Good Cop, Bad Cop: The Opposing Effects of Macrophage Activation State on Maintaining or Damaging Functional β-Cell Mass



Abstract
:1. Introduction
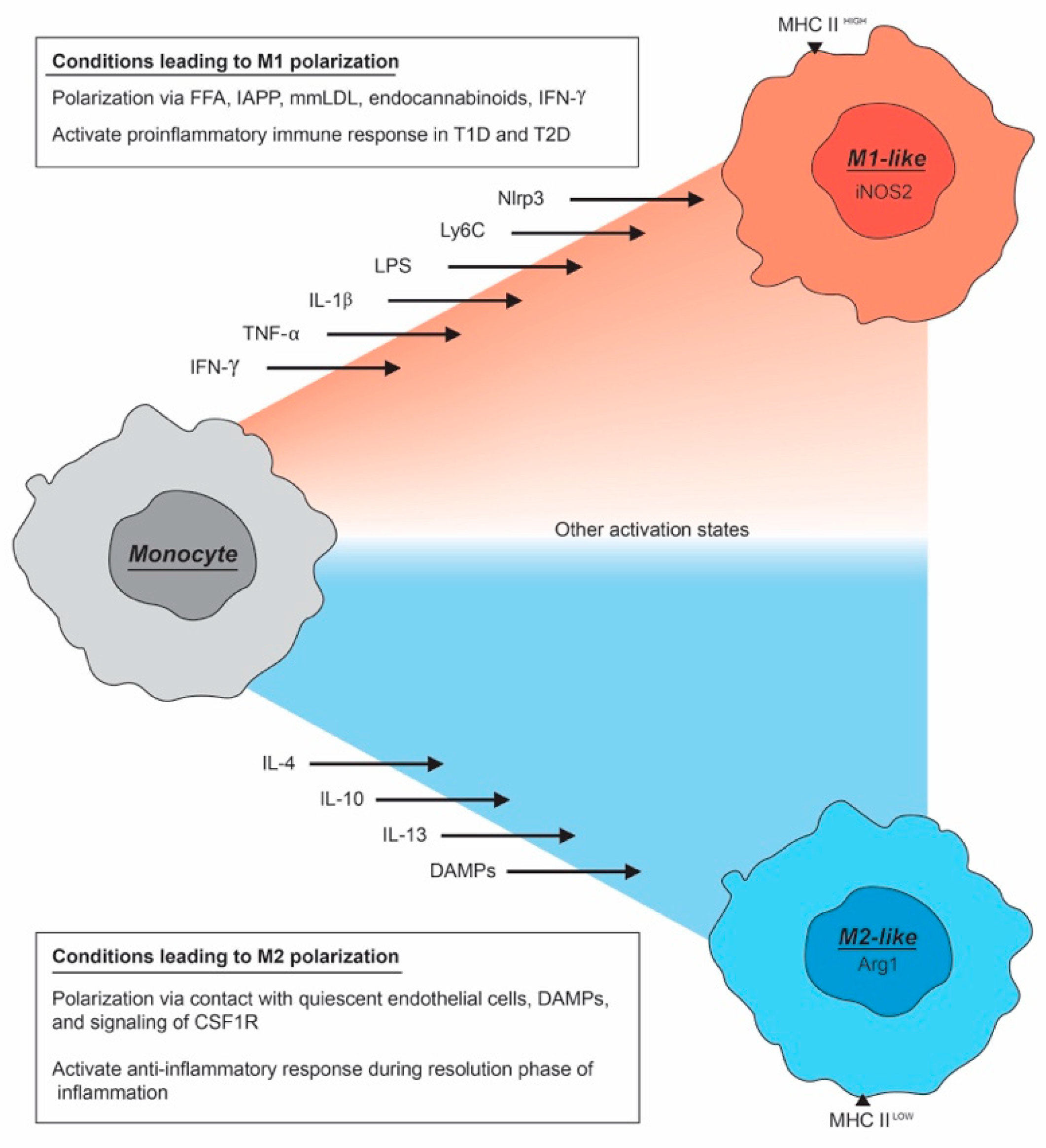
2. The Macrophage Activation Spectrum
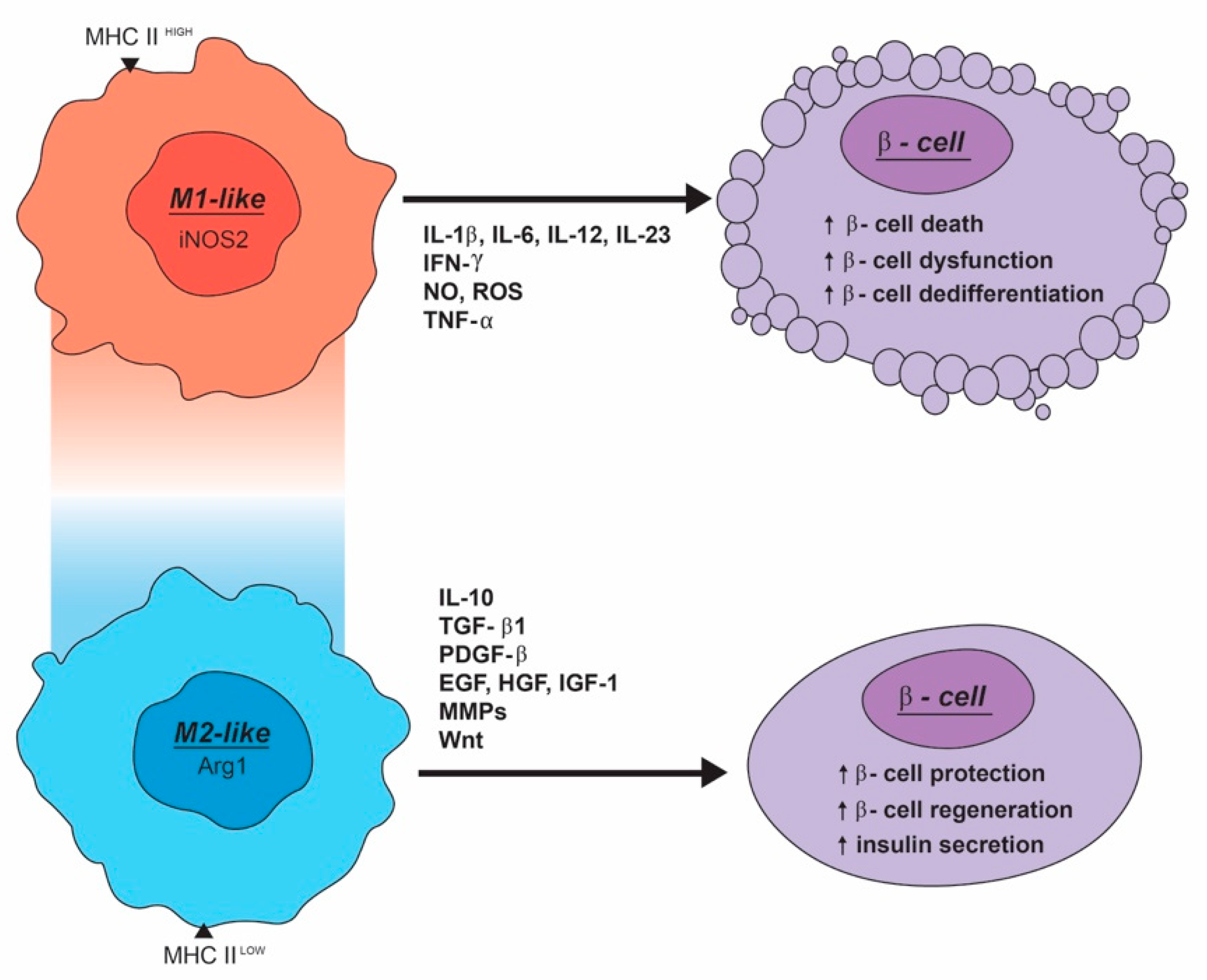
3. Macrophages Can Impair β-Cell Function and Survival
Macrophage Produced Secreted Factors That Negatively Modulate Functional β-Cell Mass
4. M2-Like Macrophage Can Enhance the Development, Maintenance, and Function of β-Cells
Macrophage Produced Secreted Factors That Positively Modulate Functional β-Cell Mass
5. Use of Macrophages to Improve Functional β-Cell Mass as a Treatment for Diabetes
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aldh1a2 | Aldehyde dehydrogenase 1 family, member A2 |
| ARG1 | Arginase 1 |
| ATF3 | Activating transcription factor 3 |
| ATF4 | Activating transcription factor 4 |
| ATP | Adenosine triphosphate |
| BET | Bromodomain and extraterminal domain family |
| BIP | Binding immunoglobulin protein |
| CCL17 | Chemokine (C-C motif) ligand 17 |
| CCL20 | Chemokine (C-C motif) ligand 20 |
| CCL24 | Chemokine (C-C motif) ligand 24 |
| CCL5 | Chemokine (C-C motif) ligand 5 |
| CHI3L1 | Chitinase 3 Like 1 |
| CHOP | CCAAT/enhancer binding protein (C/EBP) homologous protein |
| CSF-1 | Macrophage colony-stimulating factor |
| CSF-1R | Colony-stimulating 1 receptor |
| CSF-2 | Granulocyte-macrophage colony-stimulating factor |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 |
| CXCL10 | Chemokine (C-X-C motif) ligand 10 (see also IP-10) |
| CXCL2 | Chemokine (C-X-C motif) ligand 2 |
| CXCL8 | Chemokine (C-X-C motif) ligand 8 |
| CXCL9 | Chemokine (C-X-C motif) ligand 9 |
| CXCR2 | Chemokine (C-X-C motif) receptor 2 |
| Db/db | Diabetic mouse model |
| DNA | Deoxyribonucleic acid |
| DP-BB | BioBreeding Diabetes Prone rat |
| ECM | Extracellular matrix |
| EGF | Endothelial growth factor |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| Fas | Apoptosis antigen 1 |
| Foxo1 | Forkhead box protein O1 |
| GCSF | Granulocyte colony-stimulating factor |
| GK | Goto-Kakizaki rat |
| GSIS | Glucose-stimulated insulin secretion |
| HGF | Hepatocyte growth factor |
| I-BET151 | BET bromodomain inhibitor |
| IFN-γ | Interferon-gamma |
| IFNGR | Interferon gamma receptor |
| IGF | Insulin-like growth factor |
| IGF-1 | Insulin-like growth factor 1 |
| IL-1 | Interleukin-1 |
| IL-12 | Interleukin-12 |
| IL-13 | Interleukin-13 |
| IL-18 | Interleukin-18 |
| IL-1β | Interleukin-1 beta |
| IL-21 | Interleukin-21 |
| IL-33 | Interleukin-33 |
| IL-4 | Interleukin-4 |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| iNOS | Inducible nitric oxide synthase |
| iNOS2 | Inducible nitric oxide synthase 2 |
| IP-10 | Chemokine (C-X-C motif) ligand 10 (see also CXCL10) |
| IRF-1 | Interferon regulatory factor 1 |
| Isl-1 | Insulin gene enhancer protein 1 |
| JAK/STAT | Janus kinase/signal transducer and activator of transcription |
| JNK | c-Jun N-terminal kinase |
| Kcnj11 | Potassium Inwardly Rectifying Channel Subfamily J Member 11 |
| LPS | Lipopolysaccharide |
| mAb | Monoclonal antibody |
| MafA | MAF BZIP Transcription Factor A |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein-1 |
| M-CSF | Macrophage colony-stimulating factor |
| MerTK | Tyrosine-protein kinase MER |
| Min6 | Mouse insulinoma cell line |
| MIP1α | Macrophage inflammatory protein-1 alpha |
| MMP | Metalloproteinase |
| MMP9 | Metalloproteinase 9 |
| mRNA | Messenger ribonucleic acid |
| NAD | Nicotinamide adenine dinucleotide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NOD-, LRR- and pyrin domain containing 3 |
| NO | Nitric oxide |
| NOD | Non-obese diabetic |
| NOS | Nitrogen species |
| NOS2 | Nitric oxide synthase |
| p27CDKN1B | Cyclin-dependent kinase inhibitor 1B |
| PARP | Poly (ADP-ribose) polymerase |
| PDGF | Platelet-derived growth factor |
| PDGFR | Platelet-derived growth factor receptor |
| PDX1 | Pancreatic and duodenal homeobox 1 |
| PI3K-AKT | Phosphatidylinositol 3-kinase-protein kinase B signaling pathway |
| RARβ | Retinoic acid receptor beta |
| ROS | Reactive oxygen species |
| SMAD2 | Mothers against decapentaplegic homolog 2 |
| SMAD7 | Mothers against decapentaplegic homolog 7 |
| Sox9 | SRY-Box Transcription Factor 9 |
| STAT1 | Signal transducer and activator of transcription 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| STZ | Streptozotocin |
| T1D | Type 1 diabetes |
| T2D | Type 2 diabetes |
| TGFβ | Transforming growth factor alpha |
| TGF-β1 | Transforming growth factor beta 1 |
| Th1 | T-helper type 1 cell |
| Th2 | T-helper type 2 cell |
| TNF-a | Tumor necrosis factor alpha |
| TNFR | Tumor necrosis factor receptor |
| Ucn3 | Urocortin-3 |
| VEGF | Vascular endothelial growth factor |
| VEGF-A | Vascular endothelial growth factor A |
| Wnt3a | Wingless-Type MMTV Integration Site Family, Member 3A |
| XBP1 | X-box binding protein-1 |
| YM1 | Synonym for chitinase-like 3 |
| ZDF | Zucker diabetic fatty rat |
Note
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pr. 2019, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallone, R.; Eizirik, D.L. Presumption of innocence for beta cells: Why are they vulnerable autoimmune targets in type 1 diabetes? Diabetologia 2020, 63, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Efrat, S. Beta-Cell Dedifferentiation in Type 2 Diabetes: Concise Review. Stem Cells 2019, 37, 1267–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugliese, A. Insulitis in the pathogenesis of type 1 diabetes. Pediatr. Diabetes 2016, 17, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Fu, W.X.; Lee, Y.S.; Olefsky, J.M. The role of macrophages in obesity-associated islet inflammation and beta-cell abnormalities. Nat. Rev. Endocrinol. 2020, 16, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Saunders, D.; Powers, A.C. Replicative capacity of beta-cells and type 1 diabetes. J. Autoimmun. 2016, 71, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Ley, K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017, 199, 2191–2193. [Google Scholar] [CrossRef]
- Poltavets, A.S.; Vishnyakova, P.A.; Elchaninov, A.V.; Sukhikh, G.T.; Fatkhudinov, T.K. Macrophage Modification Strategies for Efficient Cell Therapy. Cells 2020, 9, 1535. [Google Scholar] [CrossRef]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Van Gassen, N.; Staels, W.; Van Overmeire, E.; De Groef, S.; Sojoodi, M.; Heremans, Y.; Leuckx, G.; Van de Casteele, M.; Van Ginderachter, J.A.; Heimberg, H.; et al. Concise Review: Macrophages: Versatile Gatekeepers During Pancreatic beta-Cell Development, Injury, and Regeneration. Stem Cells Transl. Med. 2015, 4, 555–563. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Jaburek, M.; Holendova, B.; Plecita-Hlavata, L. Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity. Molecules 2018, 23, 1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hume, D.A. The Many Alternative Faces of Macrophage Activation. Front. Immunol. 2015, 6, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Dalmas, E.; Sauter, N.S.; Boni-Schnetzler, M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, T.; Godlewski, G.; Cinar, R.; Bertola, A.; Szanda, G.; Liu, J.; Tam, J.; Han, T.; Mukhopadhyay, B.; Skarulis, M.C.; et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat. Med. 2013, 19, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Yang, H. Modulation of macrophage activation and programming in immunity. J. Cell Physiol. 2013, 228, 502–512. [Google Scholar] [CrossRef]
- Aamodt, K.I.; Powers, A.C. Signals in the pancreatic islet microenvironment influence beta-cell proliferation. Diabetes Obes. Metab. 2017, 19, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, C.J.; Leibovich, S.J. Regulation of Macrophage Polarization and Wound Healing. Adv. Wound Care 2012, 1, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinberg, S.; Hasko, G.; Wu, D.; Leibovich, S.J. Suppression of PLCbeta2 by endotoxin plays a role in the adenosine A(2A) receptor-mediated switch of macrophages from an inflammatory to an angiogenic phenotype. Am. J. Pathol. 2009, 175, 2439–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burg, A.R.; Tse, H.M. Redox-Sensitive Innate Immune Pathways During Macrophage Activation in Type 1 Diabetes. Antioxid. Redox Signal. 2018, 29, 1373–1398. [Google Scholar] [CrossRef] [PubMed]
- Oschilewski, U.; Kiesel, U.; Kolb, H. Administration of silica prevents diabetes in BB-rats. Diabetes 1985, 34, 197–199. [Google Scholar] [CrossRef] [Green Version]
- Hanninen, A.; Jalkanen, S.; Salmi, M.; Toikkanen, S.; Nikolakaros, G.; Simell, O. Macrophages, T cell receptor usage, and endothelial cell activation in the pancreas at the onset of insulin-dependent diabetes mellitus. J. Clin. Investig. 1992, 90, 1901–1910. [Google Scholar] [CrossRef] [Green Version]
- Gaglia, J.L.; Guimaraes, A.R.; Harisinghani, M.; Turvey, S.E.; Jackson, R.; Benoist, C.; Mathis, D.; Weissleder, R. Noninvasive imaging of pancreatic islet inflammation in type 1A diabetes patients. J. Clin. Investig. 2011, 121, 442–445. [Google Scholar] [CrossRef] [Green Version]
- Nunemaker, C.S. Considerations for Defining Cytokine Dose, Duration, and Milieu That Are Appropriate for Modeling Chronic Low-Grade Inflammation in Type 2 Diabetes. J. Diabetes Res. 2016, 2016, 2846570. [Google Scholar] [CrossRef] [Green Version]
- Juhas, U.; Ryba-Stanislawowska, M.; Brandt-Varma, A.; Mysliwiec, M.; Mysliwska, J. Monocytes of newly diagnosed juvenile DM1 patients are prone to differentiate into regulatory IL-10(+) M2 macrophages. Immunol. Res. 2019, 67, 58–69. [Google Scholar] [CrossRef] [Green Version]
- El Hadri, K.; Mahmood, D.F.; Couchie, D.; Jguirim-Souissi, I.; Genze, F.; Diderot, V.; Syrovets, T.; Lunov, O.; Simmet, T.; Rouis, M. Thioredoxin-1 promotes anti-inflammatory macrophages of the M2 phenotype and antagonizes atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1445–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padgett, L.E.; Burg, A.R.; Lei, W.; Tse, H.M. Loss of NADPH oxidase-derived superoxide skews macrophage phenotypes to delay type 1 diabetes. Diabetes 2015, 64, 937–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [Green Version]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Masters, S.L.; Mielke, L.A.; Cornish, A.L.; Sutton, C.E.; O’Donnell, J.; Cengia, L.H.; Roberts, A.W.; Wicks, I.P.; Mills, K.H.; Croker, B.A. Regulation of interleukin-1beta by interferon-gamma is species specific, limited by suppressor of cytokine signalling 1 and influences interleukin-17 production. EMBO Rep. 2010, 11, 640–646. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef] [Green Version]
- Westwell-Roper, C.Y.; Ehses, J.A.; Verchere, C.B. Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1beta production and beta-cell dysfunction. Diabetes 2014, 63, 1698–1711. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Islet-associated macrophages in type 2 diabetes. Diabetologia 2009, 52, 1686–1688. [Google Scholar] [CrossRef] [Green Version]
- Kamata, K.; Mizukami, H.; Inaba, W.; Tsuboi, K.; Tateishi, Y.; Yoshida, T.; Yagihashi, S. Islet amyloid with macrophage migration correlates with augmented beta-cell deficits in type 2 diabetic patients. Amyloid 2014, 21, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Collier, J.J.; Sparer, T.E.; Karlstad, M.D.; Burke, S.J. Pancreatic islet inflammation: An emerging role for chemokines. J. Mol. Endocrinol. 2017, 59, R33–R46. [Google Scholar] [CrossRef] [Green Version]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.Y.; Lee, K.; Maxwell, E.L.; Liang, C.; Laybutt, D.R. Macrophage alterations in islets of obese mice linked to beta cell disruption in diabetes. Diabetologia 2019, 62, 993–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Yuan, T.; Maedler, K. Macrophage-associated pro-inflammatory state in human islets from obese individuals. Nutr. Diabetes 2019, 9, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmastro, M.M.; Piganelli, J.D. Oxidative stress and redox modulation potential in type 1 diabetes. Clin. Dev. Immunol. 2011, 2011, 593863. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Batdorf, H.M.; Burk, D.H.; Martin, T.M.; Mendoza, T.; Stadler, K.; Alami, W.; Karlstad, M.D.; Robson, M.J.; Blakely, R.D.; et al. Pancreatic deletion of the interleukin-1 receptor disrupts whole body glucose homeostasis and promotes islet beta-cell de-differentiation. Mol. Metab. 2018, 14, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Stadler, K.; Lu, D.; Gleason, E.; Han, A.; Donohoe, D.R.; Rogers, R.C.; Hermann, G.E.; Karlstad, M.D.; Collier, J.J. IL-1beta reciprocally regulates chemokine and insulin secretion in pancreatic beta-cells via NF-kappaB. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E715–E726. [Google Scholar] [CrossRef] [Green Version]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thevenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Welsh, N.; Cnop, M.; Kharroubi, I.; Bugliani, M.; Lupi, R.; Marchetti, P.; Eizirik, D.L. Is there a role for locally produced interleukin-1 in the deleterious effects of high glucose or the type 2 diabetes milieu to human pancreatic islets? Diabetes 2005, 54, 3238–3244. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Mandrup-Poulsen, T. A choice of death—The signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 2001, 44, 2115–2133. [Google Scholar] [CrossRef]
- Ablamunits, V.; Henegariu, O.; Hansen, J.B.; Opare-Addo, L.; Preston-Hurlburt, P.; Santamaria, P.; Mandrup-Poulsen, T.; Herold, K.C. Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade: Evidence of improved immune regulation. Diabetes 2012, 61, 145–154. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, C.M.; Lu, C.; Corbin, K.L.; Sharma, P.R.; Dula, S.B.; Carter, J.D.; Ramadan, J.W.; Xin, W.; Lee, J.K.; Nunemaker, C.S. Circulating levels of IL-1B+IL-6 cause ER stress and dysfunction in islets from prediabetic male mice. Endocrinology 2013, 154, 3077–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkart, V.; Wang, Z.Q.; Radons, J.; Heller, B.; Herceg, Z.; Stingl, L.; Wagner, E.F.; Kolb, H. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat. Med. 1999, 5, 314–319. [Google Scholar] [CrossRef]
- Westwell-Roper, C.; Dai, D.L.; Soukhatcheva, G.; Potter, K.J.; van Rooijen, N.; Ehses, J.A.; Verchere, C.B. IL-1 blockade attenuates islet amyloid polypeptide-induced proinflammatory cytokine release and pancreatic islet graft dysfunction. J. Immunol. 2011, 187, 2755–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzakri, K.; Ribaux, P.; Tomas, A.; Parnaud, G.; Rickenbach, K.; Halban, P.A. Rab GTPase-activating protein AS160 is a major downstream effector of protein kinase B/Akt signaling in pancreatic beta-cells. Diabetes 2008, 57, 1195–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawalich, W.S.; Zawalich, K.C. Interleukin 1 is a potent stimulator of islet insulin secretion and phosphoinositide hydrolysis. Am. J. Physiol. 1989, 256, E19–E24. [Google Scholar] [CrossRef] [PubMed]
- Cucak, H.; Mayer, C.; Tonnesen, M.; Thomsen, L.H.; Grunnet, L.G.; Rosendahl, A. Macrophage Contact Dependent and Independent TLR4 Mechanisms Induce beta-Cell Dysfunction and Apoptosis in a Mouse Model of Type 2 Diabetes. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Ehses, J.A.; Lacraz, G.; Giroix, M.H.; Schmidlin, F.; Coulaud, J.; Kassis, N.; Irminger, J.C.; Kergoat, M.; Portha, B.; Homo-Delarche, F.; et al. IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proc. Natl. Acad. Sci. USA 2009, 106, 13998–14003. [Google Scholar] [CrossRef] [Green Version]
- Sauter, N.S.; Thienel, C.; Plutino, Y.; Kampe, K.; Dror, E.; Traub, S.; Timper, K.; Bedat, B.; Pattou, F.; Kerr-Conte, J.; et al. Angiotensin II induces interleukin-1beta-mediated islet inflammation and beta-cell dysfunction independently of vasoconstrictive effects. Diabetes 2015, 64, 1273–1283. [Google Scholar] [CrossRef] [Green Version]
- Novak, I.; Solini, A. P2X receptor-ion channels in the inflammatory response in adipose tissue and pancreas-potential triggers in onset of type 2 diabetes? Curr. Opin. Immunol. 2018, 52, 1–7. [Google Scholar] [CrossRef]
- Cnop, M.; Abdulkarim, B.; Bottu, G.; Cunha, D.A.; Igoillo-Esteve, M.; Masini, M.; Turatsinze, J.V.; Griebel, T.; Villate, O.; Santin, I.; et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 2014, 63, 1978–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diana, J.; Lehuen, A. Macrophages and beta-cells are responsible for CXCR2-mediated neutrophil infiltration of the pancreas during autoimmune diabetes. EMBO Mol. Med. 2014, 6, 1090–1104. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef]
- Ward, M.G.; Li, G.; Hao, M. Apoptotic beta-cells induce macrophage reprogramming under diabetic conditions. J. Biol. Chem. 2018, 293, 16160–16173. [Google Scholar] [CrossRef] [Green Version]
- Riera-Borrull, M.; Cuevas, V.D.; Alonso, B.; Vega, M.A.; Joven, J.; Izquierdo, E.; Corbi, A.L. Palmitate Conditions Macrophages for Enhanced Responses toward Inflammatory Stimuli via JNK Activation. J. Immunol. 2017, 199, 3858–3869. [Google Scholar] [CrossRef]
- Hajmrle, C.; Smith, N.; Spigelman, A.F.; Dai, X.; Senior, L.; Bautista, A.; Ferdaoussi, M.; MacDonald, P.E. Interleukin-1 signaling contributes to acute islet compensation. JCI Insight 2016, 1, e86055. [Google Scholar] [CrossRef]
- Zawalich, W.S.; Dierolf, B.; Zawalich, K.C. Interleukin-1 induces time-dependent potentiation in isolated rat islets: Possible involvement of phosphoinositide hydrolysis. Endocrinology 1989, 124, 720–726. [Google Scholar] [CrossRef]
- Dasu, M.R.; Devaraj, S.; Jialal, I. High glucose induces IL-1beta expression in human monocytes: Mechanistic insights. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E337–E346. [Google Scholar] [CrossRef] [Green Version]
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Boni-Schnetzler, M.; Donath, M.Y. The Role of Inflammation in beta-cell Dedifferentiation. Sci. Rep. 2017, 7, 6285. [Google Scholar] [CrossRef]
- Oshima, M.; Knoch, K.P.; Diedisheim, M.; Petzold, A.; Cattan, P.; Bugliani, M.; Marchetti, P.; Choudhary, P.; Huang, G.C.; Bornstein, S.R.; et al. Virus-like infection induces human beta cell dedifferentiation. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Ibarra Urizar, A.; Prause, M.; Wortham, M.; Sui, Y.; Thams, P.; Sander, M.; Christensen, G.L.; Billestrup, N. Beta-cell dysfunction induced by non-cytotoxic concentrations of Interleukin-1beta is associated with changes in expression of beta-cell maturity genes and associated histone modifications. Mol. Cell. Endocrinol. 2019, 496, 110524. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Xu, G.; Fujii, N.; Kim, S.; Bonner-Weir, S.; Weir, G.C. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J. Biol. Chem. 2002, 277, 30010–30018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinas, G.A.; Hansen, B.S.; Linde, S.; Kastern, W.; Molvig, J.; Mandruppoulsen, T.; Dinarello, C.A.; Nielsen, J.H.; Nerup, J. Interleukin-1 Dose-Dependently Affects the Biosynthesis of (Pro)Insulin in Isolated Rat Islets of Langerhans. Diabetologia 1987, 30, 474–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Kristiansen, O.P.; Mandrup-Poulsen, T. Interleukin-6 and diabetes: The good, the bad, or the indifferent? Diabetes 2005, 54, S114–S124. [Google Scholar] [CrossRef] [Green Version]
- Brozzi, F.; Nardelli, T.R.; Lopes, M.; Millard, I.; Barthson, J.; Igoillo-Esteve, M.; Grieco, F.A.; Villate, O.; Oliveira, J.M.; Casimir, M.; et al. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 2015, 58, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Maedler, K.; Spinas, G.A.; Lehmann, R.; Sergeev, P.; Weber, M.; Fontana, A.; Kaiser, N.; Donath, M.Y. Glucose induces beta-cell apoptosis via upregulation of the Fas receptor in human islets. Diabetes 2001, 50, 1683–1690. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Rebello, O.; Savino, R.; Terracciano, R.; Schuster-Klein, C.; Guardiola, B.; Maedler, K. TLR4 triggered complex inflammation in human pancreatic islets. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 86–97. [Google Scholar] [CrossRef]
- Nackiewicz, D.; Dan, M.; He, W.; Kim, R.; Salmi, A.; Rutti, S.; Westwell-Roper, C.; Cunningham, A.; Speck, M.; Schuster-Klein, C.; et al. TLR2/6 and TLR4-activated macrophages contribute to islet inflammation and impair beta cell insulin gene expression via IL-1 and IL-6. Diabetologia 2014, 57, 1645–1654. [Google Scholar] [CrossRef]
- Baltrusch, S. Mitochondrial network regulation and its potential interference with inflammatory signals in pancreatic beta cells. Diabetologia 2016, 59, 683–687. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.Y.; Cao, Z.H.; Hu, X.B.; Li, G.Q.; Dong, S.F.; Xu, G.L.; Zhang, K.Q. LIGHT/IFN-gamma triggers beta cells apoptosis via NF-kappaB/Bcl2-dependent mitochondrial pathway. J. Cell Mol. Med. 2016, 20, 1861–1871. [Google Scholar] [CrossRef]
- Cao, Z.H.; -Zheng, Q.Y.; Li, G.Q.; Hu, X.B.; Feng, S.L.; Xu, G.L.; Zhang, K.Q. STAT1-mediated down-regulation of Bcl-2 expression is involved in IFN-gamma/TNF-alpha-induced apoptosis in NIT-1 cells. PLoS ONE 2015, 10, e0120921. [Google Scholar] [CrossRef] [Green Version]
- Gysemans, C.A.; Ladriere, L.; Callewaert, H.; Rasschaert, J.; Flamez, D.; Levy, D.E.; Matthys, P.; Eizirik, D.L.; Mathieu, C. Disruption of the gamma-interferon signaling pathway at the level of signal transducer and activator of transcription-1 prevents immune destruction of beta-cells. Diabetes 2005, 54, 2396–2403. [Google Scholar] [CrossRef] [Green Version]
- Baldeon, M.E.; Chun, T.; Gaskins, H.R. Interferon-alpha and interferon-gamma differentially affect pancreatic beta-cell phenotype and function. Am. J. Physiol. 1998, 275, C25–C32. [Google Scholar] [CrossRef]
- Freudenburg, W.; Gautam, M.; Chakraborty, P.; James, J.; Richards, J.; Salvatori, A.S.; Baldwin, A.; Schriewer, J.; Buller, R.M.L.; Corbett, J.A.; et al. Reduction in ATP Levels Triggers Immunoproteasome Activation by the 11S (PA28) Regulator during Early Antiviral Response Mediated by IFN beta in Mouse Pancreatic beta-Cells. PLoS ONE 2013, 8, e52408. [Google Scholar] [CrossRef] [Green Version]
- Heitmeier, M.R.; Scarim, A.L.; Corbett, J.A. Interferon-gamma increases the sensitivity of islets of Langerhans for inducible nitric-oxide synthase expression induced by interleukin 1. J. Biol. Chem. 1997, 272, 13697–13704. [Google Scholar] [CrossRef] [Green Version]
- Oleson, B.J.; Corbett, J.A. Dual Role of Nitric Oxide in Regulating the Response of beta Cells to DNA Damage. Antioxid. Redox Sign. 2018, 29, 1432–1445. [Google Scholar] [CrossRef]
- Hughes, K.J.; Meares, G.P.; Hansen, P.A.; Corbett, J.A. FoxO1 and SIRT1 Regulate beta-Cell Responses to Nitric Oxide. J. Biol. Chem. 2011, 286, 8338–8348. [Google Scholar] [CrossRef] [Green Version]
- Dahlen, E.; Dawe, K.; Ohlsson, L.; Hedlund, G. Dendritic cells and macrophages are the first and major producers of TNF-alpha in pancreatic islets in the nonobese diabetic mouse. J. Immunol. 1998, 160, 3585–3593. [Google Scholar]
- Odegaard, J.I.; Chawla, A. Connecting Type 1 and Type 2 Diabetes through Innate Immunity. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Janjuha, S.; Singh, S.P.; Tsakmaki, A.; Mousavy Gharavy, S.N.; Murawala, P.; Konantz, J.; Birke, S.; Hodson, D.J.; Rutter, G.A.; Bewick, G.A.; et al. Age-related islet inflammation marks the proliferative decline of pancreatic beta-cells in zebrafish. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.A.; Paszek, P.; Woodcock, D.J.; Nelson, D.E.; Horton, C.A.; Wang, Y.; Spiller, D.G.; Rand, D.A.; White, M.R.; Harper, C.V. Physiological levels of TNFalpha stimulation induce stochastic dynamics of NF-kappaB responses in single living cells. J. Cell Sci. 2010, 123, 2834–2843. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, K.; Manabe, I. Macrophages and islet inflammation in type 2 diabetes. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 152–158. [Google Scholar] [CrossRef]
- Morgan, D.; Oliveira-Emilio, H.R.; Keane, D.; Hirata, A.E.; Santos da Rocha, M.; Bordin, S.; Curi, R.; Newsholme, P.; Carpinelli, A.R. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 2007, 50, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Flodstrom, M.; Karlsen, A.E.; Welsh, N. The harmony of the spheres: Inducible nitric oxide synthase and related genes in pancreatic beta cells. Diabetologia 1996, 39, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Lopez, L.; Blandino-Rosano, M.; Perez-Arana, G.; Cebada-Aleu, A.; Lechuga-Sancho, A.; Aguilar-Diosdado, M.; Segundo, C. Nitric Oxide Is a Mediator of Antiproliferative Effects Induced by Proinflammatory Cytokines on Pancreatic Beta Cells. Mediat. Inflamm. 2013, 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunnet, L.G.; Aikin, R.; Tonnesen, M.F.; Paraskevas, S.; Blaabjerg, L.; Storling, J.; Rosenberg, L.; Billestrup, N.; Maysinger, D.; Mandrup-Poulsen, T. Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta-cells. Diabetes 2009, 58, 1807–1815. [Google Scholar] [CrossRef] [Green Version]
- Pavlovic, D.; Chen, M.C.; Gysemans, C.A.; Mathieu, C.; Eizirik, D.L. The role of interferon regulatory factor-1 in cytokine-induced mRNA expression and cell death in murine pancreatic beta-cells. Eur. Cytokine Netw. 1999, 10, 403–412. [Google Scholar]
- Gysemans, C.; Callewaert, H.; Moore, F.; Nelson-Holte, M.; Overbergh, L.; Eizirik, D.L.; Mathieu, C. Interferon regulatory factor-1 is a key transcription factor in murine beta cells under immune attack. Diabetologia 2009, 52, 2374–2384. [Google Scholar] [CrossRef] [Green Version]
- Grieco, F.A.; Moore, F.; Vigneron, F.; Santin, I.; Villate, O.; Marselli, L.; Rondas, D.; Korf, H.; Overbergh, L.; Dotta, F.; et al. IL-17A increases the expression of proinflammatory chemokines in human pancreatic islets. Diabetologia 2014, 57, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Parkash, J.; Chaudhry, M.A.; Rhoten, W.B. Tumor necrosis factor-alpha-induced changes in insulin-producing beta-cells. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2005, 286, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Geutskens, S.B.; Otonkoski, T.; Pulkkinen, M.A.; Drexhage, H.A.; Leenen, P.J. Macrophages in the murine pancreas and their involvement in fetal endocrine development in vitro. J. Leukoc. Biol. 2005, 78, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Banaei-Bouchareb, L.; Gouon-Evans, V.; Samara-Boustani, D.; Castellotti, M.C.; Czernichow, P.; Pollard, J.W.; Polak, M. Insulin cell mass is altered in Csf1op/Csf1op macrophage-deficient mice. J. Leukoc. Biol. 2004, 76, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Banaei-Bouchareb, L.; Peuchmaur, M.; Czernichow, P.; Polak, M. A transient microenvironment loaded mainly with macrophages in the early developing human pancreas. J. Endocrinol. 2006, 188, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosser, R.E.; Maulis, M.F.; Moulle, V.S.; Dunn, J.C.; Carboneau, B.A.; Arasi, K.; Pappan, K.; Poitout, V.; Gannon, M. High-fat diet-induced beta-cell proliferation occurs prior to insulin resistance in C57Bl/6J male mice. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E573–E582. [Google Scholar] [CrossRef]
- Woodland, D.C.; Liu, W.; Leong, J.; Sears, M.L.; Luo, P.; Chen, X. Short-term high-fat feeding induces islet macrophage infiltration and beta-cell replication independently of insulin resistance in mice. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E763–E771. [Google Scholar] [CrossRef] [Green Version]
- Criscimanna, A.; Coudriet, G.M.; Gittes, G.K.; Piganelli, J.D.; Esni, F. Activated macrophages create lineage-specific microenvironments for pancreatic acinar- and beta-cell regeneration in mice. Gastroenterology 2014, 147, 1106–1118.e1111. [Google Scholar] [CrossRef] [Green Version]
- Riley, K.G.; Pasek, R.C.; Maulis, M.F.; Dunn, J.C.; Bolus, W.R.; Kendall, P.L.; Hasty, A.H.; Gannon, M. Macrophages are essential for CTGF-mediated adult beta-cell proliferation after injury. Mol. Metab. 2015, 4, 584–591. [Google Scholar] [CrossRef]
- Xiao, X.; Guo, P.; Prasadan, K.; Shiota, C.; Peirish, L.; Fischbach, S.; Song, Z.; Gaffar, I.; Wiersch, J.; El-Gohary, Y.; et al. Pancreatic cell tracing, lineage tagging and targeted genetic manipulations in multiple cell types using pancreatic ductal infusion of adeno-associated viral vectors and/or cell-tagging dyes. Nat. Protoc. 2014, 9, 2719–2724. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Preuett, B.; Krishna, P.; Xiao, X.; Shiota, C.; Wiersch, J.; Gaffar, I.; Tulachan, S.; El-Gohary, Y.; Song, Z.; et al. Barrier function of the coelomic epithelium in the developing pancreas. Mech. Dev. 2014, 134, 67–79. [Google Scholar] [CrossRef]
- Xiao, X.; Gaffar, I.; Guo, P.; Wiersch, J.; Fischbach, S.; Peirish, L.; Song, Z.; El-Gohary, Y.; Prasadan, K.; Shiota, C.; et al. M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc. Natl. Acad. Sci. USA 2014, 111, E1211–E1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Prasadan, K.; Guo, P.; El-Gohary, Y.; Fischbach, S.; Wiersch, J.; Gaffar, I.; Shiota, C.; Gittes, G.K. Pancreatic duct cells as a source of VEGF in mice. Diabetologia 2014, 57, 991–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessem, J.S.; Jensen, J.N.; Pelli, H.; Dai, X.M.; Zong, X.H.; Stanley, E.R.; Jensen, J.; DeGregori, J. Critical roles for macrophages in islet angiogenesis and maintenance during pancreatic degeneration. Diabetes 2008, 57, 1605–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinert, R.B.; Cai, Q.; Hong, J.Y.; Plank, J.L.; Aamodt, K.; Prasad, N.; Aramandla, R.; Dai, C.; Levy, S.E.; Pozzi, A.; et al. Vascular endothelial growth factor coordinates islet innervation via vascular scaffolding. Development 2014, 141, 1480–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brissova, M.; Aamodt, K.; Brahmachary, P.; Prasad, N.; Hong, J.Y.; Dai, C.; Mellati, M.; Shostak, A.; Poffenberger, G.; Aramandla, R.; et al. Islet microenvironment, modulated by vascular endothelial growth factor-A signaling, promotes beta cell regeneration. Cell Metab. 2014, 19, 498–511. [Google Scholar] [CrossRef] [Green Version]
- Smeets, S.; Stange, G.; Leuckx, G.; Roelants, L.; Cools, W.; De Paep, D.L.; Ling, Z.; De Leu, N.; In’t Veld, P. Evidence of Tissue Repair in Human Donor Pancreas After Prolonged Duration of Stay in Intensive Care. Diabetes 2020, 69, 401–412. [Google Scholar] [CrossRef]
- Cao, X.; Han, Z.B.; Zhao, H.; Liu, Q. Transplantation of mesenchymal stem cells recruits trophic macrophages to induce pancreatic beta cell regeneration in diabetic mice. Int. J. Biochem. Cell Biol. 2014, 53, 372–379. [Google Scholar] [CrossRef]
- Criscimanna, A.; Speicher, J.A.; Houshmand, G.; Shiota, C.; Prasadan, K.; Ji, B.; Logsdon, C.D.; Gittes, G.K.; Esni, F. Duct cells contribute to regeneration of endocrine and acinar cells following pancreatic damage in adult mice. Gastroenterology 2011, 141, 1451–1462.e6. [Google Scholar] [CrossRef] [Green Version]
- Dalmas, E.; Lehmann, F.M.; Dror, E.; Wueest, S.; Thienel, C.; Borsigova, M.; Stawiski, M.; Traunecker, E.; Lucchini, F.C.; Dapito, D.H.; et al. Interleukin-33-Activated Islet-Resident Innate Lymphoid Cells Promote Insulin Secretion through Myeloid Cell Retinoic Acid Production. Immunity 2017, 47, 928–942.e927. [Google Scholar] [CrossRef] [Green Version]
- El-Gohary, Y.; Tulachan, S.; Wiersch, J.; Guo, P.; Welsh, C.; Prasadan, K.; Paredes, J.; Shiota, C.; Xiao, X.; Wada, Y.; et al. A smad signaling network regulates islet cell proliferation. Diabetes 2014, 63, 224–236. [Google Scholar] [CrossRef] [Green Version]
- El-Gohary, Y.; Tulachan, S.; Guo, P.; Welsh, C.; Wiersch, J.; Prasadan, K.; Paredes, J.; Shiota, C.; Xiao, X.; Wada, Y.; et al. Smad signaling pathways regulate pancreatic endocrine development. Dev. Biol. 2013, 378, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; Seo, J.B.; Yang, B.H.; Wollam, J.; Riopel, M.; et al. Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting beta Cell Proliferation and Function in Obesity. Cell Metab. 2019, 29, 457–474.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikoba, N.; Kumagai, K.; Kanmura, S.; Nakamura, Y.; Ono, M.; Eguchi, H.; Kamibayashiyama, T.; Oda, K.; Mawatari, S.; Tanoue, S.; et al. HGF-MET Signaling Shifts M1 Macrophages Toward an M2-Like Phenotype Through PI3K-Mediated Induction of Arginase-1 Expression. Fron. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ocana, A.; Takane, K.K.; Syed, M.A.; Philbrick, W.M.; Vasavada, R.C.; Stewart, A.F. Hepatocyte growth factor overexpression in the islet of transgenic mice increases beta cell proliferation, enhances islet mass, and induces mild hypoglycemia. J. Biol. Chem. 2000, 275, 1226–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Perez, J.C.; Ernst, S.; Demirci, C.; Casinelli, G.P.; Mellado-Gil, J.M.D.; Rausell-Palamos, F.; Vasavada, R.C.; Garcia-Ocana, A. Hepatocyte Growth Factor/c-Met Signaling Is Required for beta-Cell Regeneration. Diabetes 2014, 63, 216–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nackiewicz, D.; Dan, M.X.; Speck, M.; Chow, S.Z.; Chen, Y.C.; Pospisilik, J.A.; Verchere, C.B.; Ehses, J.A. Islet Macrophages Shift to a Reparative State following Pancreatic Beta-Cell Death and Area Major Source of Islet Insulin-like Growth Factor-1. Iscience 2020, 23. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, A.; Kaminski, E.R.; Morgan, N.G. Pre-incubation with interleukin-4 mediates a direct protective effect against the loss of pancreatic beta-cell viability induced by proinflammatory cytokines. Clin. Exp. Immunol. 2007, 148, 583–588. [Google Scholar] [CrossRef]
- Souza, K.L.A.; Gurgul-Convey, E.; Elsner, M.; Lenzen, S. Interaction between pro-inflammatory and anti-inflammatory cytokines in insulin-producing cells. J. Endocrinol. 2008, 197, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Pfleger, C.; Meierhoff, G.; Kolb, H.; Schloot, N.C.; Grp, P.S. Association of T-cell reactivity with beta-cell function in recent onset type 1 diabetes patients. J. Autoimmun. 2010, 34, 127–135. [Google Scholar] [CrossRef]
- Weitz, J.R.; Jacques-Silva, C.; Qadir, M.M.F.; Umland, O.; Pereira, E.; Qureshi, F.; Tamayo, A.; Dominguez-Bendala, J.; Rodriguez-Diaz, R.; Almaca, J.; et al. Secretory Functions of Macrophages in the Human Pancreatic Islet Are Regulated by Endogenous Purinergic Signaling. Diabetes 2020, 69, 1206–1218. [Google Scholar] [CrossRef]
- Fu, W.; Farache, J.; Clardy, S.M.; Hattori, K.; Mander, P.; Lee, K.; Rioja, I.; Weissleder, R.; Prinjha, R.K.; Benoist, C.; et al. Epigenetic modulation of type-1 diabetes via a dual effect on pancreatic macrophages and beta cells. eLife 2014, 3, e04631. [Google Scholar] [CrossRef] [PubMed]
- Parsa, R.; Andresen, P.; Gillett, A.; Mia, S.; Zhang, X.M.; Mayans, S.; Holmberg, D.; Harris, R.A. Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes 2012, 61, 2881–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Wang, Y.; Cao, Q.; Lee, V.W.; Zheng, G.; Sun, Y.; Tan, T.K.; Wang, Y.; Alexander, S.I.; Harris, D.C. Transfused macrophages ameliorate pancreatic and renal injury in murine diabetes mellitus. Nephron. Exp. Nephrol. 2011, 118, e87–e99. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Effectors | Target |
|---|---|
| IL-1β | Initiates β-cell apoptosis through ERK signaling pathways [18,64] Decreases insulin mRNA levels [57,58,59,60,61,62,63] Impairs GSIS [57,58,59,60,61,62,63] Increases IL-6 release in β-cell [18] Transcriptional changes of 3068 genes associated with inflammation, cell death, antigen presentation, and cytokines/chemokines [65] Contributes to increased ER stress [68] Increases Fas expression [77] |
| IL-6 | Impairs GSIS [76,77] Decreases Ins1, Ins2, and PDX1 mRNA levels in the islet [79] |
| IFN-γ | Participates with IL-1β to activate NF-κB genes, leading to NO and cytokine production leading to ER stress [65] Participates with IL-1β to cause transcriptional changes to 3068 genes associated with inflammation, cell death, antigen presentation, and cytokines/chemokines [65] Impairs β-cell insulin secretion [84,85] |
| TNF-α | Contributes to increased ER stress [68] Participates in NF-κB pathway activation [92] Activates proapoptotic and proinflammatory pathways through NF-κB [49,91,93] Increases iNOS and NADPH oxidase activity, leading to increased ROS production and mitochondrial damage [94,95,96,97] Induces intrinsic apoptosis [97] Increases the expression of cytokines CXCL1, CXCL8, CCL20, CCL2, and CXCL10, which promote immune cell infiltration of the islet [100] Induces Ca2+ influx in β-cells, impairing insulin secretion [101] |
| Effectors | Target |
|---|---|
| WNT3A | Increases β-cell proliferation and survival via Wnt/β-catenin pathway [107,117,118] |
| RETINOIC ACID | Increased expression of RARβ and increased insulin production and secretion [119] |
| TGFβ1 | Induces upregulation of SMAD7, which is responsible for increased β-cell proliferation [111] Induces upregulation of SMAD2, which is a SMAD7 inhibitor [111] |
| EGF | Inhibits SMAD2 nuclear localization, working in conjunction with TGFβ1 to induce β-cell proliferation [111] |
| PDGF | Induces β-cell proliferation [122] |
| IGF-1 | Promotes β-cell survival by maintaining GSIS [126] |
| IL-10 | Induces upregulation of anti-apoptotic genes, promoting greater β-cell survival [127] Increases iNOS levels, decreasing NO levels in β-cells [128] Increases insulin secretion [129] |
| MMP9 | Promotes islet vascularization and β-cell expansion [113] |
| Rodent or Human Islets | References |
|---|---|
| Rodent | [18,25,26,31,32,33,34,37,38,40,42,45,46,47,50,51,52,55,56,58,62,64,67,71,73,79,86,89,94,98,99,102,103,106,107,108,111,113,115,116,118,119,121,123,124,127,132,133] |
| Human | [27,28,30,36,39,43,48,61,63,69,77,78,100,104,117,130,131] |
| Both | [5,17,20,59,66,76,93,97,120] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jensen, D.M.; Hendricks, K.V.; Mason, A.T.; Tessem, J.S. Good Cop, Bad Cop: The Opposing Effects of Macrophage Activation State on Maintaining or Damaging Functional β-Cell Mass. Metabolites 2020, 10, 485. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo10120485
Jensen DM, Hendricks KV, Mason AT, Tessem JS. Good Cop, Bad Cop: The Opposing Effects of Macrophage Activation State on Maintaining or Damaging Functional β-Cell Mass. Metabolites. 2020; 10(12):485. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo10120485
Chicago/Turabian StyleJensen, Daelin M., Kyle V. Hendricks, Austin T. Mason, and Jeffery S. Tessem. 2020. "Good Cop, Bad Cop: The Opposing Effects of Macrophage Activation State on Maintaining or Damaging Functional β-Cell Mass" Metabolites 10, no. 12: 485. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo10120485