1. Introduction
Nuclear energy has been regarded as the most promising energy for the future, and its development has attracted significant attention. Uranium dioxide (UO
2) is the preferred fuel for light–water reactors (LWRs), and its properties play an important role in the reliability and safety of nuclear reactors. Over the past century, numerous experimental and theoretical studies have been conducted on the structure and properties of uranium dioxide (UO
2) under irradiation. Xenon (Xe) is the product of uranium fission during reactor operations [
1,
2]. Xe released into the helium-filled gap of wrapping materials can reduce the thermal conductivity of the fueled gap and increase the pressure of the ventilation system. In addition, Xe release can generate stress on the cladding, resulting in the mechanical degradation of structural materials in fuel components [
3,
4]. Fission gas also tends to accumulate and form bubbles in the interior of nuclear fuel, resulting in expansion, microstructural change, and performance degradation, which affect the safety of nuclear reactors [
5,
6,
7,
8]. To improve the physical, chemical, and thermodynamic properties of nuclear fuel in the ground state and reduce the release of fission gas during fuel irradiation, many dopants, such as niobium (Nb) [
9,
10], magnesium (Mg) [
11,
12], titanium (Ti) [
13,
14], chromium (Cr) [
15,
16] and zirconium (Zr) [
17,
18] have been studied in the past decades. Several studies have reported that Zr doping can improve the performance of UO
2 nuclear fuels. Zr doping can reduce the swelling rate of UO
2 fuel and improve its high-temperature water corrosion resistance [
19]. Ti doping can enhance the sinterability and grain size of UO
2 [
20]. Thorium (Th) doping can improve the antioxidant capacity of UO
2 and guarantee long-term storage [
21]. However, few studies have been conducted on the effects of Ti, Th, and Zr on Xe solubility in UO
2 fuel. Therefore, further research on the doping effect of these metals is required to effectively analyze nuclear fuel performance.
There is a lack of sufficient experimental data owing to the high cost of radioactive UO
2 fuel and related irradiation, and existing data are decades old. However, with the advancement of computers, using computational methods as an effective complement to experiments can enhance the understanding of fission gas behavior in UO
2-based nuclear materials. For instance, density-functional-theory (DFT)-based first-principles calculations [
22,
23] have been widely used in many atomic-scale simulations of UO
2 nuclear materials. In particular, the stable site occupation and diffusion behavior of fission gas Xe in UO
2 have been extensively studied. During the initial stages of the fuel life cycle, noble gas atoms can occupy the empty octahedral sites in the UO
2 lattice [
24]. However, as the fission reaction of UO
2 progresses and bombardment continues, Frenkel defects consisting of uranium and oxygen vacancies are formed in UO
2. Subsequently, these Frenkel defects form Schottky defects in the UO
2 lattice through diffusion and other methods [
25,
26]. The release of fission gas in UO
2 is characterized by point defects, dislocations, voids, and bubbles, which further leads to nuclear fuel swelling. Yu et al. [
27] studied the stable occupation of Xe in UO
2 using the DFT + U method and found that Xe was more likely to occupy the Schottky defect position. Nerikar et al. [
28] analyzed the stability of charged defects by considering various defect charge states. However, they found that the formation energies of the neutral complexes highly correlate with the experimental values than those of the charged complexes.
In this study, the UO2 model was validated using the DFT + U method, and the Ueff value is thus determined. Additionally, the occupation matrix control (OMCs) scheme was adopted to ensure that the subsequent calculations regarding the defect system can converge to the ground state. The effects of Th, Zr, and Ti on the dissolution and nucleation of Xe atoms in UO2 were also studied. The electron charge density diagram of the doped system was comprehensively analyzed, and the Xe aggregation behavior in uranium vacancies was studied.
2. Computational Details
The DFT calculations in this study were performed on VASP (the Vienna ab-initio simulation package) [
29]. The electron wave function was calculated using the projector-augmented-wave (PAW) method [
30,
31]. The generalized gradient approximation (GGA) [
32] describes the exchange-correlation potential between electrons in the Perdew–Burke-Ernzerhof form. The cutoff energy of the plane wave was 500 eV according to the convergence tests. The Brillouin zone was sampled using the Monkhorst–Pack [
33] method with a 4 × 4 × 4
k-point mesh. The distribution function of the integral in the inverted space was a Gaussian function, and the Gaussian smearing was set to 0.05 eV. Convergence was achieved when the total energies converged within 1 × 10
−5 eV and the Hellmann–Feynman forces on each ion were lower than 0.01 eVÅ
−1. Complete geometric optimization of the UO
2 supercells was allowed during relaxation, without any volume or symmetry restrictions. The relaxation of the magnetic moment was limited to collinear. Spin-orbit coupling was not considered because the expected effect on the results was negligible [
34].
UO
2 is a conventional Mott insulator [
35]; however, if the DFT-based plane-wave pseudopotential method with local density approximation (LDA) or GGA [
36,
37] is used, it will be predicted as a metal. Owing to the strong correlation effect between the 5f electrons of the U atom in UO
2, traditional LDA or GGA cannot adequately describe the electronic structure. Therefore, the DFT + U method is commonly used for correction calculation [
38]. Dudarev et al. [
39] and Yu et al. [
27] studied the basic structural properties of UO
2 using DFT + U, and the calculated results correlated well with the experimental data, indicating the accuracy of the method. Although point defects and defect clusters in bulk UO
2 and the stable occupation of Xe in UO
2 have been extensively studied using DFT + U, the results vary widely. This is mainly attributed to the existence of metastable states [
40]. OMC and U-ramping are employed to achieve accurate DFT + U calculations and ensure that the results converge to the ground state of the system. Solomon et al. [
41] found that the values calculated using the OMC method were lower than those obtained using the U-ramping method. Therefore, in this study, the occupation matrix of the U atom is initialized with the occupation matrix obtained in the UO
2 ground state in all subsequent calculations, and the entire effect related to the inert gas is captured in the subsequent unconstrained structural optimization.
In UO
2, Jahn–Teller distortion is directly related to the direction of the magnetic moment of the uranium atom. The experimental results show that UO
2 is a nonlinear 3
k AFM system, and the magnetic moment direction of the U atom is <111> [
42]. The 3
k AFM configuration is challenging to implement in the calculation; however, the existing results show that the 1
k AFM system can provide accurate results, and the calculation results are similar to those of the 3
k AFM system [
42]. Therefore, the 1
k AFM system was used to describe the magnetic order of UO
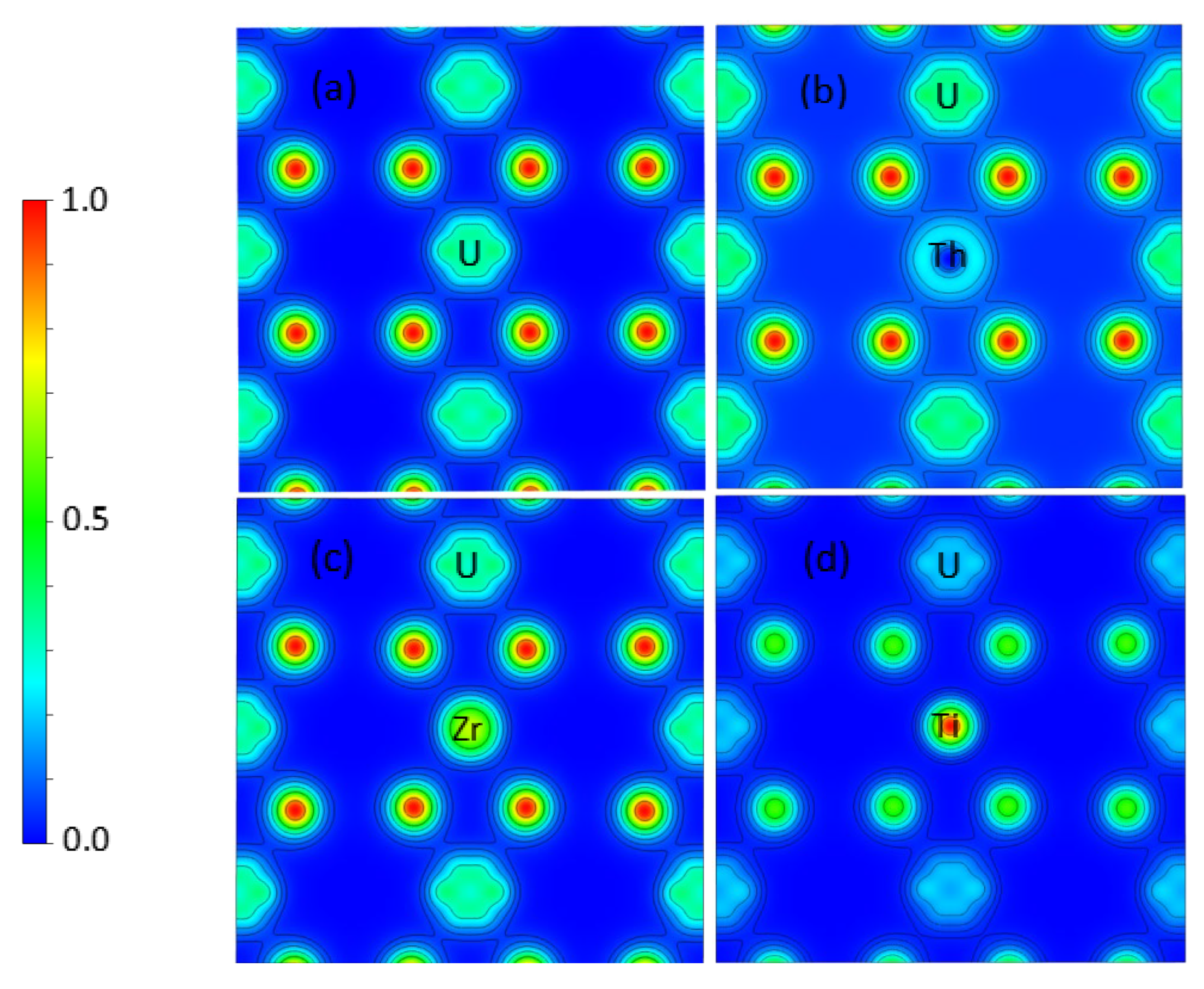
2 in this study. The defects were constructed based on a
UO
2 supercell, including 32 U atoms and 64 O atoms. There are many inherent defects in UO
2, including interstitial atoms, oxygen vacancies, uranium vacancies, divacancies (consisting of a uranium vacancy and the nearest oxygen vacancy), and Schottky defects (composed of one uranium vacancy and two nearest oxygen vacancies). There were three different configurations, as shown in
Figure 1. In the figure, the point vacancies and vacancy clusters are represented by Int, Vo, Vu, Di, SD1, SD2, and SD3.
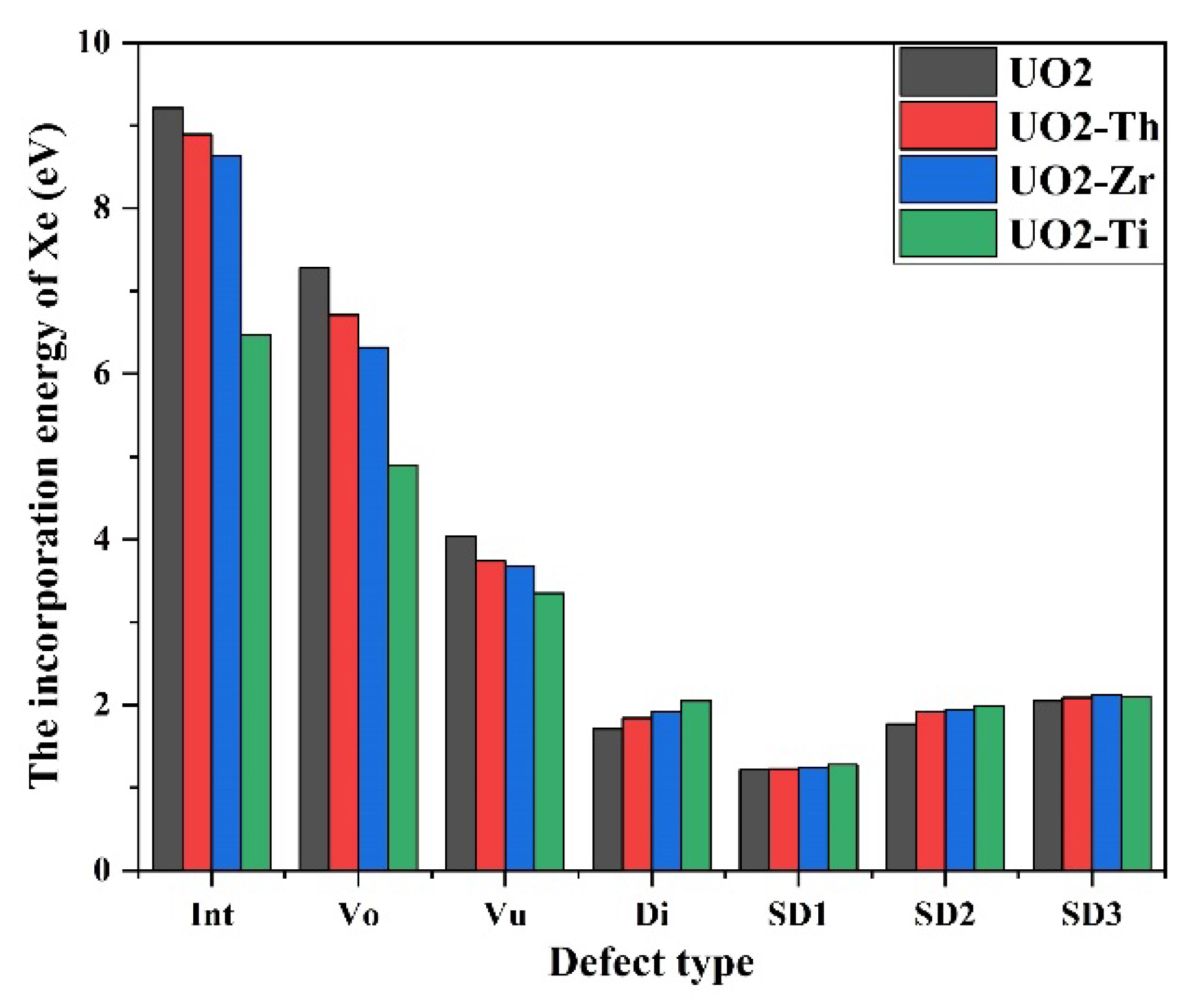
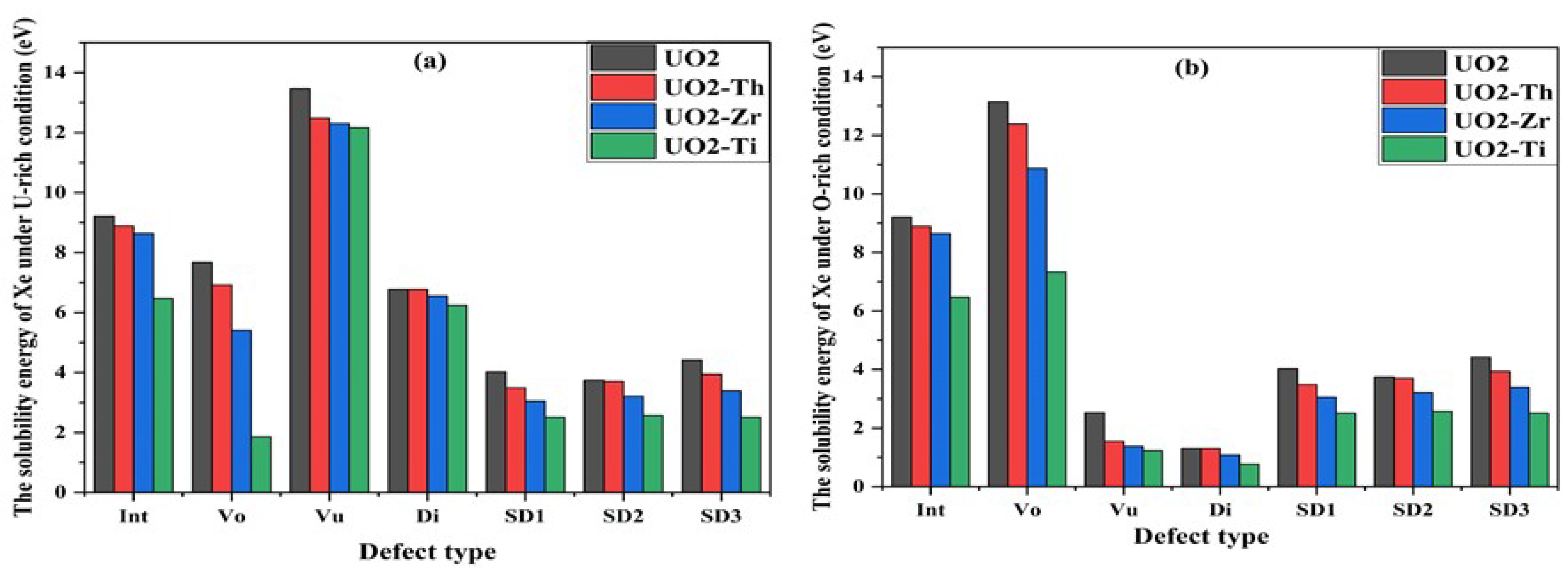
To investigate the interaction between dopants and Xe, the influence of dopants on the solution energy of Xe was evaluated. The solution energy is defined as the sum of the formation and incorporation energies, as first proposed by Grimes and Catlow [
43]. The incorporation energy (
) is defined as
where
is the total energy of the system after Xe is adsorbed on a vacancy defect,
is the total energy when there is a vacancy defect in the cell, and
is the chemical potential of an isolated Xe atom. The solution energy (
) of Xe in the cell is calculated as
where
is the solution energy required for Xe to dissolve into a vacancy defect.
and
are the numbers of U and O atoms missing from the defect-containing unit cell relative to the defect-free original unit cell, respectively,
and
are the chemical potentials of U and O atoms, respectively, and
is the energy of the original cell.
As the nuclear fuel reaction progresses, uranium atoms are continuously consumed, and the oxide with the lowest uranium chemical potential undergoes a phase transition from UO
2 to UO
3. This may imply that the gradual transition of UO
2 to a highly oxidized phase prompts a corresponding increase in the chemical potential of oxygen in uranium oxide. Therefore, the chemical potential of each atom must be carefully selected based on environmental conditions and possible phases to determine the energy associated with the UO
2 defect. The energy of the atomic chemical potential can be calculated using DFT as:
The chemical potential of uranium and oxygen, and the chemical potential of UO
2, can be expressed as:
is the total energy per unit formula. Equation (4) is applied at equilibrium. Thus, if one of the chemical potentials becomes critically low, the oxide would decompose into α-U and oxygen molecules, which are the reference states used for each element. Therefore, to maintain the oxide form, the range of chemical potentials is limited. When α-U begins to form as a result of decomposition, its chemical potential reaches a maximum, which is denoted as the U-rich limit, from the following expression:
DFT + U was used to calculate the total energy of and the value obtained was −8.48 eV. According to Formula (4), we can obtain = −10.41 eV under the U-rich condition.
Because the chemical potentials of U and O atoms are unknown, α-U and O
2 molecules are considered the reference states, respectively, and combined with
where the calculated
is −4.94 eV. Hence, we can obtain
= −19.41 eV under O-rich. To study the aggregation behavior of Xe atoms in vacancies, the binding energy (
) must be calculated.
is defined as
where
represents the energy of
n Xe atoms on uranium vacancies,
. represents the energy of a Xe atom at the interstitial site, and
represents the energy of
n + 1 Xe atoms on uranium vacancies.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}