
Metabolomics-Guided Elucidation of Plant Abiotic Stress Responses in the 4IR Era: An Overview

 and
and
Abstract
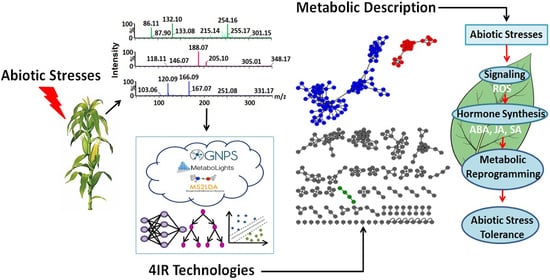
:
1. Introduction—A Dawn of a New Era and a Prime to Plant Defenses
1.1. The Fourth Industrial Revolution (4IR) Era
1.2. Plant Defense Mechanisms—Current Models
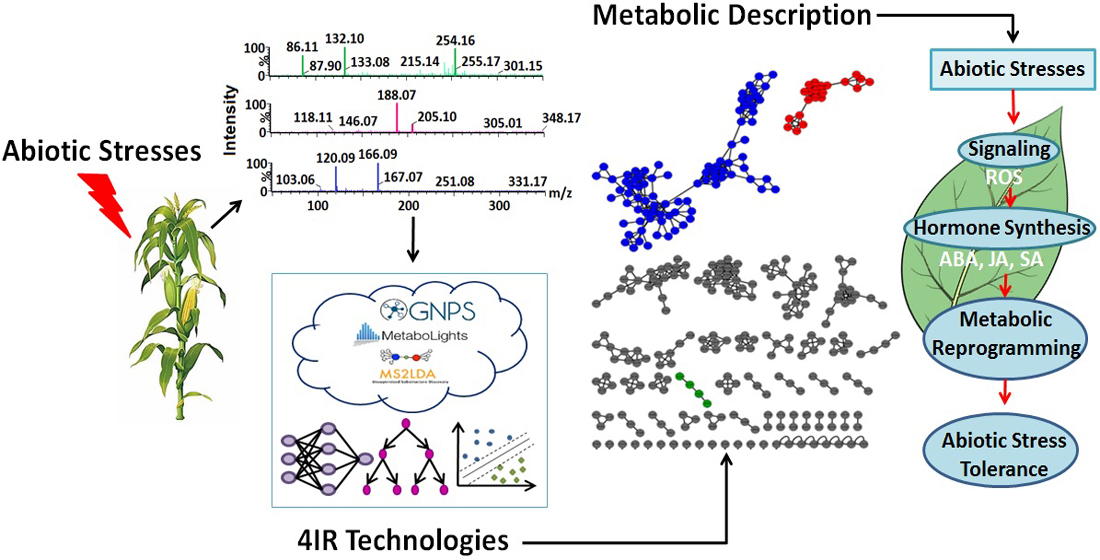
2. 4IR Technologies and Plant Metabolomics
2.1. Automation in Sample Preparation
2.2. Automation and Analytical Intelligence in Analytical Platforms
2.2.1. Mass Spectrometry (MS)-Based Platforms
2.2.1.1. Orthogonal Separations
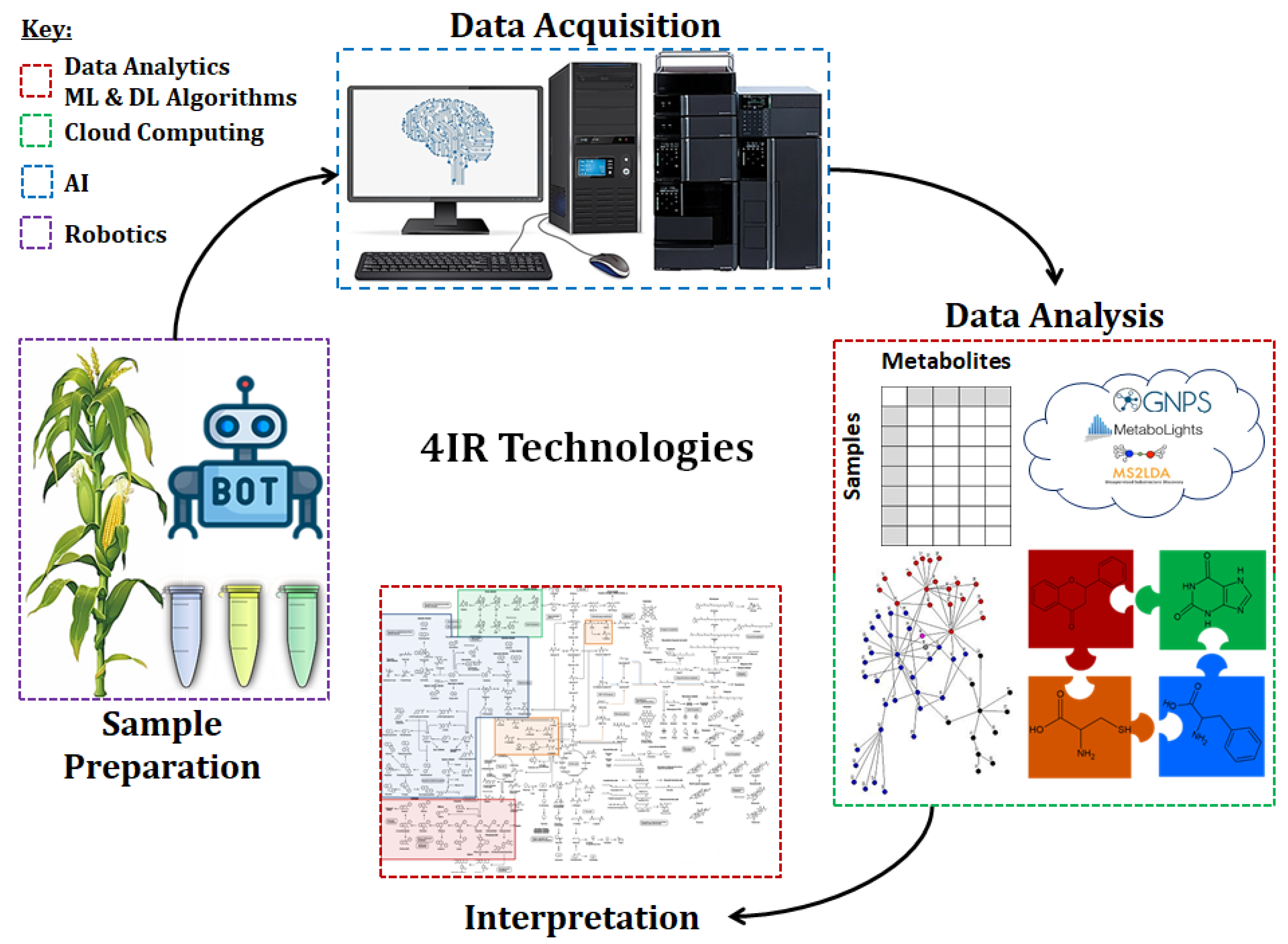
2.2.1.2. Spatial Metabolomics: Mass Spectrometry Imaging
2.2.1.3. Lab-On-Chip and Microfluidic Devices
2.2.1.4. Virtual Metabolomics Mass Spectrometer
2.2.2. Nuclear Magnetic Resonance (NMR)-Based Platforms
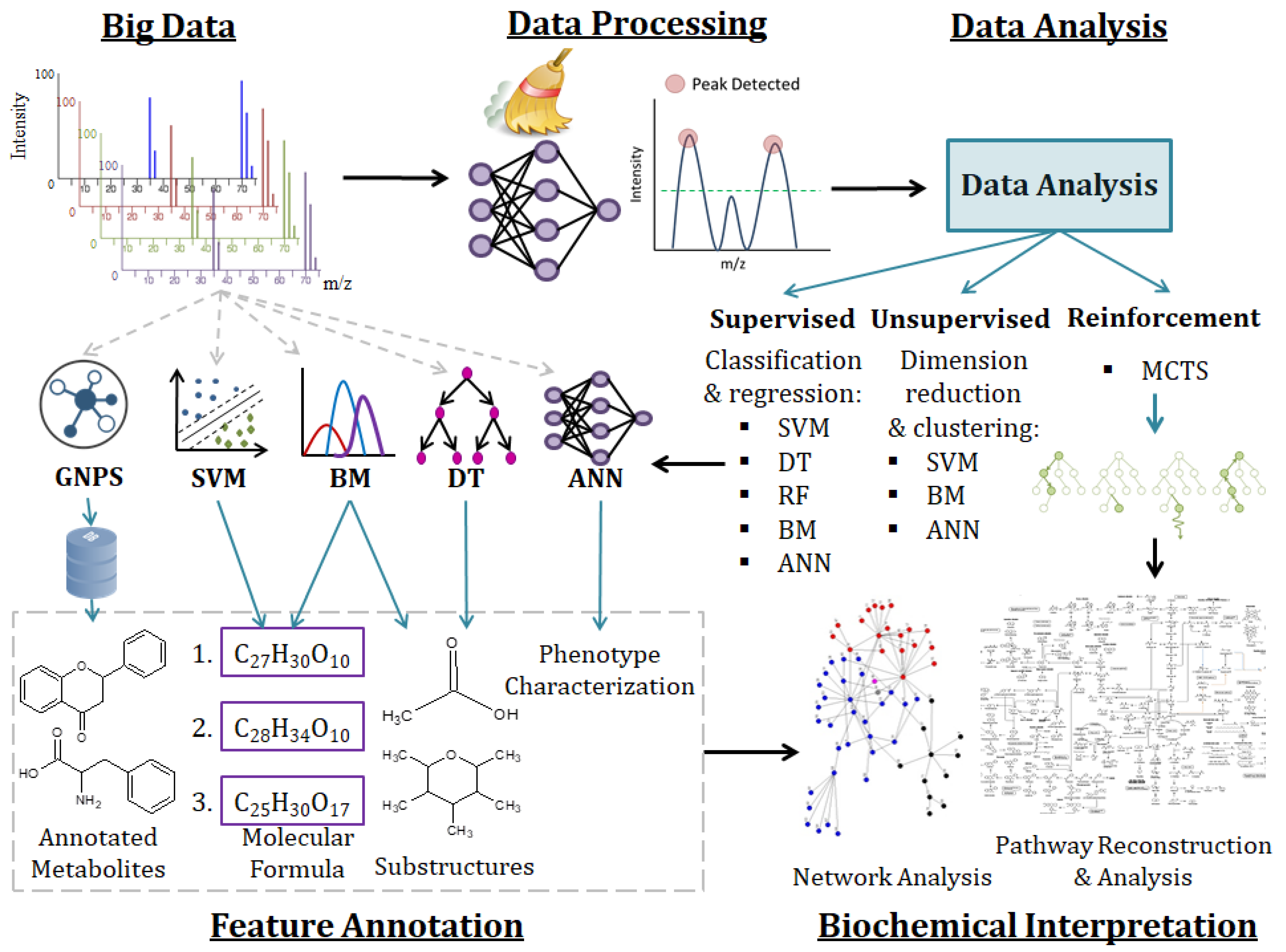
2.3. Machine Learning Methods for Metabolomic Data Mining and Interpretation
2.3.1. Support Vector Machines
2.3.2. Decision Trees
2.3.3. Ensemble Learning
2.3.4. Bayesian Models
2.3.5. Artificial Neural Networks
2.3.6. Machine Learning for Pathway Modeling
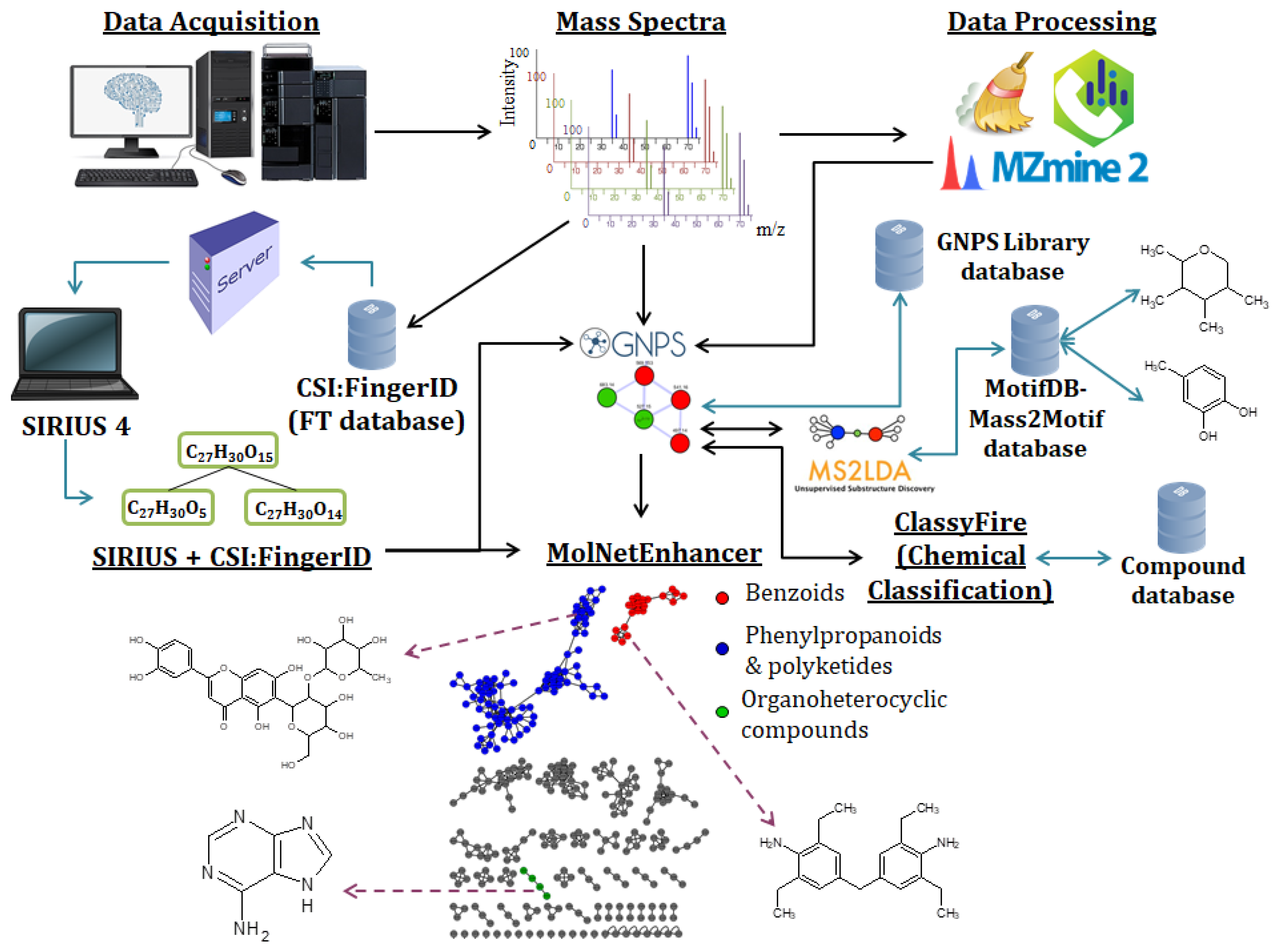
2.4. Large-Scale Metabolite Annotation
2.4.1. Spectral Similarity and Substructure Based Annotation
2.4.2. Structure-Based Annotation
2.4.3. Spectral Similarity Scoring for Library Matching and Correlation of Spectra
2.4.4. Chemical Compound Class-Based Annotation
2.4.5. Large-Scale and Repository-Wide Metabolomics Analyses
3. Metabolomics and Plant Responses to Abiotic Stresses—Current Frameworks
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xu, M.; David, J.M.; Kim, S.H. The fourth industrial revolution: Opportunities and challenges. Int. J. Financ. Res. 2018, 9, 90–95. [Google Scholar] [CrossRef] [Green Version]
- Ndung’u, N.; Signé, L. The Fourth Industrial Revolution and digitization will transform Africa into a global powerhouse. Foresight Afr. 2020, 2020, 60–73. [Google Scholar]
- Koh, L.; Orzes, G.; Jia, F. The fourth industrial revolution (Industry 4.0): Technologies disruption on operations and supply chain management. Int. J. Oper. Prod. Manag. 2019, 39, 817–828. [Google Scholar] [CrossRef]
- Mendez, K.M.; Pritchard, L.; Reinke, S.N.; Broadhurst, D.I. Toward collaborative open data science in metabolomics using Jupyter Notebooks and cloud computing. Metabolomics 2019, 15, s11306–s113019. [Google Scholar] [CrossRef] [Green Version]
- Tugizimana, F.; Engel, J.; Salek, R.; Dubery, I.; Piater, L.; Burgess, K. The Disruptive 4IR in the Life Sciences: Metabolomics; Springer: Berlin/Heidelberg, Germany, 2020; Volume 674, ISBN 9783030482305. [Google Scholar]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Kale, N.S.; Haug, K.; Conesa, P.; Jayseelan, K.; Moreno, P.; Rocca-Serra, P.; Nainala, V.C.; Spicer, R.A.; Williams, M.; Li, X.; et al. MetaboLights: An open-access database repository for metabolomics data. Curr. Protoc. Bioinform. 2016, 2016, 14.13.1–14.13.18. [Google Scholar] [CrossRef]
- Van Der Hooft, J.J.J.; Wandy, J.; Barrett, M.P.; Burgess, K.E.V.; Rogers, S. Topic modeling for untargeted substructure exploration in metabolomics. Proc. Natl. Acad. Sci. USA 2016, 113, 13738–13743. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.B.; Ernst, M.; van der Hooft, J.J.J.; da Silva, R.R.; Park, J.; Medema, M.H.; Sung, S.H.; Dorrestein, P.C. Comprehensive mass spectrometry-guided phenotyping of plant specialized metabolites reveals metabolic diversity in the cosmopolitan plant family Rhamnaceae. Plant J. 2019, 98, 1134–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kell, D.B.; Brown, M.; Davey, H.M.; Dunn, W.B.; Spasic, I.; Oliver, S.G. Metabolic footprinting and systems biology: The medium is the message. Nat. Rev. Microbiol. 2005, 3, 557–565. [Google Scholar] [CrossRef]
- Rai, A.; Yamazaki, M.; Saito, K. A new era in plant functional genomics. Curr. Opin. Syst. Biol. 2019, 15, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Fernie, A.R.; Bachem, C.W.B.; Helariutta, Y.; Neuhaus, H.E.; Prat, S.; Ruan, Y.L.; Stitt, M.; Sweetlove, L.J.; Tegeder, M.; Wahl, V.; et al. Synchronization of developmental, molecular and metabolic aspects of source–sink interactions. Nat. Plants 2020, 6, 55–66. [Google Scholar] [CrossRef]
- Drobek, M.; Frąc, M.; Cybulska, J. Plant biostimulants: Importance of the quality and yield of horticultural crops and the improvement of plant tolerance to abiotic stress—A review. Agronomy 2019, 9, 335. [Google Scholar] [CrossRef] [Green Version]
- Llorens, E.; González-Hernández, A.I.; Scalschi, L.; Fernández-Crespo, E.; Camañes, G.; Vicedo, B.; García-Agustín, P. Priming Mediated Stress and Cross-Stress Tolerance in Plants: Concepts and Opportunities; Elsevier: Amsterdam, The Netherlands, 2020; ISBN 9780128178935. [Google Scholar]
- Janni, M.; Gullì, M.; Maestri, E.; Marmiroli, M.; Valliyodan, B.; Nguyen, H.T.; Marmiroli, N.; Foyer, C. Molecular and genetic bases of heat stress responses in crop plants and breeding for increased resilience and productivity. J. Exp. Bot. 2020, 71, 3780–3802. [Google Scholar] [CrossRef]
- Ramegowda, V.; Da Costa, M.V.J.; Harihar, S.; Karaba, N.N.; Sreeman, S.M. Abiotic and Biotic Stress Interactions in Plants: A Cross-Tolerance Perspective; Elsevier: Amsterdam, The Netherlands, 2020; ISBN 9780128178935. [Google Scholar]
- Nobori, T.; Tsuda, K. The plant immune system in heterogeneous environments. Curr. Opin. Plant Biol. 2019, 50, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Razzaq, A.; Sadia, B.; Raza, A.; Hameed, M.K.; Saleem, F. Metabolomics: A way forward for crop improvement. Metabolites 2019, 9, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Boukhari, M.E.M.; Barakate, M.; Bouhia, Y.; Lyamlouli, K. Trends in seaweed extract based biostimulants: Manufacturing process and beneficial effect on soil-plant systems. Plants 2020, 9, 359. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Redondo-Gómez, S. Abiotic and Biotic Stress Tolerance in Plants. In Molecular Stress Physiology of Plants; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–440. ISBN 9788132208075. [Google Scholar]
- Obata, T.; Fernie, A.R. The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 2012, 69, 3225–3243. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J. Review Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Saud, S.; Li, X.; Chen, Y.; Zhang, L.; Fahad, S.; Hussain, S.; Sadiq, A.; Chen, Y. Silicon application increases drought tolerance of Kentucky bluegrass by improving plant water relations and morphophysiological functions. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef]
- Tátrai, Z.A.; Sanoubar, R.; Pluhár, Z.; Mancarella, S.; Orsini, F.; Gianquinto, G. Morphological and Physiological Plant Responses to Drought Stress in Thymus citriodorus. Int. J. Agron. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Tugizimana, F.; Piater, L.; Dubery, I. Plant metabolomics: A new frontier in phytochemical analysis. S. Afr. J. Sci. 2013, 109, 1–11. [Google Scholar] [CrossRef]
- Castro-moretti, F.R.; Gentzel, I.N.; Mackey, D.; Alonso, A.P. Metabolomics as an emerging tool for the study of plant–pathogen interactions. Metabolites 2020, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawid, C.; Hille, K. Functional metabolomics-a useful tool to characterize stress-induced metabolome alterations opening new avenues towards tailoring food crop quality. Agronomy 2018, 8, 138. [Google Scholar] [CrossRef] [Green Version]
- Monge, M.E.; Dodds, J.N.; Baker, E.S.; Edison, A.S.; Fernaacutendez, F.M. Challenges in Identifying the Dark Molecules of Life. Annu. Rev. Anal. Chem. 2019, 12, 177–199. [Google Scholar] [CrossRef] [PubMed]
- Wolfender, J.L.; Nuzillard, J.M.; Van Der Hooft, J.J.J.; Renault, J.H.; Bertrand, S. Accelerating Metabolite Identification in Natural Product Research: Toward an Ideal Combination of Liquid Chromatography-High-Resolution Tandem Mass Spectrometry and NMR Profiling, in Silico Databases, and Chemometrics. Anal. Chem. 2019, 91, 704–742. [Google Scholar] [CrossRef] [PubMed]
- Rinschen, M.M.; Ivanisevic, J.; Giera, M.; Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol. 2019, 20, 353–367. [Google Scholar] [CrossRef]
- Beisken, S.; Eiden, M.; Salek, R.M. Getting the right answers: Understanding metabolomics challenges. Expert Rev. Mol. Diagn. 2015, 15, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Tugizimana, F.; Mhlongo, M.I.; Piater, L.A.; Dubery, I.A. Metabolomics in plant priming research: The way forward? Int. J. Mol. Sci. 2018, 19, 1759. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.A.; De Souza, L.P.; Serag, A.; Fernie, A.R.; Farag, M.A.; Ezzat, S.M.; Alseekh, S. Metabolomics in the context of plant natural products research: From sample preparation to metabolite analysis. Metabolites 2020, 10, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.W.; Li, Q.H.; Xu, Z.D.; Dou, J.J. Mass spectrometry-based metabolomics in health and medical science: A systematic review. RSC Adv. 2020, 10, 3092–3104. [Google Scholar] [CrossRef] [Green Version]
- Damiani, C.; Gaglio, D.; Sacco, E.; Alberghina, L.; Vanoni, M. Systems metabolomics: From metabolomic snapshots to design principles. Curr. Opin. Biotechnol. 2020, 63, 190–199. [Google Scholar] [CrossRef]
- Kell, D.B.; Oliver, S.G. The metabolome 18 years on: A concept comes of age. Metabolomics 2016, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tugizimana, F.; Djami-Tchatchou, A.T.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. Metabolomic analysis of defense-related reprogramming in sorghum bicolor in response to Colletotrichum sublineolum infection reveals a functional metabolic web of phenylpropanoid and flavonoid pathways. Front. Plant Sci. 2019, 9, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Filla, L.A.; Sanders, K.L.; Filla, R.T.; Edwards, J.L. Automated sample preparation in a microfluidic culture device for cellular metabolomics. Analyst 2016, 141, 3858–3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.G.; Hu, J.; Wu, X.; Xu, Y.J. The Recent Developments in Sample Preparation for Mass Spectrometry-Based Metabolomics. Crit. Rev. Anal. Chem. 2017, 47, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.; Park, J.M.; Duong, V.A.; Kwon, D.; Jeon, J.; Han, M.; Cho, B.K.; Choi, H.K.; Lee, C.G.; Kang, H.G.; et al. An automated high-throughput sample preparation method using double-filtration for serum metabolite LC-MS analysis. Anal. Methods 2019, 11, 4060–4065. [Google Scholar] [CrossRef]
- Roopashree, K.M.; Naik, D. Advanced method of secondary metabolite extraction and quality analysis. J. Pharmacogn. Phytochem. 2019, 8, 1829–1842. [Google Scholar]
- Rigobello-Masini, M.; Pereira, E.A.O.; Abate, G.; Masini, J.C. Solid-Phase Extraction of Glyphosate in the Analyses of Environmental, Plant, and Food Samples. Chromatographia 2019, 82, 1121–1138. [Google Scholar] [CrossRef]
- Van Der Hooft, J.J.J.; Akermi, M.; Unlu, F.Y.; Mihaleva, V.; Roldan, V.G.; Bino, R.J.; De Vos, R.C.H.; Vervoort, J. Structural annotation and elucidation of conjugated phenolic compounds in black, green, and white tea extracts. J. Agric. Food Chem. 2012, 60, 8841–8850. [Google Scholar] [CrossRef]
- Raks, V.; Al-Suod, H.; Buszewski, B. Isolation, Separation, and Preconcentration of Biologically Active Compounds from Plant Matrices by Extraction Techniques. Chromatographia 2018, 81, 189–202. [Google Scholar] [CrossRef]
- Bladergroen, M.R.; van der Burgt, Y.E.M. Solid-Phase Extraction Strategies to Surmount Body Fluid Sample Complexity in High-Throughput Mass Spectrometry-Based Proteomics. J. Anal. Methods Chem. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitnikov, D.G.; Monnin, C.S.; Vuckovic, D. Systematic Assessment of Seven Solvent and Solid-Phase Extraction Methods for Metabolomics Analysis of Human Plasma by LC-MS. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Miggiels, P.; Wouters, B.; van Westen, G.J.P.; Dubbelman, A.C.; Hankemeier, T. Novel technologies for metabolomics: More for less. TrAC Trends Anal. Chem. 2019, 120, 115323. [Google Scholar] [CrossRef]
- Reyes-Garcés, N.; Gionfriddo, E. Recent developments and applications of solid phase microextraction as a sample preparation approach for mass-spectrometry-based metabolomics and lipidomics. TrAC Trends Anal. Chem. 2019, 113, 172–181. [Google Scholar] [CrossRef]
- Mousavi, F.; Bojko, B.; Pawliszyn, J. High-Throughput Solid-Phase Microextraction–Liquid Chromatography–Mass Spectrometry for Microbial Untargeted Metabolomics. In Microbial Metabolomics: Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2018; Volume 1859, pp. 133–152. ISBN 9781493987573. [Google Scholar]
- Dugheri, S.; Mucci, N.; Bonari, A.; Marrubini, G.; Cappelli, G.; Ubiali, D.; Campagna, M.; Montalti, M.; Arcangeli, G. Liquid Phase Microextraction Techniques Combined with Chromatography Analysis: A Review. Acta Chromatogr. 2020, 32, 69–79. [Google Scholar] [CrossRef]
- He, Y.; Concheiro-Guisan, M. Microextraction sample preparation techniques in forensic analytical toxicology. Biomed. Chromatogr. 2019, 33. [Google Scholar] [CrossRef] [Green Version]
- Drouin, N.; Rudaz, S.; Schappler, J. Sample preparation for polar metabolites in bioanalysis. Analyst 2018, 143, 16–20. [Google Scholar] [CrossRef] [Green Version]
- Sramkova, I.H.; Horstkotte, B.; Fikarova, K.; Sklenarova, H.; Solich, P. Direct-immersion single-drop microextraction and in-drop stirring microextraction for the determination of nanomolar concentrations of lead using automated Lab-In-Syringe technique. Talanta 2018, 184, 162–172. [Google Scholar] [CrossRef]
- Kellogg, J.J.; Wallace, E.D.; Graf, T.N.; Oberlies, N.H.; Cech, N.B. Conventional and accelerated-solvent extractions of green tea (camellia sinensis) for metabolomics-based chemometrics. J. Pharm. Biomed. Anal. 2017, 145, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Pinu, F.R.; Villas-Boas, S.G.; Aggio, R. Analysis of intracellular metabolites from microorganisms: Quenching and extraction protocols. Metabolites 2017, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Njila, M.I.N.; Mahdi, E.; Lembe, D.; Nde, Z.; Doriane, N. Review on Extraction and Isolation of Plant Secondary Metabolites. In Proceedings of the 7th International Conference on Agricultural, Chemical, Biological and Environmental Sciences (ACBES-2017), Kuala Lumpur, Malasya, 22–24 May 2017. [Google Scholar] [CrossRef]
- Sanchez-Camargo, A.d.P.; Parada-Alonso, F.; Ibanez, E.; Cifuentes, A. Recent applications of on-line supercritical fluid extraction coupled to advanced analytical techniques for compounds extraction and identification. J. Sep. Sci. 2019, 42, 243–257. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, I.; Javad, S.; Yousaf, Z.; Iqbal, S.; Jabeen, K. Microwave assisted extraction of phytochemicals an efficient and modern approach for botanicals and pharmaceuticals. Pak. J. Pharm. Sci. 2019, 32, 223–230. [Google Scholar]
- Lozano-Grande, M.A.; Dávila-Ortiz, G.; García-Dávila, J.; Ríos-Cortés, G.; Espitia-Rangel, E.; Martínez-Ayala, A.L. Optimisation of Microwave-Assisted Extraction of Squalene from Amaranthus spp. Seeds. J. Microw. Power Electromagn. Energy 2019, 53, 243–258. [Google Scholar] [CrossRef]
- Melgar, B.; Dias, M.I.; Barros, L.; Ferreira, I.C.F.R.; Rodriguez-Lopez, A.D.; Garcia-Castello, E.M. Ultrasound and Microwave Assisted Extraction of Opuntia Fruit Peels Biocompounds: Optimization and Comparison Using RSM-CCD. Molecules 2019, 24, 3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramasamy, J.; Kandasamy, R.; Palanisamy, S.; Nadesan, S. Optimization of Ultrasonic-Assisted Extraction of Flavonoids and Anti-oxidant Capacity from the Whole Plant of Andrographis echioides (L.) Nees by Response Surface Methodology and Chemical Composition Analysis. Pharmacogn. Mag. 2019, 15, 547–556. [Google Scholar] [CrossRef]
- Alcantara, C.; Zugcic, T.; Abdelkebir, R.; Garcaa-Perez, J.V.; Jambrak, A.R.; Lorenzo, J.M.; Collado, M.C.; Granato, D.; Barba, F.J. Effects of ultrasound-assisted extraction and solvent on the phenolic profile, bacterial growth, and anti-inflammatory/antioxidant activities of mediterranean olive and fig leaves extracts. Molecules 2020, 25, 1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugheri, S.; Mucci, N.; Bonari, A.; Marrubini, G.; Cappelli, G.; Ubiali, D.; Campagna, M.; Montalti, M.; Arcangeli, G. Solid phase microextraction techniques used for gas chromatography: A review. Acta Chromatogr. 2020, 32, 1–9. [Google Scholar] [CrossRef]
- Khaled, A.; Gionfriddo, E.; Acquaro, V.; Singh, V.; Pawliszyn, J. Development and validation of a fully automated solid phase microextraction high throughput method for quantitative analysis of multiresidue veterinary drugs in chicken tissue. Anal. Chim. Acta 2019, 1056, 34–46. [Google Scholar] [CrossRef]
- Medina, D.A.V.; Santos-Neto, A.J.; Cerda, V.; Maya, F. Automated dispersive liquid-liquid microextraction based on the solidification of the organic phase. Talanta 2018, 189, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Ekezie, F.G.C.; Sun, D.W.; Cheng, J.H. Acceleration of microwave-assisted extraction processes of food components by integrating technologies and applying emerging solvents: A review of latest developments. Trends Food Sci. Technol. 2017, 67, 160–172. [Google Scholar] [CrossRef]
- Llompart, M.; Celeiro, M.; Dagnac, T. Microwave-assisted extraction of pharmaceuticals, personal care products and industrial contaminants in the environment. TrAC Trends Anal. Chem. 2019, 116, 136–150. [Google Scholar] [CrossRef]
- Chemat, F.; Rombaut, N.; Sicaire, A.G.; Meullemiestre, A.; Fabiano-Tixier, A.S.; Abert-Vian, M. Ultrasound assisted extraction of food and natural products. Mechanisms, techniques, combinations, protocols and applications. A review. Ultrason. Sonochem. 2017, 34, 540–560. [Google Scholar] [CrossRef]
- Panzella, L.; Moccia, F.; Nasti, R.; Marzorati, S.; Verotta, L.; Napolitano, A. Bioactive Phenolic Compounds from Agri-Food Wastes: An Update on Green and Sustainable Extraction Methodologies. Front. Nutr. 2020, 7, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Risticevic, S.; Souza-Silva, E.A.; Gionfriddo, E.; DeEll, J.R.; Cochran, J.; Hopkins, W.S.; Pawliszyn, J. Application of in vivo solid phase microextraction (SPME) in capturing metabolome of apple (Malus × domestica Borkh.) fruit. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabir, A.; Locatelli, M.; Ulusoy, H.I. Recent trends in microextraction techniques employed in analytical and bioanalytical sample preparation. Separations 2017, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.H.; Han, S.B.; Wang, Y. Development of an improved sample preparation platform for acidic endogenous hormones in plant tissues using electromembrane extraction. J. Chromatogr. A 2018, 1535, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gallo-Molina, A.C.; Castro-Vargas, H.I.; Garzón-Méndez, W.F.; Martínez Ramírez, J.A.; Rivera Monroy, Z.J.; King, J.W.; Parada-Alfonso, F. Extraction, isolation and purification of tetrahydrocannabinol from the Cannabis sativa L. plant using supercritical fluid extraction and solid phase extraction. J. Supercrit. Fluids 2019, 146, 208–216. [Google Scholar] [CrossRef]
- Belwal, T.; Pandey, A.; Bhatt, I.D.; Rawal, R.S. Optimized microwave assisted extraction (MAE) of alkaloids and polyphenols from Berberis roots using multiple-component analysis. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tsiaka, T.; Fotakis, C.; Lantzouraki, D.Z.; Tsiantas, K.; Siapi, E.; Sinanoglou, V.J.; Zoumpoulakis, P. Expanding the Role of Sub-Exploited DOE-High Energy Extraction and Metabolomic Profiling towards Agro-Byproduct Valorization: The Case of Carotenoid-Rich Apricot Pulp. Molecules 2020, 25, 2702. [Google Scholar] [CrossRef]
- Fleischer, H.; Drews, R.R.; Janson, J.; Chinna Patlolla, B.R.; Chu, X.; Klos, M.; Thurow, K. Application of a Dual-Arm Robot in Complex Sample Preparation and Measurement Processes. J. Lab. Autom. 2016, 21, 671–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elpa, D.P.; Prabhu, G.R.D.; Wu, S.P.; Tay, K.S.; Urban, P.L. Automation of mass spectrometric detection of analytes and related workflows: A review. Talanta 2020, 207, 120304. [Google Scholar] [CrossRef]
- Joshi, S.; Chu, X.; Fleischer, H.; Roddelkopf, T.; Klos, M.; Thurow, K. Analysis of measurement process design for a dual-arm robot using graphical user interface. IEEE Int. Instrum. Meas. Technol. Conf. 2019, 1–6. [Google Scholar] [CrossRef]
- Alseekh, S.; Fernie, A.R. Metabolomics 20 years on: What have we learned and what hurdles remain? Plant J. 2018, 94, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Nandania, J.; Peddinti, G.; Pessia, A.; Kokkonen, M.; Velagapudi, V. Validation and automation of a high-throughput multitargeted method for semiquantification of endogenous metabolites from different biological matrices using tandem mass spectrometry. Metabolites 2018, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivia, W.R.; Raedschelders, K.; Gomez, M.; Van Eyk, J.E. Automated Metabolite Extraction for Plasma Using the Agilent Bravo Platform. 2019. Available online: https://www.agilent.com/cs/library/technicaloverviews/public/technicaloverview-metabolomics-sample-prep-bravo-5994-0685en-agilent.pdf (accessed on 27 February 2020).
- Akita, S.; Watanabe, K. New Analytical Intelligence Concept—Support for Automating Analytical Operations; TR C190-E2; Shimadzu Corp.: Kyoto, Japan, 2019; pp. 1–4. [Google Scholar]
- Chen, D.; Wang, Z.; Guo, D.; Orekhov, V.; Qu, X. Review and Prospect: Deep Learning in Nuclear Magnetic Resonance Spectroscopy. Chem. A Eur. J. 2020, 26, 10391–10401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, A.M.; Ribeiro-Barros, A.I.; Antonio, C. Experimental design and sample preparation in forest tree metabolomics. Metabolites 2019, 9, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Su, X.; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite Measurement: Pitfalls to Avoid and Practices to Follow. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef]
- Stoll, D.R.; Carr, P.W. Two-Dimensional Liquid Chromatography: A State of the Art Tutorial. Anal. Chem. 2017, 89, 519–531. [Google Scholar] [CrossRef]
- Keppler, E.A.H.; Jenkins, C.L.; Davis, T.J.; Bean, H.D. Advances in the application of comprehensive two-dimensional gas chromatography in metabolomics. TrAC Trends Anal. Chem. 2018, 109, 275–286. [Google Scholar] [CrossRef]
- Fouque, K.J.D.; Fernandez-Lima, F. Recent advances in biological separations using trapped ion mobility spectrometry—Mass spectrometry. TrAC Trends Anal. Chem. 2019, 116, 308–315. [Google Scholar] [CrossRef]
- De Raad, M.; Fischer, C.R.; Northen, T.R. High-throughput platforms for metabolomics. Curr. Opin. Chem. Biol. 2016, 30, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Dodds, J.N.; Baker, E.S. Ion Mobility Spectrometry: Fundamental Concepts, Instrumentation, Applications, and the Road Ahead. J. Am. Soc. Mass Spectrom. 2019, 30, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.D.; Zhou, Z.W.; Zhu, Z.J. The Application of Ion Mobility-Mass Spectrometry in Untargeted Metabolomics: From Separation to Identification. J. Anal. Test. 2020, 4, 163–174. [Google Scholar] [CrossRef]
- Purves, R.W. Enhancing Biological LC-MS Analyses Using Ion Mobility Spectrometry; Elsevier Ltd.: Amsterdam, The Netherlands, 2018; Volume 79. [Google Scholar]
- Yang, X.; Wei, S.; Liu, B.; Guo, D.; Zheng, B.; Feng, L.; Liu, Y.; Tomas-Barberan, F.A.; Luo, L.; Huang, D. A novel integrated non-targeted metabolomic analysis reveals significant metabolite variations between different lettuce (Lactuca sativa. L) varieties. Hortic. Res. 2018, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- McCullagh, M.; Pereira, C.A.M.; Yariwake, J.H. Use of ion mobility mass spectrometry to enhance cumulative analytical specificity and separation to profile 6-C/8-C-glycosylflavone critical isomer pairs and known–unknowns in medicinal plants. Phytochem. Anal. 2019, 30, 424–436. [Google Scholar] [CrossRef]
- Metz, T.O.; Baker, E.S.; Schymanski, E.L.; Renslow, R.S.; Thomas, D.G.; Causon, T.J.; Webb, I.K.; Hann, S.; Smith, R.D.; Teeguarden, J.G. Integrating ion mobility spectrometry into mass spectrometry-based exposome measurements: What can it add and how far can it go? Bioanalysis 2017, 9, 81–98. [Google Scholar] [CrossRef] [Green Version]
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Schulz, B.L.; Ferro, V. Applications of ion mobility-mass spectrometry in carbohydrate chemistry and glycobiology. Molecules 2018, 23, 2557. [Google Scholar] [CrossRef] [Green Version]
- Boughton, B.A.; Thinagaran, D.; Sarabia, D.; Bacic, A.; Roessner, U. Mass spectrometry imaging for plant biology: A review. Phytochem. Rev. 2016, 15, 445–488. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Zhang, Y.; Liu, Y.; He, H.; Han, M.; Li, Y.; Zeng, M.; Wang, X. Recent advances in matrix-assisted laser desorption/ionisation mass spectrometry imaging (MALDI-MSI) for in situ analysis of endogenous molecules in plants. Phytochem. Anal. 2018, 29, 351–364. [Google Scholar] [CrossRef]
- Holzlechner, M.; Eugenin, E.; Prideaux, B. Mass spectrometry imaging to detect lipid biomarkers and disease signatures in cancer. Cancer Rep. 2019, 2. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, T. Spatial Metabolomics and Imaging Mass Spectrometry in the Age of Artificial Intelligence. Annu. Rev. Biomed. Data Sci. 2020, 3, 61–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swales, J.G.; Hamm, G.; Clench, M.R.; Goodwin, R.J.A. Mass spectrometry imaging and its application in pharmaceutical research and development: A concise review. Int. J. Mass Spectrom. 2019, 437, 99–112. [Google Scholar] [CrossRef]
- Snel, M.F. Ion Mobility Separation Mass Spectrometry Imaging, 1st ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2019; Volume 83. [Google Scholar]
- Fujii, T.; Matsuda, S.; Tejedor, M.L.; Esaki, T.; Sakane, I.; Mizuno, H.; Tsuyama, N.; Masujima, T. Direct metabolomics for plant cells by live single-cell mass spectrometry. Nat. Protoc. 2015, 10, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Foll, M.C.; Moritz, L.; Wollmann, T.; Stillger, M.N.; Vockert, N.; Werner, M.; Bronsert, P.; Rohr, K.; Grüning, B.A.; Schilling, O. Accessible and reproducible mass spectrometry imaging data analysis in Galaxy. Gigascience 2019, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dueñas, M.E.; Larson, E.A.; Lee, Y.J. Toward mass spectrometry imaging in the metabolomics scale: Increasing metabolic coverage through multiple on-tissue chemical modifications. Front. Plant Sci. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Silva, D.B.; Turatti, I.C.C.; Gouveia, D.R.; Ernst, M.; Teixeira, S.P.; Lopes, N.P. Mass Spectrometry of flavonoid vicenin-2, based sunlight barriers in Lychnophora species. Sci. Rep. 2014, 4, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, M.; Nothias, L.F.; van der Hooft, J.J.J.; Silva, R.R.; Saslis-Lagoudakis, C.H.; Grace, O.M.; Martinez-Swatson, K.; Hassemer, G.; Funez, L.A.; Simonsen, H.T.; et al. Assessing specialized metabolite diversity in the cosmopolitan plant genus Euphorbia l. Front. Plant Sci. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bokhart, M.T.; Nazari, M.; Garrard, K.P.; Muddiman, D.C. MSiReader v1.0: Evolving Open-Source Mass Spectrometry Imaging Software for Targeted and Untargeted Analyses. J. Am. Soc. Mass Spectrom. 2018, 28, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Hu, T.; Woods, E.; Allam, M.; Cai, S.; Henderson, W.; Coskun, A.F. Spatially resolved 3D metabolomic profiling in tissues. Sci. Adv. 2021, 7, 1–17. [Google Scholar] [CrossRef]
- Wang, H.; Chen, L.; Sun, L. Digital microfluidics: A promising technique for biochemical applications. Front. Mech. Eng. 2017, 12, 510–525. [Google Scholar] [CrossRef]
- Damiati, S.; Kompella, U.B.; Damiati, S.A.; Kodzius, R. Microfluidic devices for drug delivery systems and drug screening. Genes 2018, 9, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedde, R.D.; Li, H.; Borchers, C.H.; Akbari, M. Microfluidic-Mass Spectrometry Interfaces for Translational Proteomics. Trends Biotechnol. 2017, 35, 954–970. [Google Scholar] [CrossRef]
- Feng, D.; Xu, T.; Li, H.; Shi, X.; Xu, G. Single-cell Metabolomics Analysis by Microfluidics and Mass Spectrometry: Recent New Advances. J. Anal. Test. 2020, 4, 198–209. [Google Scholar] [CrossRef]
- Steckel, A.; Schlosser, G. An organic chemist’s guide to electrospray mass spectrometric structure elucidation. Molecules 2019, 24, 611. [Google Scholar] [CrossRef] [Green Version]
- Stanley, C.E.; Shrivastava, J.; Brugman, R.; Heinzelmann, E.; van Swaay, D.; Grossmann, G. Dual-flow-RootChip reveals local adaptations of roots towards environmental asymmetry at the physiological and genetic levels. New Phytol. 2018, 217, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Fenaille, F.; Barbier Saint-Hilaire, P.; Rousseau, K.; Junot, C. Data acquisition workflows in liquid chromatography coupled to high resolution mass spectrometry-based metabolomics: Where do we stand? J. Chromatogr. A 2017, 1526, 1–12. [Google Scholar] [CrossRef]
- Davies, V.; Wandy, J.; Weidt, S.; van der Hooft, J.J.J.; Miller, A.; Daly, R.; Rogers, S. Rapid Development of Improved Data-Dependent Acquisition Strategies. Anal. Chem. 2021, 93, 5676–5683. [Google Scholar] [CrossRef]
- Wandy, J.; Davies, V.; Van Der Hooft, J.J.J.; Weidt, S.; Daly, R.; Rogers, S. In silico optimization of mass spectrometry fragmentation strategies in metabolomics. Metabolites 2019, 9, 219. [Google Scholar] [CrossRef] [Green Version]
- Bailey, D.J.; McDevitt, M.T.; Westphall, M.S.; Pagliarini, D.J.; Coon, J.J. Intelligent data acquisition blends targeted and discovery methods. J. Proteome Res. 2014, 13, 2152–2161. [Google Scholar] [CrossRef]
- Van Der Hooft, J.J.J.; Vervoort, J.; Bino, R.J.; Beekwilder, J.; De Vos, R.C.H. Polyphenol identification based on systematic and robust high-resolution accurate mass spectrometry fragmentation. Anal. Chem. 2011, 83, 409–416. [Google Scholar] [CrossRef]
- Van der Hooft, J.J.J.; Vervoort, J.; Bino, R.J.; de Vos, R.C.H. Spectral trees as a robust annotation tool in LC-MS based metabolomics. Metabolomics 2012, 8, 691–703. [Google Scholar] [CrossRef]
- Van der Hooft, J.J.J.; Rankin, N. Metabolite Identification in Complex Mixtures Using Nuclear Magnetic Resonance Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2016; ISBN 9783319283883. [Google Scholar]
- Wishart, D.S. NMR metabolomics: A look ahead. J. Magn. Reson. 2019, 306, 155–161. [Google Scholar] [CrossRef]
- Emwas, A.H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Nagana Gowda, G.A.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. Nmr spectroscopy for metabolomics research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; Gogna, N.; Dorai, K. NMR-based investigation of the altered metabolic response of Bougainvillea spectabilis leaves exposed to air pollution stress during the circadian cycle. Environ. Exp. Bot. 2019, 164, 58–70. [Google Scholar] [CrossRef]
- Bornet, A.; Maucourt, M.; Deborde, C.; Jacob, D.; Milani, J.; Vuichoud, B.; Ji, X.; Dumez, J.N.; Moing, A.; Bodenhausen, G.; et al. Highly Repeatable Dissolution Dynamic Nuclear Polarization for Heteronuclear NMR Metabolomics. Anal. Chem. 2016, 88, 6179–6183. [Google Scholar] [CrossRef]
- Coutinho, I.D.; Moraes, T.B.; Mertz-Henning, L.M.; Nepomuceno, A.L.; Giordani, W.; Marcolino-Gomes, J.; Santagneli, S.; Colnago, L.A. Integrating High-Resolution and Solid-State Magic Angle Spinning NMR Spectroscopy and a Transcriptomic Analysis of Soybean Tissues in Response to Water Deficiency. Phytochem. Anal. 2017, 28, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Van der Hooft, J.J.J.; Mihaleva, V.; De Vos, R.C.H.; Bino, R.J.; Vervoort, J. A strategy for fast structural elucidation of metabolites in small volume plant extracts using automated MS-guided LC-MS-SPE-NMR. Magn. Reson. Chem. 2011, 49, S55–S60. [Google Scholar] [CrossRef]
- Lima, R.D.C.L.; Gramsbergen, S.M.; Van Staden, J.; Jager, A.K.; Kongstad, K.T.; Staerk, D. Advancing HPLC-PDA-HRMS-SPE-NMR Analysis of Coumarins in Coleonema album by Use of Orthogonal Reversed-Phase C18 and Pentafluorophenyl Separations. J. Nat. Prod. 2017, 80, 1020–1027. [Google Scholar] [CrossRef]
- Hao, J.; Liebeke, M.; Astle, W.; De Iorio, M.; Bundy, J.G.; Ebbels, T.M.D. Bayesian deconvolution and quantification of metabolites in complex 1D NMR spectra using BATMAN. Nat. Protoc. 2014, 9, 1416–1427. [Google Scholar] [CrossRef]
- Röhnisch, H.E.; Eriksson, J.; Müllner, E.; Agback, P.; Sandström, C.; Moazzami, A.A. AQuA: An Automated Quantification Algorithm for High-Throughput NMR-Based Metabolomics and Its Application in Human Plasma. Anal. Chem. 2018, 90, 2095–2102. [Google Scholar] [CrossRef]
- Cañueto, D.; Gómez, J.; Salek, R.M.; Correig, X.; Cañellas, N. rDolphin: A GUI R package for proficient automatic profiling of 1D 1 H-NMR spectra of study datasets. Metabolomics 2018, 14, 1–5. [Google Scholar] [CrossRef]
- Jonas, E.; Kuhn, S. Rapid prediction of NMR spectral properties with quantified uncertainty. J. Cheminform. 2019, 11, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Nugroho, A.E.; Morita, H. Computationally-assisted discovery and structure elucidation of natural products. J. Nat. Med. 2019, 73, 687–695. [Google Scholar] [CrossRef] [Green Version]
- Reher, R.; Kim, H.W.; Zhang, C.; Mao, H.H.; Wang, M.; Nothias, L.F.; Caraballo-Rodriguez, A.M.; Glukhov, E.; Teke, B.; Leao, T.; et al. A Convolutional Neural Network-Based Approach for the Rapid Annotation of Molecularly Diverse Natural Products. J. Am. Chem. Soc. 2020, 142, 4114–4120. [Google Scholar] [CrossRef]
- Mäkelä, V.; Vaahtera, L.; Helminen, J.; Koskela, H.; Brosché, M.; Kilpeläinen, I.; Heikkinen, S. Automated processing and statistical analysis of nmr spectra obtained from Arabidopsis thaliana extracts. bioRxiv 2019. [Google Scholar] [CrossRef]
- Peng, W.K. Clustering Nuclear Magnetic Resonance: Machine learning assistive rapid two-dimensional relaxometry mapping. Eng. Reports 2021, 1–11. [Google Scholar] [CrossRef]
- Goeddel, L.C.; Patti, G.J. Maximizing the value of metabolomic data. Bioanalysis 2012, 4, 2199–2201. [Google Scholar] [CrossRef]
- Biesecker, L.G. Hypothesis-generating research and predictive medicine. Genome Res. 2013, 23, 1051–1053. [Google Scholar] [CrossRef] [Green Version]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef]
- Boccard, J.; Rudaz, S. Harnessing the complexity of metabolomic data with chemometrics. J. Chemom. 2014, 28, 1–9. [Google Scholar] [CrossRef]
- Kell, D.B. Metabolomics, machine learning and modelling: Towards an understanding of the language of cells. Biochem. Soc. Trans. 2005, 33, 520–524. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Konter, J.; Herndon, E.; Jin, L.; Steinhoefel, G.; Sanchez, D.; Brantley, S. Quantifying an early signature of the industrial revolution from lead concentrations and isotopes in soils of Pennsylvania, USA. Anthropocene 2014, 7, 16–29. [Google Scholar] [CrossRef]
- Mendez, K.M.; Reinke, S.N.; Broadhurst, D.I. A comparative evaluation of the generalised predictive ability of eight machine learning algorithms across ten clinical metabolomics data sets for binary classification. Metabolomics 2019, 15. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Ganapathysubramanian, B.; Singh, A.K.; Sarkar, S. Machine Learning for High-Throughput Stress Phenotyping in Plants. Trends Plant Sci. 2016, 21, 110–124. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Abdullah, A.A.; Huang, M.; Nishioka, T.; Altaf-Ul-Amin, M.; Kanaya, S. Novel Approach to Classify Plants Based on Metabolite-Content Similarity. Biomed Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Willett, D.S.; Rering, C.C.; Ardura, D.A.; Beck, J.J. Application of Mathematical Models and Computation in Plant Metabolomics; Elsevier Inc.: Amsterdam, The Netherlands, 2018; ISBN 9780128123645. [Google Scholar]
- Mendez, K.M.; Broadhurst, D.I.; Reinke, S.N. The application of artificial neural networks in metabolomics: A historical perspective. Metabolomics 2019, 15, 1–14. [Google Scholar] [CrossRef]
- Heinemann, J. Machine Learning in Untargeted Metabolomics Experiments; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1859, ISBN 9781493987573. [Google Scholar]
- Nguyen, L.H.; Holmes, S. Ten quick tips for effective dimensionality reduction. PLoS Comput. Biol. 2019, 15, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Chagas-Paula, D.A.; Oliveira, T.B.; Zhang, T.; Edrada-Ebel, R.; Da Costa, F.B. Prediction of anti-inflammatory plants and discovery of their biomarkers by machine learning algorithms and metabolomic studies. Planta Med. 2015, 81, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Xue, Y.; Wei, J.; Dai, Q.; Li, P. Integrating metabolomic data with machine learning approach for discovery of Q - markers from Jinqi Jiangtang preparation against type 2 diabetes. Chin. Med. 2021, 16, 1–12. [Google Scholar] [CrossRef]
- Boutet, S.; Barreda, L.; Perreau, F.; Mouille, G.; Delannoy, E.; Magniette, M.-L.; Monti, A.; Lepiniec, L.; Zanetti, F.; Corso, M. Untargeted metabolomic analyses reveal the diversity and plasticity of the specialized metabolome in seeds of different Camelina sativa varieties. bioRxiv 2021. [Google Scholar] [CrossRef]
- Etalo, D.; de Vos, R.C.; Joosten, M.H.A.J.; Hall, R. Spatially-resolved plant metabolomics: Some potentials and limitations of Laser-Ablation Electrospray Ionization (LAESI) Mass Spectrometry metabolite imaging. Plant Physiol. 2015, 169, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Fürtauer, L.; Pschenitschnigg, A.; Scharkosi, H.; Weckwerth, W.; Nägele, T. Combined multivariate analysis and machine learning reveals a predictive module of metabolic stress response in Arabidopsis thaliana. Mol. Omics 2018, 14, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Ataş, M.; Yardimci, Y.; Temizel, A. A new approach to aflatoxin detection in chili pepper by machine vision. Comput. Electron. Agric. 2012, 87, 129–141. [Google Scholar] [CrossRef]
- Scott, I.M.; Vermeer, C.P.; Liakata, M.; Corol, D.I.; Ward, J.L.; Lin, W.; Johnson, H.E.; Whitehead, L.; Kular, B.; Baker, J.M.; et al. Enhancement of plant metabolite fingerprinting by machine learning. Plant Physiol. 2010, 153, 1506–1520. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira Almeida, R.; Valente, G.T. Predicting metabolic pathways of plant enzymes without using sequence similarity: Models from machine learning. Plant Genome 2020. [Google Scholar] [CrossRef]
- Toubiana, D.; Puzis, R.; Wen, L.; Sikron, N.; Kurmanbayeva, A.; Soltabayeva, A.; del Mar Rubio Wilhelmi, M.; Sade, N.; Fait, A.; Sagi, M.; et al. Combined network analysis and machine learning allows the prediction of metabolic pathways from tomato metabolomics data. Commun. Biol. 2019, 2. [Google Scholar] [CrossRef] [Green Version]
- Grapov, D.; Fahrmann, J.; Wanichthanarak, K.; Khoomrung, S. Rise of deep learning for genomic, proteomic, and metabolomic data integration in precision medicine. Omics A J. Integr. Biol. 2018, 22, 630–636. [Google Scholar] [CrossRef] [Green Version]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and Obstacles for Deep Learning in Biology and Medicine. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef] [Green Version]
- Bueno, P.C.P.; Lopes, N.P. Metabolomics to Characterize Adaptive and Signaling Responses in Legume Crops under Abiotic Stresses. ACS Omega 2020, 5, 1752–1763. [Google Scholar] [CrossRef] [Green Version]
- Jarrin, E.P.; Cordeiro, F.B.; Medranda, W.C.; Barrett, M.; Zambrano, M.; Regato, M. A Machine Learning-Based algorithm for the assessment of clinical metabolomic fingerprints in Zika virus disease. In Proceedings of the 2019 IEEE Latin American Conference on Computational Intelligence (LA-CCI), Guayaquil, Ecuador, 11–15 November 2019; pp. 8–13. [Google Scholar] [CrossRef]
- Silva, J.C.F.; Teixeira, R.M.; Silva, F.F.; Brommonschenkel, S.H.; Fontes, E.P.B. Machine learning approaches and their current application in plant molecular biology: A systematic review. Plant Sci. 2019, 284, 37–47. [Google Scholar] [CrossRef]
- Heinemann, J.; Mazurie, A.; Tokmina-Lukaszewska, M.; Beilman, G.J.; Bothner, B. Application of support vector machines to metabolomics experiments with limited replicates. Metabolomics 2014, 10, 1121–1128. [Google Scholar] [CrossRef]
- Gromski, P.S.; Muhamadali, H.; Ellis, D.I.; Xu, Y.; Correa, E.; Turner, M.L.; Goodacre, R. A tutorial review: Metabolomics and partial least squares-discriminant analysis—A marriage of convenience or a shotgun wedding. Anal. Chim. Acta 2015, 879, 10–23. [Google Scholar] [CrossRef]
- Gu, Q.; Han, J. Clustered support vector machines. J. Mach. Learn. Res. 2013, 31, 307–315. [Google Scholar]
- Carrizosa, E.; Nogales-Gómez, A.; Romero Morales, D. Clustering categories in support vector machines. Omega 2017, 66, 28–37. [Google Scholar] [CrossRef]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Yao, C.; Yao, S.; Yang, W.; Wu, W.; Guo, D. A metabolomics strategy for authentication of plant medicines with multiple botanical origins, a case study of Uncariae Rammulus Cum Uncis. J. Sep. Sci. 2020, 43, 1043–1050. [Google Scholar] [CrossRef]
- Du, H.; Huo, Y.; Liu, H.; Kamal, G.M.; Yang, J.; Zeng, Y.; Zhao, S.; Liu, Y. Fast nutritional characterization of different pigmented rice grains using a combination of NMR and decision tree analysis. J. Food 2019, 17, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Niazian, M.; Niedbała, G. Machine learning for plant breeding and biotechnology. Agriculture 2020, 10, 436. [Google Scholar] [CrossRef]
- Liakos, K.G.; Busato, P.; Moshou, D.; Pearson, S.; Bochtis, D. Machine learning in agriculture: A review. Sensors 2018, 18, 2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekonnen, Y.; Namuduri, S.; Burton, L.; Sarwat, A.; Bhansali, S. Review—Machine Learning Techniques in Wireless Sensor Network Based Precision Agriculture. J. Electrochem. Soc. 2020, 167, 037522. [Google Scholar] [CrossRef]
- Hummel, J.; Strehmel, N.; Selbig, J.; Walther, D.; Kopka, J. Decision tree supported substructure prediction of metabolites from GC-MS profiles. Metabolomics 2010, 6, 322–333. [Google Scholar] [CrossRef] [Green Version]
- De Bruijn, W.J.C.; van Dinteren, S.; Gruppen, H.; Vincken, J.P. Mass spectrometric characterisation of avenanthramides and enhancing their production by germination of oat (Avena sativa). Food Chem. 2019, 277, 682–690. [Google Scholar] [CrossRef]
- Dos Santos, F.A.; Sousa, I.P.; Furtado, N.A.J.C.; Da Costa, F.B. Combined OPLS-DA and decision tree as a strategy to identify antimicrobial biomarkers of volatile oils analyzed by gas chromatography–mass spectrometry. Rev. Bras. Farmacogn. 2018, 28, 647–653. [Google Scholar] [CrossRef]
- Sok, H.K.; Ooi, M.P.L.; Kuang, Y.C.; Demidenko, S. Multivariate alternating decision trees. Pattern Recognit. 2016, 50, 195–209. [Google Scholar] [CrossRef]
- Alakwaa, F.M.; Chaudhary, K.; Garmire, L.X. Deep Learning Accurately Predicts Estrogen Receptor Status in Breast Cancer Metabolomics Data. J. Proteome Res. 2018, 17, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Touw, W.G.; Bayjanov, J.R.; Overmars, L.; Backus, L.; Boekhorst, J.; Wels, M.; Sacha van Hijum, A.F.T. Data mining in the life science swith random forest: A walk in the park or lost in the jungle? Brief. Bioinform. 2013, 14, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Ansarifar, J.; Akhavizadegan, F.; Wang, L. Performance prediction of crosses in plant breeding through genotype by environment interactions. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.K.; Mo, C.; Lee, J.H.; Long, N.P.; Dong, Z.; Li, J.; Lim, J.; Kwon, S.W. The integration of multi-platform MS-based metabolomics and multivariate analysis for the geographical origin discrimination of Oryza sativa L. J. Food Drug Anal. 2018, 26, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Oza, V.H.; Aicher, J.K.; Reed, L.K. Random forest analysis of untargeted metabolomics data suggests increased use of omega fatty acid oxidation pathway in drosophila melanogaster larvae fed a medium chain fatty acid rich high-fat diet. Metabolites 2019, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Lima, E.D.O.; Navarro, L.C.; Morishita, K.N.; Kamikawa, C.M.; Rodrigues, R.G.M.; Dabaja, M.Z.; de Oliveira, D.N.; Delafiori, J.; Dias-Audibert, F.L.; Ribeiro, M.d.S.; et al. Metabolomics and Machine Learning Approaches Combined in Pursuit for More Accurate Paracoccidioidomycosis Diagnoses. mSystems 2020, 5, 1–12. [Google Scholar] [CrossRef]
- Chen, T.; Cao, Y.; Zhang, Y.; Liu, J.; Bao, Y.; Wang, C.; Jia, W.; Zhao, A. Random forest in clinical metabolomics for phenotypic discrimination and biomarker selection. Evidence-based Complement. Altern. Med. 2013. [Google Scholar] [CrossRef] [Green Version]
- Antonelli, J.; Claggett, B.L.; Henglin, M.; Kim, A.; Ovsak, G.; Kim, N.; Deng, K.; Rao, K.; Tyagi, O.; Watrous, J.D.; et al. Statistical workflow for feature selection in human metabolomics data. Metabolites 2019, 9, 143. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.S.; McGeachie, M.J.; Lee-Sarwar, K.A.; Kachroo, P.; Chu, S.H.; Virkud, Y.V.; Huang, M.; Litonjua, A.A.; Weiss, S.T.; Lasky-Su, J. Partial least squares discriminant analysis and bayesian networks for metabolomic prediction of childhood asthma. Metabolites 2018, 8, 68. [Google Scholar] [CrossRef] [Green Version]
- Gillies, C.E.; Jennaro, T.S.; Puskarich, M.A.; Sharma, R.; Ward, K.R.; Fan, X.; Jones, A.E.; Stringer, K.A. A multilevel bayesian approach to improve effect size estimation in regression modeling of metabolomics data utilizing imputation with uncertainty. Metabolites 2020, 10, 319. [Google Scholar] [CrossRef]
- Teklehaymanot, F.K.; Muma, M.; Zoubir, A.M. Bayesian Cluster Enumeration Criterion for Unsupervised Learning. IEEE Trans. Signal Process. 2018, 66, 5392–5406. [Google Scholar] [CrossRef] [Green Version]
- Del Carratore, F.; Schmidt, K.; Vinaixa, M.; Hollywood, K.A.; Greenland-Bews, C.; Takano, E.; Rogers, S.; Breitling, R. Integrated Probabilistic Annotation: A Bayesian-Based Annotation Method for Metabolomic Profiles Integrating Biochemical Connections, Isotope Patterns, and Adduct Relationships. Anal. Chem. 2019, 91, 12799–12807. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, M.; Dührkop, K.; Böcker, S. Bayesian networks for mass spectrometric metabolite identification via molecular fingerprints. Bioinformatics 2018, 34, i333–i340. [Google Scholar] [CrossRef]
- Ludwig, M.; Nothias, L.F.; Dührkop, K.; Koester, I.; Fleischauer, M.; Hoffmann, M.A.; Petras, D.; Vargas, F.; Morsy, M.; Aluwihare, L.; et al. Database-independent molecular formula annotation using Gibbs sampling through ZODIAC. Nat. Mach. Intell. 2020, 2, 629–641. [Google Scholar] [CrossRef]
- O’Brien, K.A.; Atkinson, R.A.; Richardson, L.; Kou, A.; Murray, A.J.; Harr, S.D.R.; Martin, D.S.; Levett, D.Z.H.; Mitchell, K.; Mythen, M.G.; et al. Metabolomic and lipidomic plasma profile changes in human participants ascending to Everest Base Camp. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- McGeachie, M.J.; Chang, H.H.; Weiss, S.T. CGBayesNets: Conditional Gaussian Bayesian Network Learning and Inference with Mixed Discrete and Continuous Data. PLoS Comput. Biol. 2014, 10. [Google Scholar] [CrossRef] [Green Version]
- Pomyen, Y.; Wanichthanarak, K.; Poungsombat, P.; Fahrmann, J.; Grapov, D.; Khoomrung, S. Deep metabolome: Applications of deep learning in metabolomics. Comput. Struct. Biotechnol. J. 2020, 18, 2818–2825. [Google Scholar] [CrossRef]
- O’Shea, K.; Cameron, S.J.S.; Lewis, K.E.; Lu, C.; Mur, L.A.J. Metabolomic-based biomarker discovery for non-invasive lung cancer screening: A case study. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 2682–2687. [Google Scholar] [CrossRef] [Green Version]
- Patrício, D.I.; Rieder, R. Computer vision and artificial intelligence in precision agriculture for grain crops: A systematic review. Comput. Electron. Agric. 2018, 153, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Liebal, U.W.; Phan, A.N.T.; Sudhakar, M.; Raman, K.; Blank, L.M. Machine learning applications for mass spectrometry-based metabolomics. Metabolites 2020, 10, 243. [Google Scholar] [CrossRef]
- Melnikov, A.D.; Tsentalovich, Y.P.; Yanshole, V.V. Deep Learning for the Precise Peak Detection in High-Resolution LC-MS Data. Anal. Chem. 2020, 92, 588–592. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lin, T.; Xu, J.; Luo, X.; Ying, Y. DeepSpectra: An end-to-end deep learning approach for quantitative spectral analysis. Anal. Chim. Acta 2019, 1058, 48–57. [Google Scholar] [CrossRef]
- Golhani, K.; Balasundram, S.K.; Vadamalai, G.; Pradhan, B. A review of neural networks in plant disease detection using hyperspectral data. Inf. Process. Agric. 2018, 5, 354–371. [Google Scholar] [CrossRef]
- Jia, Y.; Zhao, R.; Chen, L. Similarity-Based Machine Learning Model for Predicting the Metabolic Pathways of Compounds. IEEE Access 2020, 8, 130687–130696. [Google Scholar] [CrossRef]
- Wang, P.; Moore, B.M.; Uygun, S.; Lehti-Shiu, M.D.; Barry, C.; Shiu, S.-H. Optimizing the use of gene expression data to predict metabolic pathway memberships with unsupervised and supervised machine learning. bioRxiv 2020. [Google Scholar] [CrossRef]
- Presnell, K.V.; Alper, H.S. Systems Metabolic Engineering Meets Machine Learning: A New Era for Data-Driven Metabolic Engineering. Biotechnol. J. 2019, 14. [Google Scholar] [CrossRef]
- Volk, M.J.; Lourentzou, I.; Mishra, S.; Vo, L.T.; Zhai, C.; Zhao, H. Biosystems Design by Machine Learning. ACS Synth. Biol. 2020, 9, 1514–1533. [Google Scholar] [CrossRef]
- Nagaraja, A.A.; Fontaine, N.; Delsaut, M.; Charton, P.; Damour, C.; Offmann, B.; Grondin-Perez, B.; Cadet, F. Flux prediction using artificial neural network (ANN) for the upper part of glycolysis. PLoS ONE 2019, 14, 10–12. [Google Scholar] [CrossRef]
- Zampieri, G.; Vijayakumar, S.; Yaneske, E.; Angione, C. Machine and deep learning meet genome-scale metabolic modeling. PLoS Comput. Biol. 2019, 15, 1–24. [Google Scholar] [CrossRef]
- Costello, Z.; Martin, H.G. A machine learning approach to predict metabolic pathway dynamics from time-series multiomics data. NPJ Syst. Biol. Appl. 2018, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Volkova, S.; Matos, M.R.A.; Mattanovich, M.; de Mas, I.M. Metabolic modelling as a framework for metabolomics data integration and analysis. Metabolites 2020, 10, 303. [Google Scholar] [CrossRef]
- Baranwal, M.; Magner, A.; Elvati, P.; Saldinger, J.; Violi, A.; Violi, A.; Hero, A.O. A deep learning architecture for metabolic pathway prediction. Bioinformatics 2020, 36, 2547–2553. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Wright, S.N.; Hamblin, M.; McCloskey, D.; Alcantar, M.A.; Schrübbers, L.; Lopatkin, A.J.; Satish, S.; Nili, A.; Palsson, B.O.; et al. A White-Box Machine Learning Approach for Revealing Antibiotic Mechanisms of Action. Cell 2019, 177, 1649–1661. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M. Machine learning methods for analysis of metabolic data and metabolic pathway modeling. Metabolites 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Rana, P.; Berry, C.; Ghosh, P.; Fong, S.S. Recent advances on constraint-based models by integrating machine learning. Curr. Opin. Biotechnol. 2020, 64, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, R.; Hassanpour, N.; Liu, L.P.; Hassoun, S. Pathway activity analysis and metabolite annotation for untargeted metabolomics using probabilistic modeling. Metabolites 2020, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Liu, Y.; Ren, Z.; Du, Y.; Huang, Q.; Garmire, L.X. Lilikoi V2.0: A deep learning-enabled, personalized pathway-based R package for diagnosis and prognosis predictions using metabolomics data. Gigascience 2021, 10, 1–11. [Google Scholar] [CrossRef]
- Wang, S.; Alseekh, S.; Fernie, A.R.; Luo, J. The Structure and Function of Major Plant Metabolite Modifications. Mol. Plant 2019, 12, 899–919. [Google Scholar] [CrossRef]
- Koch, M.; Duigou, T.; Faulon, J.L. Reinforcement learning for bioretrosynthesis. ACS Synth. Biol. 2020, 9, 157–168. [Google Scholar] [CrossRef]
- McLuskey, K.; Wandy, J.; Vincent, I.; van der Hooft, J.J.J.; Rogers, S.; Burgess, K.; Daly, R. Ranking Metabolite Sets by Their Activity Levels. Metabolites 2021, 11, 103. [Google Scholar] [CrossRef]
- Scheubert, K.; Hufsky, F.; Petras, D.; Wang, M.; Nothias, L.F.; Dührkop, K.; Bandeira, N.; Dorrestein, P.C.; Böcker, S. Significance estimation for large scale metabolomics annotations by spectral matching. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Rawlinson, C.; Jones, D.; Rakshit, S.; Meka, S.; Moffat, C.S.; Moolhuijzen, P. Hierarchical clustering of MS/MS spectra from the firefly metabolome identifies new lucibufagin compounds. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Piasecka, A.; Kachlicki, P.; Stobiecki, M. Analytical methods for detection of plant metabolomes changes in response to biotic and abiotic stresses. Int. J. Mol. Sci. 2019, 20, 379. [Google Scholar] [CrossRef] [Green Version]
- Quinn, R.A.; Nothias, L.F.; Vining, O.; Meehan, M.; Esquenazi, E.; Dorrestein, P.C. Molecular Networking as a Drug Discovery, Drug Metabolism, and Precision Medicine Strategy. Trends Pharmacol. Sci. 2017, 38, 143–154. [Google Scholar] [CrossRef]
- De Souza, L.P.; Alseekh, S.; Brotman, Y.; Fernie, A.R. Network-based strategies in metabolomics data analysis and interpretation: From molecular networking to biological interpretation. Expert Rev. Proteomics 2020, 17, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Van Der Hooft, J.J.J.; Mohimani, H.; Bauermeister, A.; Dorrestein, P.C.; Duncan, K.R.; Medema, M.H. Linking genomics and metabolomics to chart specialized metabolic diversity. Chem. Soc. Rev. 2020, 49, 3297–3314. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Jia, Q.; Su, W.; Yan, Z.; Situ, W.; He, X.; Peng, W.; Yao, H. Integration of molecular networking and fingerprint analysis for studying constituents in Microctis Folium. PLoS ONE 2020, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Demarque, D.P.; Dusi, R.G.; de Sousa, F.D.M.; Grossi, S.M.; Silvério, M.R.S.; Lopes, N.P.; Espindola, L.S. Mass spectrometry-based metabolomics approach in the isolation of bioactive natural products. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron, A.T.; Gentry, E.C.; McPhail, K.L.; Nothias, L.F.; Nothias-Esposito, M.; Bouslimani, A.; Petras, D.; Gauglitz, J.M.; Sikora, N.; Vargas, F.; et al. Reproducible molecular networking of untargeted mass spectrometry data using GNPS. Nat. Protoc. 2020, 15, 1954–1991. [Google Scholar] [CrossRef] [PubMed]
- Wandy, J.; Zhu, Y.; Van Der Hooft, J.J.J.; Daly, R.; Barrett, M.P.; Rogers, S. Ms2lda.org: Web-based topic modelling for substructure discovery in mass spectrometry. Bioinformatics 2018, 34, 317–318. [Google Scholar] [CrossRef]
- Rogers, S.; Ong, C.W.; Wandy, J.; Ernst, M.; Ridder, L.; Van Der Hooft, J.J.J. Deciphering complex metabolite mixtures by unsupervised and supervised substructure discovery and semi-automated annotation from MS/MS spectra. Faraday Discuss. 2019, 218, 284–302. [Google Scholar] [CrossRef] [Green Version]
- Blaženović, I.; Kind, T.; Ji, J.; Fiehn, O. Software tools and approaches for compound identification of LC-MS/MS data in metabolomics. Metabolites 2018, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Kuhn, M.; Gavin, A.C.; Bork, P. Identification of metabolites from tandem mass spectra with a machine learning approach utilizing structural features. Bioinformatics 2020, 36, 1213–1218. [Google Scholar] [CrossRef] [Green Version]
- Nothias, L.F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef]
- Ludwig, M.; Fleischauer, M.; Dührkop, K.; Hoffmann, M.A.; Böcker, S. De Novo Molecular Formula Annotation and Structure Elucidation Using SIRIUS 4. Methods Mol. Biol. 2020, 2104, 185–207. [Google Scholar] [CrossRef] [PubMed]
- Huber, F.; Ridder, L.; Rogers, S.; van der Hooft, J.J.J. Spec2Vec: Improved mass spectral similarity scoring through learning of structural relationships. PLoS Comput. Biol. 2021, 17. [Google Scholar] [CrossRef]
- Ernst, M.; Bin Kang, K.; Caraballo-Rodríguez, A.M.; Nothias, L.F.; Wandy, J.; Wang, M.; Rogers, S.; Medema, M.H.; Dorrestein, P.C.; van der Hooft, J.J.J. MolNetEnhancer: Enhanced molecular networks by integrating metabolome mining and annotation tools. Metabolites 2019, 9, 144. [Google Scholar] [CrossRef] [Green Version]
- Huber, F.; van der Burg, S.; van der Hooft, J.J.J.; Ridder, L. MS2DeepScore—A novel deep learning similarity measure for mass fragmentation spectrum comparisons. bioRxiv 2021. [Google Scholar] [CrossRef]
- Feunang, Y.D.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Sha, B.; Schymanski, E.L.; Ruttkies, C.; Cousins, I.T.; Wang, Z. Exploring open cheminformatics approaches for categorizing per- And polyfluoroalkyl substances (PFASs). Environ. Sci. Process. Impacts 2019, 21, 1835–1851. [Google Scholar] [CrossRef] [Green Version]
- Dührkop, K.; Nothias, L.F.; Fleischauer, M.; Reher, R.; Ludwig, M.; Hoffmann, M.A.; Petras, D.; Gerwick, W.H.; Rousu, J.; Dorrestein, P.C.; et al. Systematic classification of unknown metabolites using high-resolution fragmentation mass spectra. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef]
- Kim, H.W.; Wang, M.; Leber, C.A.; Nothias, L. NPClassifier: Deep neural structural classification tool for natural products. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Hastings, J.; Glauer, M.; Memariani, A.; Neuhaus, F.; Mossakowski, T. Learning Chemistry: Exploring the suitability of machine learning for the task of structure-based chemical ontology classification. J. Cheminform. 2021, 13, 1–20. [Google Scholar] [CrossRef]
- Dührkop, K.; Nothias, L.F.; Fleischauer, M.; Ludwig, M.; Hoffmann, M.A.; Rousu, J.; Dorrestein, P.C.; Böcker, S. Classes for the masses: Systematic classification of unknowns using fragmentation spectra. bioRxiv 2020, 1–42. [Google Scholar] [CrossRef] [Green Version]
- Beniddir, M.A.; Kang, K.B.; Genta-Jouve, G.; Huber, F.; Rogers, S.; van der Hooft, J.J.J. Advances in decomposing complex metabolite mixtures using substructure- and network-based computational metabolomics approaches. Nat. Prod. Rep. 2021. [Google Scholar] [CrossRef]
- Wang, M.; Jarmusch, A.K.; Vargas, F.; Aksenov, A.A.; Gauglitz, J.M.; Weldon, K.; Petras, D.; da Silva, R.; Quinn, R.; Melnik, A.V.; et al. Mass spectrometry searches using MASST. Nat. Biotechnol. 2020, 38, 23–26. [Google Scholar] [CrossRef]
- Jarmusch, A.K.; Wang, M.; Aceves, C.M.; Advani, R.S.; Aguirre, S.; Aksenov, A.A.; Aleti, G.; Aron, A.T.; Bauermeister, A.; Bolleddu, S.; et al. ReDU: A framework to find and reanalyze public mass spectrometry data. Nat. Methods 2020, 17, 901–904. [Google Scholar] [CrossRef]
- Abdelraheem, A.; Esmaeili, N.; O’Connell, M.; Zhang, J. Progress and perspective on drought and salt stress tolerance in cotton. Ind. Crops Prod. 2019, 130, 118–129. [Google Scholar] [CrossRef]
- Ullah, N.; Yuce, M.; Neslihan Ozturk Gokce, Z.; Budak, H. Comparative metabolite profiling of drought stress in roots and leaves of seven Triticeae species. BMC Genomics 2017, 18, 1–12. [Google Scholar] [CrossRef]
- Wang, X.; Cai, X.; Xu, C.; Wang, Q.; Dai, S. Drought-responsive mechanisms in plant leaves revealed by proteomics. Int. J. Mol. Sci. 2016, 17, 1706. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.W.; Roux, M.; Petersen, M.; Mundy, J. MAP Kinase Cascades in Arabidopsis Innate Immunity. Front. Plant Sci. 2012, 3, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Pandey, G.K. Emergence of a novel calcium signaling pathway in plants: CBL-CIPK signaling network. Physiol. Mol. Biol. Plants 2008, 14, 51–68. [Google Scholar] [CrossRef] [Green Version]
- Mühlenbock, P.; Szechyńska-Hebda, M.; Płaszczyca, M.; Baudo, M.; Mullineaux, P.M.; Parker, J.E.; Karpińska, B.; Karpińskie, S. Chloroplast signaling and lesion simulating disease1 regulate crosstalk between light acclimation and immunity in Arabidopsis. Plant Cell 2008, 20, 2339–2356. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, P.; Prasad, M.N.V. Environmental Adaptations and Stress Tolerance of Plants in the Era of Climate Change; Springer: New York, NY, USA, 2012; ISBN 978-1-4614-0814-7. [Google Scholar]
- Poltronieri, P.; Bonsegna, S.; de Domenico, S.; Santino, A. Molecular Mechanisms in Plant Abiotic Stress Response. Ratarstvo i Povrtarstvo Field Veg. Crop Res. 2011, 48, 15–24. [Google Scholar] [CrossRef]
- Michaletti, A.; Naghavi, M.R.; Toorchi, M.; Zolla, L.; Rinalducci, S. Metabolomics and proteomics reveal drought-stress responses of leaf tissues from spring-wheat. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; De, B. Metabolomics analysis of rice responses to salinity stress revealed elevation of serotonin, and gentisic acid levels in leaves of tolerant varieties. Plant Signal. Behav. 2017, 12, 1–11. [Google Scholar] [CrossRef]
- Negrão, S.; Schmöckel, S.M.; Tester, M. Evaluating physiological responses of plants to salinity stress. Ann. Bot. 2017, 119, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Halfter, U.; Ishitani, M.; Zhu, J.-K. Molecular Characterization of Functional Domains in the Protein Kinase SOS2 That Is Required for Plant Salt Tolerance. Plant Cell 2007, 13, 1383. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Li, Z.; Dai, S.; Ding, H.; Wang, Q.; Li, X.; Ding, G.; Wang, P.; Guan, Y.; Liu, W. Integrative analyses of transcriptomics and metabolomics upon seed germination of foxtail millet in response to salinity. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Ma, X.; Zheng, J.; Zhang, X.; Hu, Q.; Qian, R. Salicylic acid alleviates the adverse effects of salt stress on dianthus superbus (Caryophyllaceae) by activating photosynthesis, protecting morphological structure, and enhancing the antioxidant system. Front. Plant Sci. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Misra, N.; Saxena, P. Effect of salicylic acid on proline metabolism in lentil grown under salinity stress. Plant Sci. 2009, 177, 181–189. [Google Scholar] [CrossRef]
- Conde, A.; Silva, P.; Agasse, A.; Conde, C.; Gerós, H. Mannitol transport and mannitol dehydrogenase activities are coordinated in Olea europaea under salt and osmotic stresses. Plant Cell Physiol. 2011, 52, 1766–1775. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Kaya, H.; Kawarazaki, T.; Hiraoka, G.; Senzaki, E.; Michikawa, M.; Kuchitsu, K. Protein phosphorylation is a prerequisite for the Ca 2+-dependent activation of Arabidopsis NADPH oxidases and may function as a trigger for the positive feedback regulation of Ca2+ and reactive oxygen species. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 398–405. [Google Scholar] [CrossRef] [Green Version]
- Nounjan, N.; Nghia, P.T.; Theerakulpisut, P. Exogenous proline and trehalose promote recovery of rice seedlings from salt-stress and differentially modulate antioxidant enzymes and expression of related genes. J. Plant Physiol. 2012, 169, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Kaya, C.; Sonmez, O.; Aydemir, S.; Ashraf, M.; Dikilitas, M. Exogenous application of mannitol and thiourea regulates plant growth and oxidative stress responses in salt-stressed maize (Zea mays L.). J. Plant Interact. 2013, 8, 234–241. [Google Scholar] [CrossRef]
- Siahpoosh, M.R.; Sanchez, D.H.; Schlereth, A.; Scofield, G.N.; Furbank, R.T.; van Dongen, J.T.; Kopka, J. Modification of OsSUT1 gene expression modulates the salt response of rice Oryza sativa cv. Taipei 309. Plant Sci. 2012, 182, 101–111. [Google Scholar] [CrossRef]
- Aldesuquy, H.S.; Abbas, M.A.; Hamed, S.A.A.; Elhakem, A.H.; Alsokari, S.S. Glycine betaine and salicylic acid induced modification in productivity of two different cultivars of wheat grown under water stress. J. Stress Physiol. Biochem. 2012, 8, 72–89. [Google Scholar]
- Manaa, A.; Gharbi, E.; Mimouni, H.; Wasti, S.; Aschi-Smiti, S.; Lutts, S.; Ben Ahmed, H. Simultaneous application of salicylic acid and calcium improves salt tolerance in two contrasting tomato (Solanum lycopersicum) cultivars. S. Afr. J. Bot. 2014, 95, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Cvikrová, M.; Gemperlová, L.; Martincová, O.; Vanková, R. Effect of drought and combined drought and heat stress on polyamine metabolism in proline-over-producing tobacco plants. Plant Physiol. Biochem. 2013, 73, 7–15. [Google Scholar] [CrossRef]
- Sarafraz-Ardakani, M.-R.; Khavari-Nejad, R.-A.; Moradi, F.; Najafi, F. Abscisic Acid and Cytokinin-Induced Osmotic and Antioxidant Regulation in Two Drought-Tolerant and Drought-Sensitive Cultivars of Wheat During Grain Filling Under Water Deficit in Field Conditions. Not. Sci. Biol. 2014, 6, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Sarker, U.; Oba, S. Drought stress enhances nutritional and bioactive compounds, phenolic acids and antioxidant capacity of Amaranthus leafy vegetable. BMC Plant Biol. 2018, 18, 258. [Google Scholar] [CrossRef] [Green Version]
- Varela, M.C.; Arslan, I.; Reginato, M.A.; Cenzano, A.M.; Luna, M.V. Phenolic compounds as indicators of drought resistance in shrubs from Patagonian shrublands (Argentina). Plant Physiol. Biochem. 2016, 104, 81–91. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Description | Advantages | Disadvantages | Originally Automated? (Yes/No) | Reference(s) |
|---|---|---|---|---|---|
| Solid-phase extraction (SPE) | Extracts metabolites based on their chemical and physical properties that determine their distribution between the mobile liquid phase and a solid stationary phase. Targeted metabolites are released from stationary phase by changing the mobile phase into the elution solvent. | Enhanced selectivity, rapid, reproducible and economical. | Poor metabolite coverage | No | [43,44,45,46,47] |
| Solid-phase microextraction (SPME) | Extracts a range of metabolites from a variety of matrices by the insertion of a polymer-coated fiber into either the vial headspace, liquid sample or exposed in vivo. The metabolites diffuse from the sample onto the fiber. | Enhanced sensitivity, minimum invasiveness, enhanced analysis throughput and compatibility with in vivo sampling and extraction. | Time-consuming steps in equilibration of the fibre (30 min) and sample extraction (up to 5 min), low metabolite coverage, and its expensive. | Yes | [40,48,49,50] |
| Dispersive liquid–liquid microextraction (DLLME) | Extraction solvent (i.e., water-immiscible organic solvent) is added to a dispersive solvent (i.e., water-miscible solvent), the mixture is then injected into the sample to form a homogenous solution. Induced dispersion increases surface contact between extract and the sample, thus resulting in instantaneous extraction. | Simple, cost-effective, rapid, has high extraction recovery, reduced solvent consumption and has high reproducibility. | Uses halogenated and organic solvents, requires manual/mechanical agitation of the sample for dispersion of the organic solvents in the sample solution, and time-consuming phase separation step. | No | [48,51,52] |
| Electromembrane extraction (EME) | An electrical field is applied between the sample and the acceptor compartments, separated by a membrane of organic solvent (i.e., the support liquid membrane (SLM)). Charged ionic metabolites are extracted from the sample solution, through the SLM, and into the acceptor compartment. Proteins, salts, etc. are incapable of passing the SLM, thus metabolites are recovered in the aqueous phase. | Enables large-scale automation. | Difficulty of extracting hydrophillic metabolites. | No | [48,52,53] |
| Hollow fiber liquid–liquid microextraction (HF-LLME) | Two-phase mode: The hollow fiber (HF) is soaked with the extraction solvent and exposed to the sample’s solution or headspace, an equilibrium between solvent and sample is establishes, thus resulting in the extraction of metabolites from the sample into the solvent. Three-phase mode: The center of the HF contains an aqueous phase (i.e., acceptor phase), in addition to the soaked HF pores with organic solvent. The HF is exposed to the sample where two equilibriums are established. The first is between the sample and the solvent, followed by the second between the solvent and the acceptor phase, thus metabolites are extracted from the sample into the acceptor phase of the HF through the solvent. | Highly selective and concentrate metabolites. | No | [51,52] | |
| Single drop microextraction (SDME) | Similar to HF-LLME. A syringe is used instead of a HF and only a drop of the extractant solvent is required. | Simple, cost-effective and time-saving. | Limited by partial solubility of organic solvents in water, limited extraction volume, metabolite losses due to volatility and dislodgement of the extractant solvent. | No | [51,52,54] |
| Accelerated solvent extraction (ASE)/Pressurized liquid extraction (PLE) | The solvent’s temperature is elevated beyond its boiling point to increase its solubilizing capacity and reduce its viscosity to penetrate into the sample matrix and increase the metabolites’ diffusion rate. Additionally, the elevated pressure ensures the solvent remains in the liquid phase and aids it in penetrating through the sample matrix, which maximizes solvent and metabolite contact, and result in effective extraction. | Reduced solvent usage and rapid. | No | [42,55,56] | |
| Supercritical fluid extraction (SFE) | Utilizes gas properties above their critical points as solvents to facilitate the extraction of non-polar to semi-polar metabolites from plant materials. | Enhanced sensitivity and accuracy, reduced extraction time, ideal for thermo-labile metabolites and reduced use of organic solvents. | Very expensive. | Yes | [42,56,57,58] |
| Microwave-assisted extraction (MAE) | Microwave, electromagnetic radiation with a frequency in the 0.3–300 GHz range, energy is used to extract polar metabolites from plant materials by heating the solvent. | Reduced extraction time (15-20 min), reduced solvent consumption, improved extraction yield and precision. | Operates at relatively high temperature which is problematic for thermally liable metabolites, low extraction yield for non-polar solvents and requires a centrifugation step to remove solid materials from extractant. | No | [57,59,60] |
| Ultrasound-assisted extraction (UAE) | Utilizes ultrasonic energy and solvents to extract secondary metabolites from various plant materials. | Reduced extraction time, solvent consumption, energy, thermal degradation, extraction temperature and equipment size, enhanced mass transfer, extraction yield and high extract recovery. | Low extraction efficiency. | No | [57,61,62,63] |
| Metabolomics Study | ML Method 1 | Reference |
|---|---|---|
| ML-modelling for prediction of metabolic pathways of plant enzymes. | SVM, ANN, NB, DTC | [160] |
| Identification of central and predictive molecular components of plant metabolic stress response. | SVMs, NNC, DTC | [157] |
| Untargeted metabolomics to reveal diversity of the metabolome in seeds of Camelina species. | DL, ANN | [155] |
| Characterisation of adaptive and signalling responses based on metabolite content under abiotic stresses. | SVM, DPClus | [148] |
| Detection of aflatoxin metabolite in chilli pepper using machine vision. | SVM, RF | [158] |
| Detection and resolution on plant metabolites (S.lycopersium) using mass spectrometry imaging. | DCNN | [156] |
| Discovery of Q-markers from Jinqi Jiangtang for medicinal purposes. | ANN | [154] |
| Prediction of metabolic pathways in correlation networks in the pericarp of a tomato. | ML algorithms | [161] |
| Discovery and identification of biomarkers using ML algorithms in metabolomic studies. | ANN, DL | [153] |
| Enhancement of plant metabolite fingerprinting using ML methods. | SVM, RF | [164] |
| Metabolite Group | Stress-Responsive Roles | Plant Species | References |
|---|---|---|---|
| Amino acids | ROS scavenging (proline), protein stabilisation and synthesis, redox control | Dianthus superbus, Lens esculenta | [261,262] |
| Polyols | Protection of photosynthesis systems, ROS scavenging, protein stabilisation | Rice, apple leaves, Fraxinus excelsior, Zea mays | [263,264,265,266] |
| Organic acids | Energy production, signalling molecules, antioxidant activities | Oryza sativa, Wheat | [249,267] |
| Sugars | Signalling molecules, carbon energy reserve, maintenance of redox homeostasis, osmoprotectants | Solanum lycopersicum, Triticum aestivum | [268,269] |
| Polyamines | Activation of antioxidant enzymes, regulation of ion channels activity, protein and membrane stabilisation | Tobacco, Triticum aestivum | [270,271] |
| Phenolics | Hormonal regulation, antioxidant activity, photosynthetic activity | Patagonian shrublands, Amaranthus tricolor | [272,273] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tinte, M.M.; Chele, K.H.; van der Hooft, J.J.J.; Tugizimana, F. Metabolomics-Guided Elucidation of Plant Abiotic Stress Responses in the 4IR Era: An Overview. Metabolites 2021, 11, 445. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11070445
Tinte MM, Chele KH, van der Hooft JJJ, Tugizimana F. Metabolomics-Guided Elucidation of Plant Abiotic Stress Responses in the 4IR Era: An Overview. Metabolites. 2021; 11(7):445. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11070445
Chicago/Turabian StyleTinte, Morena M., Kekeletso H. Chele, Justin J. J. van der Hooft, and Fidele Tugizimana. 2021. "Metabolomics-Guided Elucidation of Plant Abiotic Stress Responses in the 4IR Era: An Overview" Metabolites 11, no. 7: 445. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11070445