mice. Shock 2009, 32, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.J.; Call, M.; Nelson, M.; Calfee, C.S.; Esmon, C.T.; Brohi, K.; Pittet, J.F. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann. Surg. 2012, 255, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Chapman, M.P.; Moore, E.E.; Moore, H.B.; Gonzalez, E.; Gamboni, F.; Chandler, J.G.; Mitra, S.; Ghasabyan, A.; Chin, T.L.; Sauaia, A.; et al. Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J. Trauma Acute Care Surg. 2016, 80, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gando, S.; Mayumi, T.; Ukai, T. Activated protein C plays nomajor roles in the inhibition of coagulation or increased fibrinolysis in acute coagulopathy of trauma-shock: A systematic review. Thromb. J. 2018, 16, 13. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.I.; Stensballe, J.; Rasmussen, L.S.; Ostrowski, S.R. High circulating adrenaline levels at admission predict increased mortality after trauma. J. Trauma Acute Care Surg. 2012, 72, 428–436. [Google Scholar] [CrossRef]
- Ostrowski, S.R.; Henriksen, H.H.; Stensballe, J.; Gybel-Brask, M.; Cardenas, J.C.; Baer, L.A.; Cotton, B.A.; Holcomb, J.B.; Wade, C.E.; Johansson, P.I. Sympathoadrenal activation and endotheliopathy are drivers of hypocoagulability and hyperfibrinolysis in trauma: A prospective observational study of 404 severely injured patients. J. Trauma Acute Care Surg. 2017, 82, 293–301. [Google Scholar] [CrossRef]
- Johansson, P.I.; Stensballe, J.; Rasmussen, L.S.; Ostrowski, S.R. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann. Surg. 2011, 254, 194–200. [Google Scholar] [CrossRef]
- Ostrowski, S.R.; Johansson, P.I. Endothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathy. J. Trauma Acute Care Surg. 2012, 73, 60–66. [Google Scholar] [CrossRef]
- Rahbar, E.; Cardenas, J.C.; Baimukanova, G.; Usadi, B.; Bruhn, R.; Pati, S.; Ostrowski, S.R.; Johansson, P.I.; Holcomb, J.B.; Wade, C.E. Endothelial glycocalyx shedding and vascular permeability in severely injured trauma patients. J. Transl. Med. 2015, 13, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, P.I.; Henriksen, H.H.; Stensballe, J.; Gybel-Brask, M.; Cardenas, J.C.; Baer, L.A.; Cotton, B.A.; Holcomb, J.B.; Wade, C.E.; Ostrowski, S.R. Traumatic endotheliopathy: A prospective observational study of 424 severely injured patients. Ann. Surg. 2017, 265, 597–603. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Yu, W.K.; Lin, Z.L.; Tan, S.J.; Bai, X.W.; Ding, K.; Li, N. Chemical sympathectomy attenuates inflammation, glycocalyx shedding and coagulation disorders in ratswithacutetraumaticcoagulopathy. Blood Coagul. Fibrinolysis 2015, 26, 152–160. [Google Scholar] [CrossRef]
- Paydar, S.; Dalfardi, B.; Shayan, Z.; Shayan, L.; Saem, J.; Bolandparvaz, S. Early Predictive Factors of Hypofibrinogenemia in Acute Trauma Patients. J. Emerg. Trauma Shock. 2018, 11, 38–41. [Google Scholar] [CrossRef] [PubMed]
- McQuilten, Z.K.; Wood, E.M.; Bailey, M.; Cameron, P.A.; Cooper, D.J. Fibrinogen is an independent predictor of mortality in major traumapatients:a five-year statewidecohortstudy. Injury 2017, 48, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, T.; Kitamura, T.; Tanaka, K.; Saisaka, Y.; Ishihara, J.; Onishi, H.; Nojima, T.; Yamamoto, K.; Matusmoto, T.; Tokioka, T. Admission fibrinogen levels in severe trauma patients: A comparison of elderly and younger patients. Injury 2015, 46, 1779–1783. [Google Scholar] [CrossRef] [PubMed]
- Wohlauer, M.V.; Moore, E.E.; Thomas, S.; Sauaia, A.; Evans, E.; Harr, J.; Silliman, C.C.; Ploplis, V.; Castellino, F.J.; Walsh, M. Early platelet dysfunction: An unrecognized role in the acute coagulopathy of trauma. J. Am. Coll. Surg. 2012, 214, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutcher, M.E.; Redick, B.J.; McCreery, R.C.; Crane, I.M.; Greenberg, M.D.; Cachola, L.M.; Nelson, M.F.; Cohen, M.J. Characterization of plateletdysfunctionaftertrauma. J. Trauma Acute Care Surg. 2012, 73, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, M.T.; Fabian, T.C.; Shahan, C.P.; Sharpe, J.P.; Mabry, S.E.; Weinberg, J.A.; Croce, M.A.; Jennings, L.K. A prospective study of platelet function in trauma patients. J. Trauma Acute Care Surg. 2016, 80, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Sirajuddin, S.; Valdez, C.; DePalma, L.; Maluso, P.J.; Singhal, R.; Schroeder, M.; Sarani, B. Inhibition of platelet function is common following even minor injury. J. Trauma Acute Care Surg. 2016, 81, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Schnuriger, B.; Inaba, K.; Abdelsayed, G.A.; Lustenberger, T.; Eberle, B.M.; Barmparas, G.; Talving, P.; Demetriades, D. The impact of platelets on the progression of traumatic intracranial hemorrhage. J. Trauma 2010, 68, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.R.; Lindell, A.L.; Stansbury, L.G.; Dutton, R.P.; Scalea, T.M. The prevalence of abnormal results of conventional coagulation tests on admission to a trauma center. Transfusion 2009, 49, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.M.; Call, M.S.; Knudson, M.M.; Cohen, M.J.; Trauma Outcomes Group. A normal platelet count may not be enough: The impact of admission platelet count on mortality and transfusion in severely injured trauma patients. J. Trauma 2011, 71 (Suppl. 3), S337–S342. [Google Scholar] [CrossRef] [Green Version]
- Floccard, B.; Rugeri, L.; Faure, A.; Saint Denis, M.; Boyle, E.M.; Peguet, O.; Levrat, A.; Guillaume, C.; Marcotte, G.; Vulliez, A.; et al. Early coagulopathy in trauma patients: An on-scene and hospital admission study. Injury 2012, 43, 26–32. [Google Scholar] [CrossRef]
- Van Beek, J.G.; Mushkudiani, N.A.; Steyerberg, E.W.; Butcher, I.; McHugh, G.S.; Lu, J.; Marmarou, A.; Murray, G.D.; Maas, A.I.R. Prognostic value of admission laboratory parameters in traumatic brain injury: Results from the IMPACT study. J. Neurotrauma. 2007, 24, 315–328. [Google Scholar] [CrossRef]
- Szentkereszty, Z. Az akut traumás vérzés és véralvadási zavar korszerű ellátása [Up-to-date management of acute traumatic bleeding and coagulopathy]. Orv. Hetil. 2020, 161, 1599–1605. [Google Scholar] [CrossRef]
- Cole, E.; Weaver, A.; Gall, L.; West, A.; Nevin, D.; Tallach, R.; O’Neill, B.; Lahiri, S.; Allard, S.; Tai, N.; et al. A decade of damage control resuscitation: New transfusion practice, new survivors, new directions. Ann. Surg. 2019. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y.; Rosendaal, F.R.; Reifman, J. Computational analysis of the effects of reduced temperature on thrombin generation: The contributions of hypothermia to coagulopathy. Anesth. Analg. 2013, 117, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.H.; Wolberg, A.S.; Monroe, D.M.I.I.I.; Hoffman, M. The effect of temperature and pH on the activity of factor VIIa: Implications forthe efficacy of high-dose factor VIIa in hypothermic and acidotic patients. J. Trauma 2003, 55, 886–891. [Google Scholar] [CrossRef]
- Engström, M.; Schött, U.; Romner, B.; Reinstrup, P. Acidosis impairs the coagulation: A thromboelastographic study. J. Trauma 2006, 61, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Martini, W.Z.; Pusateri, A.E.; Uscilowicz, J.M.; Delgado, A.V.; Holcomb, J.B. Independent contributions of hypothermia and acidosis to coagulopathyin swine. J. Trauma 2005, 58, 1002–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, W.Z.; Holcomb, J.B. Acidosis and coagulopathy: The differential effects on fibrinogen synthesis and breakdown in pigs. Ann. Surg. 2007, 246, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Martini, W.Z.; Dubick, M.A.; Pusateri, A.E.; Park, M.S.; Ryan, K.L.; Holcomb, J.B. Does bicarbonate correct coagulation function impaired by acidosis in swine? J. Trauma 2006, 61, 99–106. [Google Scholar] [CrossRef]
- Shenkman, B.; Budnik, I.; Einav, Y.; Hauschner, H.; Andrejchin, M.; Martinowitz, U. Model of trauma-induced coagulopathy including hemodilution, fibrinolysis, acidosis, and hypothermia: Impact on blood coagulation and platelet function. J. Trauma Acute Care Surg. 2017, 82, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, A.S.; Meng, Z.H.; Monroe, D.M.I.I.I.; Hoffman, M. A systematic evaluation of the effect of temperature on coagulation enzyme activity and platelet function. J. Trauma 2004, 56, 1221–1228. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y.; Szlam, F.; Sniecinski, R.M.; Levy, J.H.; Reifman, J. Controlled Multifactorial Coagulopathy: Effects of Dilution, Hypothermia, and Acidosis on Thrombin Generation in Vitro. Anesth Analg. 2020, 130, 1063–1076. [Google Scholar] [CrossRef]
- Martini, W.Z. Coagulopathy by hypothermia and acidosis: Mechanisms of thrombin generation and fibrinogen availability. J. Trauma 2009, 67, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Robertis, E.; Kozek-Langenecker, S.A.; Tufano, R.; Romano, G.M.; Piazza, O.; Zito Marinosci, G. Coagulopathy induced by acidosis, hypothermia and hypocalcaemia in severe bleeding. Minerva Anestesiol. 2015, 81, 65–75. [Google Scholar] [PubMed]
- Brohi, K.; Cohen, M.J.; Ganter, M.T.; Schultz, M.J.; Levi, M.; Mackersie, R.C.; Pittet, J.F. Acute coagulopathy of trauma: Hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J. Trauma 2008, 64, 1211–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, J.O.; Scarpelini, S.; Pinto, R.; Tien, H.C.; Callum, J.; Rizoli, S.B. Hypoperfusion in severely injured trauma patients is associated with reduced coagulation factor activity. J. Trauma 2011, 71, S435–S440. [Google Scholar] [CrossRef]
- Lechleuthner, A.; Lefering, R.; Bouillon, B.; Lentke, E.; Vorweg, M.; Tiling, T. Prehospital detection of uncontrolled haemorrhage in blunt trauma. Eur. J. Emerg. Med. 1994, 1, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Gando, S.; Sawamura, A.; Hayakawa, M. Trauma, Shock and disseminated intravascular coagulation: Lessons from the classical literature. Ann. Surg. 2011, 254, 10–19. [Google Scholar] [CrossRef]
- Adrie, C.; Laurent, I.; Monchi, M.; Cariou, A.; Dhainaou, J.F.; Spaulding, C. Post resuscitation disease after cardiac arrest: A sepsis-like syndrome? Curr. Opin. Crit. Care 2004, 10, 208–212. [Google Scholar] [CrossRef]
- Johansson, P.I.; Ostrowski, S.R. Acute coagulopathy of trauma: Balancing progressive catecholamine induced endothelial activation and damage by fluid phase anticoagulation. Med. Hypotheses 2010, 75, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Neumar, R.W.; Nolan, J.P.; Adrie, C.; Aibiki, M.; Berg, R.A.; Bottiger, B.W.; Callaway, C.; Clark, R.S.; Geocadin, R.G.; Jauch, E.C.; et al. Post-cardiac arrest syndrome: Epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation 2008, 118, 2452–2483. [Google Scholar] [PubMed] [Green Version]
- Opal, S.M.; van der Poll, T. Endothelial barrier dysfunction in septic shock. J. Intern. Med. 2015, 277, 277–293. [Google Scholar] [CrossRef] [Green Version]
- Holcomb, J.B. A novel and potentially unifying mechanism for shock induced early coagulopathy. Ann. Surg. 2011, 254, 201–202. [Google Scholar] [CrossRef]
- Cohen, J.; Vincent, J.L.; Adhikari, N.K.; Machado, F.R.; Angus, D.C.; Calandra, T.; Jaton, K.; Giulieri, S.; Delaloye, J.; Opal, S.; et al. Sepsis: A roadmap for future research. Lancet Infect Dis. 2015, 15, 581–614. [Google Scholar] [CrossRef]
- Adrie, C.; Adib-Conquy, M.; Laurent, I.; Monchi, M.; Vinsonneau, C.; Fitting, C.; Fraisse, F.; Dinh-Xuan, A.T.; Carli, P.; Spaulding, C.; et al. Successful cardiopulmonary resuscitation after cardiac arrest as a “sepsis-like” syndrome. Circulation 2002, 106, 562–568. [Google Scholar] [CrossRef]
- Xiao, W.; Mindrinos, M.N.; Seok, J.; Cuschieri, J.; Cuenca, A.G.; Gao, H.; Hayden, D.L.; Hennessy, L.; Moore, E.E.; Minei, J.P.; et al. A genomic storm in critically injured humans. J. Exp. Med. 2011, 208, 2581–2590. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Dong, J.F. Coagulopathy induced by traumatic brain injury: Systemic manifestation of a localized injury. Blood 2018, 131, 2001–2006. [Google Scholar] [CrossRef]
- Johansson, P.; Stensballe, J.; Ostrowski, S. Shock induced endotheliopathy (SHINE) in acute critical illness—A unifying pathophysiologic mechanism. Crit. Care 2017, 21, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, S.C.; Smith, D.H. Coagulopathy in traumatic brain injury. Neurocrit. Care 2004, 1, 479–488. [Google Scholar] [CrossRef]
- Mitra, B.; Cameron, P.A.; Mori, A.; Fitzgerald, M. Acute coagulopathy and early deaths post major trauma. Injury 2012, 43, 22–25. [Google Scholar] [CrossRef]
- Baskett, P.J. Recommendations for uniform reporting of data following major trauma--the Utstein style. A report of a working party of the International Trauma Anaesthesia and Critical Care Society (ITACCS). Resuscitation 1999, 42, 81–100. [Google Scholar] [CrossRef]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Horng, S.; Therattil, A.; Moyon, S.; Gordon, A.; Kim, K.; Argaw, A.T.; Hara, Y.; Mariani, J.N.; Sawai, S.; Flodby, P.; et al. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J. Clin. Investig. 2017, 127, 3136–3151. [Google Scholar] [CrossRef]
- Cristante, E.; McArthur, S.; Mauro, C.; Maggioli, E.; Romero, I.A.; Wylezinska-Arridge, M.; Couraud, P.O.; Lopez-Tremoleda, J.; Christian, H.C.; Weksler, B.B.; et al. Identification of an essential endogenous regulator of blood-brain barrier integrity, and its pathological and therapeutic implications. Proc. Natl. Acad. Sci. USA 2013, 110, 832–841. [Google Scholar] [CrossRef] [Green Version]
- Haseloff, R.F.; Dithmer, S.; Winkler, L.; Wolburg, H.; Blasig, I.E. Transmembrane proteins of the tight junctions at the blood-brain barrier: Structural and functional aspects. Semin Cell Dev. Biol. 2015, 38, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Wójciak-Stothard, B.; Potempa, S.; Eichholtz, T.; Ridley, A.J. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J. Cell Sci. 2001, 114, 1343–1355. [Google Scholar] [PubMed]
- Tsukita, S.; Furuse, M. The structure and function of claudins, cell adhesion molecules at tight junctions. Ann. N Y Acad. Sci. 2000, 915, 129–135. [Google Scholar] [CrossRef]
- Maegele, M. Coagulopathy after traumatic brain injury: Incidence, pathogenesis, and treatment options. Transfusion 2013, 53 (Suppl. 1), 28S–37S. [Google Scholar] [CrossRef] [PubMed]
- Lustenberger, T.; Talving, P.; Kobayashi, L.; Inaba, K.; Lam, L.; Plurad, D.; Demetriades, D. Time course of coagulopathy in isolated severe traumatic brain injury. Injury 2010, 41, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Nakae, R.; Takayama, Y.; Kuwamoto, K.; Naoe, Y.; Sato, H.; Yokota, H. Time course of coagulation and fibrinolytic parameters in patients with traumatic brain injury. J. Neurotrauma 2016, 33, 688–695. [Google Scholar] [CrossRef]
- Aurrand-Lions, M.; Johnson-Leger, C.; Wong, C.; Du Pasquier, L.; Imhof, B.A. Heterogeneity of endothelial junctions is reflected by differential expression and specific subcellular localization of the three JAM family members. Blood 2001, 98, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Fleck, R.A.; Rao, L.V.; Rapaport, S.I.; Varki, N. Localization of human tissue factor antigen by immunostaining with monospecific, polyclonal anti-human tissue factor antibody. Thromb. Res. 1990, 59, 421–437. [Google Scholar] [CrossRef]
- Eddleston, M.; de la Torre, J.C.; Oldstone, M.B.; Loskutoff, D.J.; Edgington, T.S.; Mackman, N. Astrocytes are the primary source of tissue factor in the murine central nervous system. A role for astrocytes in cerebral hemostasis. J Clin Investig. 1993, 92, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karri, J.; Cardenas, J.C.; Matijevic, N.; Wang, Y.W.; Choi, S.; Zhu, L.; Cotton, B.A.; Kitagawa, R.; Holcomb, J.B.; Wade, C.E. Early fibrinolysis associated with hemorrhagic progression following traumatic brain injury. Shock 2017, 48, 644–650. [Google Scholar] [CrossRef]
- Hijazi, N.; Abu Fanne, R.; Abramovitch, R.; Yarovoi, S.; Higazi, M.; Abdeen, S.; Basheer, M.; Maraga, E.; Cines, D.B.; Higazi, A.A.R. Endogenous plasminogen activators mediate progressive intracerebral hemorrhage after traumatic brain injury in mice. Blood 2015, 125, 2558–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Darlington, D.N.; Cap, A.P. Procoagulant and fibrinolytic activity after polytrauma in rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R323–R329. [Google Scholar] [CrossRef] [PubMed]
- Ploplis, V.A.; Donahue, D.L.; Sandoval-Cooper, M.J.; MorenoCaffaro, M.; Sheets, P.; Thomas, S.G.; Walsh, M.; Castellino, F.J. Systemic platelet dysfunction is the result of local dysregulated coagulation and platelet activation in the brain in a rat model of isolated traumatic brain injury. J. Neurotrauma 2014, 31, 1672–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prodan, C.I.; Vincent, A.S.; Dale, G.L. Coated-platelet levels increase with number of injuries in patients with mild traumatic brain injury. J. Neurotrauma 2016, 33, 818–824. [Google Scholar] [CrossRef]
- Savioli, G.; Ceresa, I.F.; Ciceri, L.; Sciutti, F.; Belliato, M.; Iotti, G.A.; Luzzi, S.; Del Maestro, M.; Mezzini, G.; Lafe, E.; et al. Mild head trauma in elderly patients: Experience of an emergency department. Heliyon 2020, 7, e04226. [Google Scholar] [CrossRef]
- Morel, N.; Morel, O.; Petit, L.; Hugel, B.; Cochard, J.F.; Freyssinet, J.M.; Sztark, F.; Dabadie, P. Generation of procoagulant microparticles in cerebrospinal fluid and peripheral blood after traumatic brain injury. J. Trauma 2008, 64, 698–704. [Google Scholar] [CrossRef]
- Broekhuizen, L.N.; Mooij, H.L.; Kastelein, J.J.; Stroes, E.S.; Vink, H.; Nieuwdorp, M. Endothelial glycocalyx as potential diagnostic and therapeutic target in cardiovascular disease. Curr. Opin. Lipidol. 2009, 20, 57–62. [Google Scholar] [CrossRef]
- Johansson, P.I.; Sørensen, A.M.; Perner, A.; Welling, K.L.; Wanscher, M.; Larsen, C.F.; Ostrowski, S.R. Elderly trauma patients have high circulating noradrenaline levels but attenuated release of adrenaline, platelets and leukocytes in response to increasing injury severity. Crit. Care Med. 2012, 40, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.R.; Pedersen, S.H.; Jensen, J.S.; Mogelvang, R.; Johansson, P.I. Acute myocardial infarction is associated with endothelial glycocalyx and cell damage and a parallel increase in circulating catecholamines. Crit. Care 2013, 17, R32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, M.A.; Differding, J.; Thorborg, P.; Mayberry, J.C.; Mullins, R.J. Hypercoagulability is most prevalent early after injury and in female patients. J. Trauma 2005, 58, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, M.; Lefering, R.; Probst, C.; Paffrath, T.; Schneider, M.M.; Maegele, M.; Sakka, S.G.; Bouillon, B.; Wafaisade, A. Epidemiology and risk factors of multipleorgan failure after multiple trauma: An analysis of 31,154 patients from the TraumaRegister DGU. J. Trauma Acute Care Surg. 2014, 76, 921–928. [Google Scholar] [CrossRef]
- Savioli, G.; Ceresa, I.F.; Luzzi, S.; Gragnaniello, C.; Giotta Lucifero, A.; Del Maestro, M.; Marasco, S.; Manzoni, F.; Ciceri, L.; Gelfi, E.; et al. Rates of Intracranial Hemorrhage in Mild Head Trauma Patients Presenting to Emergency Department and Their Management: A Comparison of Direct Oral Anticoagulant Drugs with Vitamin K Antagonists. Medicina 2020, 56, 308. [Google Scholar] [CrossRef]
- Hess, J.R.; Lawson, J.H. The coagulopathy of trauma versus disseminated intravascular coagulation. J. Trauma 2006, 60, S12–S19. [Google Scholar] [CrossRef]
- Spahn, D.R.; Rossaint, R. Coagulopathy and blood component transfusion in trauma. Br. J. Anaesth. 2005, 95, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussmann, B.; Lefering, R.; Waydhas, C.; Touma, A.; Kauther, M.D.; Ruchholtz, S.; Lendemans, S. Does increased prehospital replacement volume lead to a poor clinical course and an increased mortality? A matched-pair analysis of 1896 patients of the Trauma Registry of the German Society for Trauma Surgery who were managed by an emergency doctor at the accident site. Injury 2013, 44, 611–617. [Google Scholar]
- Palmeri, D.; van Zante, A.; Huang, C.C.; Hemmerich, S.; Rosen, S.D. Vascular endothelial junction-associated molecule, a novel member of the immunoglobulin superfamily, is localized to intercellular boundaries of endothelial cells. J. Biol. Chem. 2000, 275, 19139–19145. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Salsbery, B.; Wang, M.; Yuan, H.; Yang, J.; Zhao, Z.; Wu, X.; Zhang, Y.; Konkle, B.A.; Thiagarajan, P.; et al. Brain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injury. Blood 2015, 125, 2151–2159. [Google Scholar] [CrossRef] [Green Version]
- Keskpaik, T.; Starkopf, J.; Kirsimägi, Ü.; Mihnovitš, V.; Lomp, A.; Raamat, E.M.; Saar, S.; Talving, P. The role of elevated high-sensitivity cardiac troponin on outcomes following severe blunt chest trauma. Injury 2020, 51, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Kalbitz, M.; Pressmar, J.; Stecher, J.; Weber, B.; Weiss, M.; Schwarz, S.; Miltner, E.; Gebhard, F.; Huber-Lang, M. The Role of Troponin in Blunt Cardiac Injury After Multiple Trauma in Humans. World J Surg. 2017, 41, 162–169. [Google Scholar] [CrossRef] [PubMed]
- McCully, B.H.; Connelly, C.R.; Fair, K.A.; Holcomb, J.B.; Fox, E.E.; Wade, C.E.; Bulger, E.M.; Schreiber, M.A.; del Junco, D.J.; Matijevic, N.; et al. Onset of coagulation function recovery is delayed in severely injured trauma patients with venous thromboembolism. J. Am. Coll. Surg. 2017, 225, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Tompkins, R.G. Genomics of injury: The glue grant experience. J. Trauma Acute Care Surg. 2015, 78, 671–686. [Google Scholar] [CrossRef] [Green Version]
- Lord, J.M.; Midwinter, M.J.; Chen, Y.F.; Belli, A.; Brohi, K.; Kovacs, E.J.; Konderman, L.; Kubes, P.; Lilford, R.J. The systemic immune response to trauma: An overview of pathophysiology and treatment. Lancet 2014, 384, 1455–1465. [Google Scholar] [CrossRef] [Green Version]
- Bortolotti, P.; Faure, E.; Kipnis, E. Inflammasomes in tissue damages and immune disorders after trauma. Front. Immunol. 2018, 9, 1900. [Google Scholar] [CrossRef] [Green Version]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Minei, J.P.; Cuschieri, J.; Sperry, J.; Moore, E.E.; West, M.A.; Harbrecht, B.G.; O’Keefe, G.E.; Cohen, M.J.; Moldawer, L.L.; Tompkins, R.G.; et al. The changing pattern and implications of multiple organ failure after blunt injury with hemorrhagic shock. Crit. Care Med. 2012, 40, 1129–1135. [Google Scholar] [CrossRef] [Green Version]
- Levi, M.; van der Poll, T. Coagulation and sepsis. Thromb. Res. 2017, 149, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Dhainaut, J.F.; Yan, S.B.; Joyce, D.E.; Pettilä, V.; Basson, B.; Brandt, J.T.; Sundin, D.P.; Levi, M. Treatment effects of drotrecogin alfa (activated) in patients with severe sepsis with or without overt disseminated intravascular coagulation. J. Thromb. Haemost. 2004, 2, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Van Haren, R.M.; Valle, E.J.; Thorson, C.M.; Jouria, J.M.; Busko, A.M.; Guarch, G.A.; Namias, N.; Livingstone, A.S.; Proctor, K.G. Hypercoagulability and other risk factors in traumaintensive care unit patients with venous thromboembolism. J. Trauma Acute Care Surg. 2014, 76, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.R.; Espina, C.; Guedj, T.; Buaron, R.; Harrois, A.; Figueiredo, S.; Duranteau, J. High level of venous thromboembolism in critically ill trauma patients despite early and well-driven thromboprophylaxis protocol. Ann. Intensive Care 2017, 7, 97. [Google Scholar] [CrossRef]
- Skrifvars, M.B.; Bailey, M.; Presneill, J.; French, C.; Nichol, A.; Little, L.; Duranteau, J.; Huet, O.; Haddad, S.; Arabi, Y.; et al. Venous thromboembolic events in critically ill traumatic brain injury patients. Intensive Care Med. 2017, 43, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Van Gent, J.M.; Calvo, R.Y.; Zander, A.L.; Olson, E.J.; Sise, C.B.; Sise, M.J.; Shackford, S.R. Risk factors for deep vein thrombosis and pulmonary embolism after traumatic injury: A competing risks analysis. J. Trauma Acute Care Surg. 2017, 83, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Sumislawski, J.J.; Kornblith, L.Z.; Conroy, A.S.; Callcut, R.A.; Cohen, M.J. Dynamic coagulability after injury: Is delaying venous thromboembolism chemoprophylaxis worth the wait? J. Trauma Acute Care Surg. 2018, 85, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Mutschler, M.; Nienaber, U.; Brockamp, T.; Wafaisade, A.; Wyen, H.; Peiniger, S.; Paffrath, T.; Bouillon, B.; Maegele, M. TraumaRegister DGU A critical reappraisal of the ATLS classification of hypovolaemic shock: Does it really reflect clinical reality? Resuscitation 2013, 84, 309–313. [Google Scholar] [CrossRef]
- Rugeri, L.; Levrat, A.; David, J.S.; Delecroix, E.; Floccard, B.; Gros, A.; Allaouchiche, B.; Negrier, C. Diagnosis of early coagulation abnormalities in trauma patients by rotation thrombelastography. J. Thromb. Haemost. 2007, 5, 289–295. [Google Scholar] [CrossRef]
- Mutschler, M.; Paffrath, T.; Wolfl, C.; Probst, C.; Nienaber, U.; Schipper, I.B.; Bouillon, B.; Maegele, M. The ATLS((R)) classification of hypovolaemic shock: A well established teaching tool on the edge? Injury 2014, 45 (Suppl. 3), S35–S38. [Google Scholar] [CrossRef]
- Rossaint, R.; Bouillon, B.; Cerny, V.; Coats, T.J.; Duranteau, J.; Fernández-Mondéjar, E.; Hunt, B.J.; Komadina, R.; Nardi, G.; Neugebauer, E. Management of bleeding following major trauma: An updated European guideline. Crit. Care 2010, 14, R52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, J.W.; Pittet, J.F.; Pierce, B. Trauma-induced coagulopathy. Curr. Anesthesiol. Rep. 2014, 4, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, E.; Moore, E.E.; Moore, H.B. Management of Trauma-Induced Coagulopathy with Thrombelastography. Crit. Care Clin. 2017, 33, 119–134. [Google Scholar] [CrossRef] [Green Version]
- Baksaas-Aasen, K.; Van Dieren, S.; Balvers, K.; Juffermans, N.P.; Næss, P.A.; Rourke, C.; Eaglestone, S.; Ostrowski, S.R.; Stensballe, J.; Stanworth, S.; et al. Data-driven Development of ROTEM and TEG Algorithms for the Management of Trauma Hemorrhage: A Prospective Observational Multicenter Study. Ann. Surg. 2019, 270, 1178–1185. [Google Scholar] [CrossRef]
- Gonzalez, E.; Moore, E.E.; Moore, H.B.; Chapman, M.P.; Chin, T.L.; Ghasabyan, A.; Wohlauer, M.V.; Barnett, C.C.; Bensard, D.D.; Biffl, W.L.; et al. Goal-directed Hemostatic Resuscitation of Trauma-induced Coagulopathy: A Pragmatic Randomized Clinical Trial Comparing a Viscoelastic Assay to Conventional Coagulation Assays. Ann. Surg. 2016, 263, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Wikkelsø, A.; Wetterslev, J.; Møller, A.M.; Afshari, A. Thromboelastography (TEG) or thromboelastometry (ROTEM) to monitor haemostatic treatment versus usual care in adults or children with bleeding. Cochrane Database Syst. Rev. 2016, 22, CD007871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maegele, M. The European Perspective on the Management of Acute Major Hemorrhage and Coagulopathy after Trauma: Summary of the 2019 Updated European Guideline. J. Clin. Med. 2021, 10, 362. [Google Scholar] [CrossRef]
- Savioli, G.; Ceresa, I.F.; Maggioni, P.; Lava, M.; Ricevuti, G.; Manzoni, F.; Oddone, E.; Bressan, M.A. Impact of ED Organization with a Holding Area and a Dedicated Team on the Adherence to International Guidelines for Patients with Acute Pulmonary Embolism: Experience of an Emergency Department Organized in Areas of Intensity of Care. Medicines 2020, 7, 60. [Google Scholar] [CrossRef]
- Savioli, G.; Ceresa, I.F.; Manzoni, F.; Ricevuti, G.; Bressan, M.A.; Oddone, E. Role of a Brief Intensive Observation Area with a Dedicated Team of Doctors in the Management of Acute Heart Failure Patients: A Retrospective Observational Study. Medicina 2020, 56, 251. [Google Scholar] [CrossRef]
- Ceresa, I.F.; Savioli, G.; Angeli, V.; Novelli, V.; Muzzi, A.; Grugnetti, G.; Cobianchi, L.; Manzoni, F.; Klersy, C.; Lago, P.; et al. Preparing for the Maximum Emergency with a Simulation: A Table-Top Test to Evaluate Bed Surge Capacity and Staff Compliance with Training. Open Access Emerg Med. 2020, 12, 377–387. [Google Scholar] [CrossRef]
- Savioli, G.; Ceresa, I.F.; Novara, E.; Persiano, T.; Grulli, F.; Ricevuti, G.; Bressan, M.A.; Oddone, E. Brief Intensive Observation areas in the management of acute heart failure in elderly patients leading to high stabilisation rate and less admissions. J. Gerontol. Geriatr. 2021, in press. [Google Scholar]
- Neal, M.D.; Moore, E.E.; Walsh, M.; Thomas, S.; Callcut, R.A.; Kornblith, L.Z.; Schreiber, M.; Ekeh, A.P.; Singer, A.J.; Lottenberg, L.; et al. A comparison between the TEG 6s and TEG 5000 analyzers to assess coagulation in trauma patients. J. Trauma Acute Care Surg. 2020, 88, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Spahn, D.R. TEG(R)- or ROTEM(R)-based individualized goal-directed coagulation algorithms: Don’t wait–act now! Crit. Care 2014, 18, 637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenni, M.; Worn, M.; Bruesch, M.; Spahn, D.R.; Ganter, M.T. Successful rotational thromboelastometry-guided treatment of traumatic haemorrhage, hyperfibrinolysis and coagulopathy. Acta Anaesthesiol. Scand. 2010, 54, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashuk, J.L.; Moore, E.E.; Johnson, J.L.; Haenel, J.; Wilson, M.; Moore, J.B.; Cothren, C.C.; Biffl, W.L.; Banerjee, A.; Sauaia, A. Postinjury life threatening coagulopathy: Is 1:1 fresh frozen plasma:packed red blood cells the answer? J. Trauma 2008, 65, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Nienaber, U.; Innerhofer, P.; Westermann, I.; Schöchl, H.; Attal, R.; Breitkopf, R.; Maegele, M. The impact of fresh frozen plasma vs coagulation factor concentrates on morbidity and mortality in trauma-associated haemorrhage and massive transfusion. Injury 2011, 42, 697–701. [Google Scholar] [CrossRef]
- Riskin, D.J.; Tsai, T.C.; Riskin, L.; Hernandez-Boussard, T.; Purtill, M.; Maggio, P.M.; Spain, D.A.; Brundage, S.I. Massive transfusion protocols: The role of aggressive resuscitation versus product ratio in mortality reduction. J. Am. Coll. Surg. 2009, 209, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Schöchl, H.; Nienaber, U.; Hofer, G.; Voelckel, W.; Jambor, C.; Scharbert, G.; Kozek-Langenecker, S.; Solomon, C. Goal-directed coagulation management of major trauma patients using thromboelastometry (ROTEM)-guided administration of fibrinogen concentrate and prothrombin complex concentrate. Crit. Care 2010, 14, R55. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.F.; Gorlinger, K.; Meininger, D.; Herrmann, E.; Bingold, T.; Moritz, A.; Cohn, L.H.; Zacharowski, K. Point-of-care testing: A prospective, randomized clinical trial of efficacy in coagulopathic cardiac surgery patients. Anesthesiology 2012, 117, 531–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, Y.; Nakajima, Y.; Tanaka, K.A.; Sessler, D.I.; Maeda, S.; Iida, J.; Ogawa, S.; Mizobe, T. Thromboelastometry-guided intraoperative haemostatic management reduces bleeding and red cell transfusion after paediatric cardiac surgery. Br. J. Anaesth. 2015, 114, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karkouti, K.; McCluskey, S.A.; Callum, J.; Freedman, J.; Selby, R.; Timoumi, T.; Roy, D.; Rao, V. Evaluation of a novel transfusion algorithm employing point-of-care coagulation assays in cardiac surgery: A retrospective cohort study with interrupted time-series analysis. Anesthesiology 2015, 122, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Görlinger, K.; Dirkmann, D.; Hanke, A.A.; Kamler, M.; Kottenberg, E.; Thielmann, M.; Jakob, H.; Peters, J. First-line therapy with coagulation factor concentrates combined with point-of-care coagulation testing is associated with decreased allogeneic blood transfusion in cardiovascular surgery: A retrospective, single-center cohort study. Anesthesiology 2011, 115, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Theusinger, O.M.; Wanner, G.A.; Emmert, M.Y.; Billeter, A.; Eismon, J.; Seifert, B.; Simmen, H.P.; Spahn, D.R.; Baulig, W. Hyperfibrinolysis diagnosed by rotational thromboelastometry (ROTEM) is associated with higher mortality in patients with severe trauma. Anesth. Analg. 2011, 113, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Levrat, A.; Gros, A.; Rugeri, L.; Inaba, K.; Floccard, B.; Negrier, C.; David, J.S. Evaluation of rotation thrombelastography for the diagnosis of hyperfibrinolysis in trauma patients. Br. J. Anaesth. 2008, 100, 792–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Menyar, A.; Ramzee, A.F.; Asim, M.; Di Somma, S.; Al-Thani, H. Comparative analysis for the implication of serum cardiac troponin measurements by conventional versus high-sensitivity assays in patients with traumatic brain injury. Minerva Cardioangiol. 2020, 68, 27–33. [Google Scholar] [CrossRef] [PubMed]
- El-Menyar, A.; Asim, M.; Ramzee, A.F.; Nabir, S.; Ahmed, M.N.; Al-Thani, A.; Al-Abdulmalek, A.; Al-Thani, H. Bio-Shock Index: Proposal and Rationale for a New Predictive Tool for In-Hospital Mortality in Patients with Traumatic Brain Injury. World Neurosurg. 2019, 132, e169–e177. [Google Scholar] [CrossRef]
- Jackson, M.R.; Olson, D.W.; Beckett, W.C., Jr.; Olsen, S.B.; Robertson, F.M. Abdominal vascular trauma: A review of 106 injuries. Am. Surg. 1992, 58, 622–626. [Google Scholar]
- Rossaint, R.; Bouillon, B.; Cerny, V.; Coats, T.J.; Duranteau, J.; Fernández-Mondéjar, E.; Filipescu, D.; Hunt, B.J.; Komadina, R.; Nardi, G.; et al. The European guideline on management of major bleeding and coagulopathy following trauma: Fourth edition. Crit. Care 2016, 20, 100. [Google Scholar] [CrossRef]
- Johnson, J.W.; Gracias, V.H.; Schwab, C.W.; Reilly, P.M.; Kauder, D.R.; Shapiro, M.B.; Dabrowski, G.P.; Rotondo, M.F. Evolution in damage control for exsanguinating penetrating abdominal injury. J. Trauma 2001, 51, 261–269. [Google Scholar] [CrossRef]
- Billy, L.J.; Amato, J.J.; Rich, N.M. Aortic injuries in Vietnam. Surgery 1971, 70, 385–391. [Google Scholar]
- American College of Surgeons. Advanced Trauma Life Support (ATLS®), 10th ed.; American College of Surgeons: Chicago, IL, USA, 2018. [Google Scholar]
- Derakhshanfar, H.; Vafaei, A.; Tabatabaey, A.; Noori, S. Prevalence and Associated Factors of Acute Traumatic Coagulopathy; a Cross Sectional Study. Emergency 2017, 5, e58. [Google Scholar] [PubMed]
- Campanella, R.; Guarnaccia, L.; Cordiglieri, C.; Trombetta, E.; Caroli, M.; Carrabba, G.; La Verde, N.; Rampini, P.; Gaudino, C.; Costa, A.; et al. Tumor-Educated Platelets and Angiogenesis in Glioblastoma: Another Brick in the Wall for Novel Prognostic and Targetable Biomarkers, Changing the Vision from a Localized Tumor to a Systemic Pathology. Cells 2020, 9, 294. [Google Scholar] [CrossRef] [Green Version]
- Brohi, K. Trauma induced coagulopathy. J. R. Army Med. Corps 2009, 155, 320–322. [Google Scholar] [CrossRef]
- Johansson, P.I.; Sorensen, A.M.; Perner, A.; Welling, K.L.; Wanscher, M.; Larsen, C.F.; Ostrowski, S.R. Disseminated intravascular coagulation or acute coagulopathy of trauma shock early after trauma? An observational study. Crit. Care 2011, 15, R272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frith, D.; Brohi, K. The pathophysiology of trauma-induced coagulopathy. Curr. Opin. Crit. Care 2012, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Bocci, M.G.; Nardi, G.; Veronesi, G.; Rondinelli, M.B.; Palma, A.; Fiore, V.; De Candia, E.; Bianchi, M.; Maresca, M.; Barelli, R.; et al. Early coagulation support protocol: A valid approach in real-life management of major trauma patients. Results from two Italian centres. Injury 2019, 50, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Cianci, P.; Fersini, A.; Tartaglia, N.; Altamura, A.; Lizzi, V.; Stoppino, L.P.; Macarini, L.; Ambrosi, A.; Neri, V. Spleen assessment after laparoscopic transperitoneal left adrenalectomy: Preliminary results. Surg. Endosc. 2016, 30, 1503–1507. [Google Scholar] [CrossRef] [Green Version]
- Cianci, P.; Fersini, A.; Tartaglia, N.; Ambrosi, A.; Neri, V. Are there differences between the right and left laparoscopic adrenalectomy? Our experience. Ann. Ital. Chir. 2016, 87, 242–246. [Google Scholar] [PubMed]
- Cianci, P.; Tartaglia, N.; Altamura, A.; Di Lascia, A.; Fersini, A.; Neri, V.; Ambrosi, A. Cervical Esophagotomy for Foreign Body Extraction: A Case Report and Extensive Literature Review of the Last 20 Years. Am. J. Case Rep. 2018, 19, 400–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lascia, A.; Tartaglia, N.; Fersini, A.; Petruzzelli, F.; Ambrosi, A. Endoscopy for treating minor post-cholecystectomy biliary fistula A review of the literature. Ann. Ital. Chir. 2018, 89, 270–277. [Google Scholar] [PubMed]
- Tartaglia, N.; Petruzzelli, F.; Vovola, F.; Fersini, A.; Ambrosi, A. Antegrade cholecystectomy before ligating the elements. A technique that reduces complications. Ann. Ital. Chir. 2019, 90, 162–164. [Google Scholar] [PubMed]
- Thorn, S.; Güting, H.; Maegele, M.; Gruen, R.L.; Mitra, B. Early Identification of Acute Traumatic Coagulopathy Using Clinical Prediction Tools: A Systematic Review. Medicina 2019, 55, 653. [Google Scholar] [CrossRef] [Green Version]
- Curry, N.; Hopewell, S.; Doree, C.; Hyde, C.; Brohi, K.; Stanworth, S. The acute management of trauma hemorrhage: A systematic review of randomized controlled trials. Crit. Care 2011, 15, R92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spahn, D.R.; Bouillon, B.; Cerny, V.; Duranteau, J.; Filipescu, D.; Hunt, B.J.; Komadina, R.; Maegele, M.; Nardi, G.; Riddez, L.; et al. The European guideline on management of major bleeding and coagulopathy following trauma: Fifth edition. Crit. Care 2019, 23, 98. [Google Scholar] [CrossRef] [Green Version]
- Lucifero, A.G.; Luzzi, S.; Gragnaniello, C.; Savioli, G.; Tartaglia, N.; Ambrosi, A. Hand-assisted laparoscopic vs. mini-laparot omy technique for ventriculoperitoneal shunt. A meta-analysis of three thousand patients. Ann. Ital. Chir. 2020, 91, 1. [Google Scholar]
- Savioli, G.; Ceresa, I.F.; Macedonio, S.; Gerosa, S.; Belliato, M.; Luzzi, S.; Lucifero, A.G.; Manzoni, F.; Ricevuti, G.; Bressan, M.A. Major Trauma in Elderly Patients: Worse Mortality and Outcomes in An Italian Trauma Center. J. Emergencies Trauma Shock 2020, in press. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physiological Criteria Anatomical Criteria Dynamic Criteria |
|---|
| Ejection from a vehicle Penetrating head/neck/throat/abdomen/pelvic/armpit/groin trauma Systolic blood pressure < 90 mmHg |
| Motorcycle crash with separation of the rider Amputations above the wrist or ankle Respiratory distress or respiratory rate of <10 or >29 breaths/min |
| Died in the same vehicle Chest trauma with flap/flail chest State of consciousness (GCS < 13) |
| CRASH intrusion >30 cm at the patient area Neurological injury with paralysis of even a single limb |
| Fall from height >2 m Fractures of two or more long bones |
| Pedestrian thrown or run over or hit at a speed >10 km/h Suspected unstable fracture ring of pelvis: Suspected unstable fracture |
| High-energy impact |
| (Speed > 65 km/h) Open or depressed skull fracture |
| Vehicle crash Burn >20% of the body surface or airway/face |
| Extrication time > 20 min |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savioli, G.; Ceresa, I.F.; Caneva, L.; Gerosa, S.; Ricevuti, G. Trauma-Induced Coagulopathy: Overview of an Emerging Medical Problem from Pathophysiology to Outcomes. Medicines 2021, 8, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines8040016
Savioli G, Ceresa IF, Caneva L, Gerosa S, Ricevuti G. Trauma-Induced Coagulopathy: Overview of an Emerging Medical Problem from Pathophysiology to Outcomes. Medicines. 2021; 8(4):16. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines8040016
Chicago/Turabian StyleSavioli, Gabriele, Iride Francesca Ceresa, Luca Caneva, Sebastiano Gerosa, and Giovanni Ricevuti. 2021. "Trauma-Induced Coagulopathy: Overview of an Emerging Medical Problem from Pathophysiology to Outcomes" Medicines 8, no. 4: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines8040016