Mitochondrial Dysfunction in the Transition from NASH to HCC


1
Institut de Recherches Cliniques de Montréal (IRCM), Montreal, Quebec, QC H2W 1R7, Canada
2
Faculty of Medicine, University of Montreal, Montreal, Quebec, QC H3G 2M1, Canada
3
Division of Experimental Medicine, McGill University, Montreal, Quebec, QC H4A 3J1, Canada
*
Author to whom correspondence should be addressed.
Metabolites 2019, 9(10), 233; https://0-doi-org.brum.beds.ac.uk/10.3390/metabo9100233
Submission received: 27 August 2019
/
Revised: 26 September 2019
/
Accepted: 11 October 2019
/
Published: 16 October 2019
(This article belongs to the Special Issue Mitochondria and Metabolism in Disorders)
{kind=link}
{kind=link}
Abstract
:The liver constantly adapts to meet energy requirements of the whole body. Despite its remarkable adaptative capacity, prolonged exposure of liver cells to harmful environmental cues (such as diets rich in fat, sugar, and cholesterol) results in the development of chronic liver diseases (including non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH)) that can progress to hepatocellular carcinoma (HCC). The pathogenesis of these diseases is extremely complex, multifactorial, and poorly understood. Emerging evidence suggests that mitochondrial dysfunction or maladaptation contributes to detrimental effects on hepatocyte bioenergetics, reactive oxygen species (ROS) homeostasis, endoplasmic reticulum (ER) stress, inflammation, and cell death leading to NASH and HCC. The present review highlights the potential contribution of altered mitochondria function to NASH-related HCC and discusses how agents targeting this organelle could provide interesting treatment strategies for these diseases.
1. Introduction
The global epidemic of obesity correlates with the rising prevalence of the metabolic syndrome, a cluster of metabolic abnormalities increasing the risk of cardiovascular disease and type 2 diabetes mellitus (T2DM) [1]. Nonalcoholic fatty liver disease (NAFLD) is the hepatic manifestation of the metabolic syndrome and is now the most common chronic liver disease in Western countries, affecting approximately 30% of the general population [2]. Importantly, about 70% of obese patients with T2DM have NAFLD [3,4,5]. While most studies report that NAFLD is significantly more prevalent in men [6], following menopause, this relationship reverses and women become equally (if not more) susceptible to NAFLD [7]. NAFLD represents a wide spectrum of liver diseases ranging from simple steatosis (triacylglycerol infiltration in >5% of hepatocytes) to a more severe necro-inflammatory form called nonalcoholic steatohepatitis (NASH) resulting in fibrosis [8,9,10]. Hepatocellular carcinoma (HCC) is more prevalent within the setting of NASH than NAFLD (2.4%–12.8% versus 0%–3%) [11], but the precise mechanism(s) that give rise to cancer within this altered metabolic environment remain unknown.
There was a 9% annual increase in HCC associated with NAFLD between 2004 and 2009 [12] and a study surveying 18 million subjects from 2002 to 2008 found NAFLD/NASH to be the leading co-morbidity in 38.2% of HCC cases [13]. Hepatocellular carcinoma is the fifth leading cause of new cancer cases, the second leading cause of cancer death in men and sixth in women, worldwide [14]. Liver resection, orthotopic transplantation, and systemic chemotherapy are the only currently accepted therapeutic options for HCC [15]. Unfortunately, HCC is minimally responsive to chemotherapy and most cases present only at a late stage, often making patients unsuitable candidates for transplantation [16,17,18]. Emerging data suggest that NASH and metabolic syndrome have a higher proportion of HCCs manifesting in the absence of cirrhosis [19,20,21,22], strikingly different compared to HCC of other etiologies, which require a cirrhotic step. Consequently, NASH-related HCC is less likely to be diagnosed by surveillance compared to HCC secondary to viral hepatitis [22]. Obesity-associated NASH is currently the third leading cause for liver transplantation and may surpass hepatitis C and alcohol consumption as the most common indication for liver transplantation in the developed world [23]. A better understanding of the molecular mechanisms underlying the relationship between hepatic metabolism and carcinogenesis is needed and would greatly contribute to the development of new therapeutic strategies.
The liver is a central organ responsible for carbohydrate, lipid, and protein metabolism. It is also one of the richest organs in terms of number and density of mitochondria, serving as a critical site for multiple metabolic pathways including β-oxidation, tricarboxylic acid (TCA) cycle, ketogenesis, respiratory activity, and adenosine triphosphate (ATP) synthesis, providing metabolic fuels for itself and the rest of the body [24]. Given the importance of mitochondria to hepatic function, it is not surprising that evidence points toward inadequate mitochondrial adaptation as a likely central player in the pathological transition from NASH to HCC [3,25,26,27]. Metabolic reprogramming is a key feature of hepatocellular carcinoma and mitochondria defects are well documented in HCC [28,29]. What remains a mystery is how and why mitochondria fail to adapt to metabolic challenges and whether targeting this aspect of the disease would prevent or reverse the progression to cancer. Deciphering if there is a role for mitochondria in the progression of NASH to HCC is also critical and offers a new horizon for therapeutic options. Herein, we review what is currently known about altered hepatic mitochondrial function in NASH and related liver cancer.
2. Mitochondrial Dysfunction in Non-Alcoholic Steatohepatitis (NASH)
2.1. Mitochondrial Adaptation and Flexibility
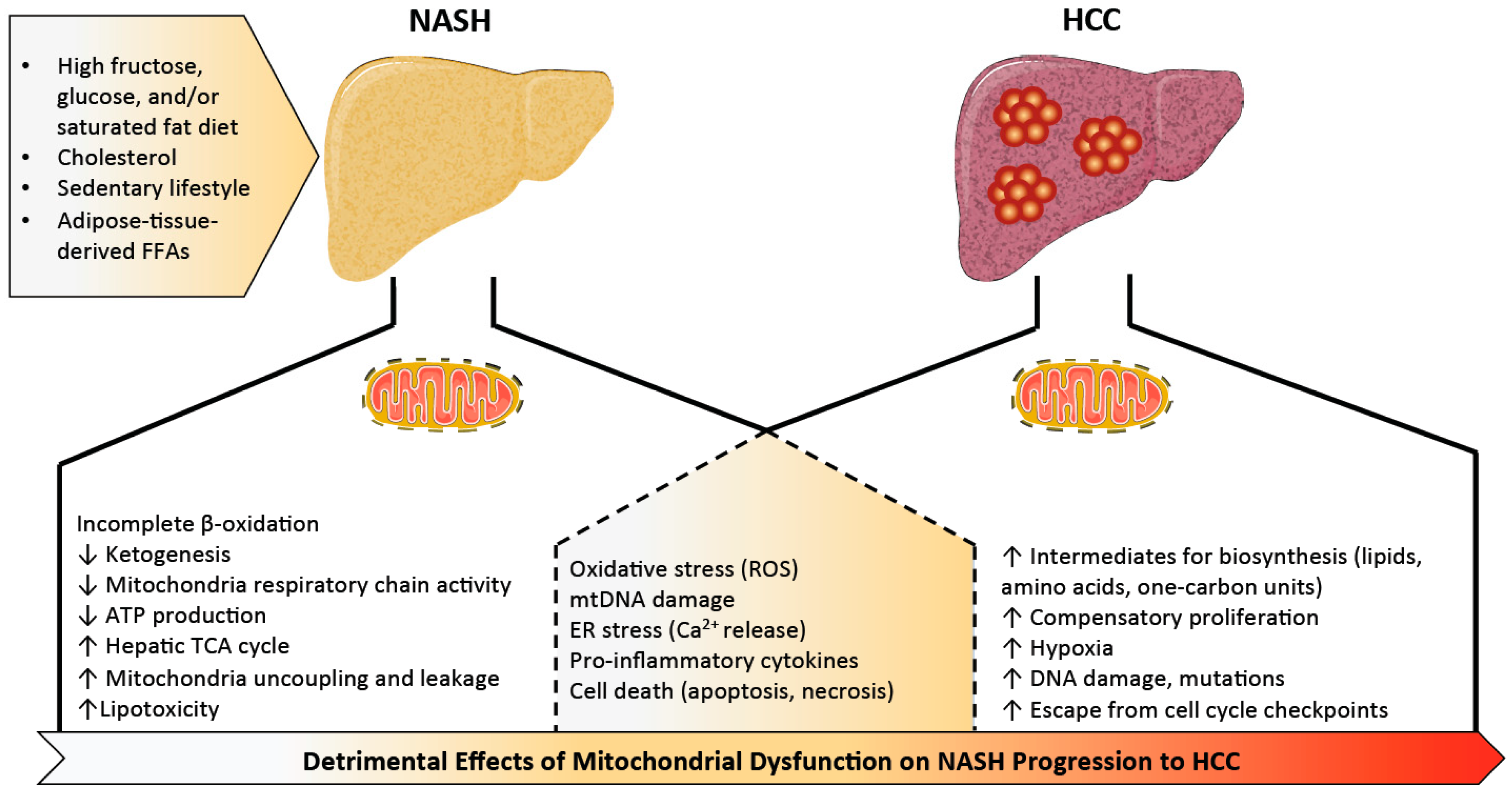
Increased mitochondrial mass and biogenesis in liver tissue is reported in NAFLD and NASH patients [27], supporting the theory that mitochondria are highly adaptive and increase in number and capacity in environments of substrate excess. However, in contrast with the often-increased mitochondrial activity (i.e., β-oxidation, mitochondrial respiration, TCA cycle, and ketogenesis) observed in early stages of NAFLD in response to hepatic insulin resistance and free fatty acid overload [30,31,32,33,34,35], mice and humans with NASH exhibit blunted ketogenesis [31,36], lower maximal respiration, mitochondrial uncoupling, and leakage [31,37]. This more severe stage of the disease is also associated with an overactive mitochondrial TCA cycle potentially to meet the high energy demand [30]. Impaired coupling of substrate oxidation and ATP production is in line with previous findings in obese subjects and mice [38,39,40,41,42,43]. This suggests that mitochondrial adaptation and flexibility become compromised once (or possibly just before) NAFLD progresses to steatohepatitis. Adaptation or remodeling of mitochondrial energetics in the pathogenesis of simple steatosis to NASH is elegantly reviewed by Sunny and colleagues [3], and we will focus specifically on aspects of mitochondrial dysfunction that seem to be important for the NASH to HCC transition (Figure 1).
2.2. The Delicate Balance of Mitochondrial Reactive Oxygen Species
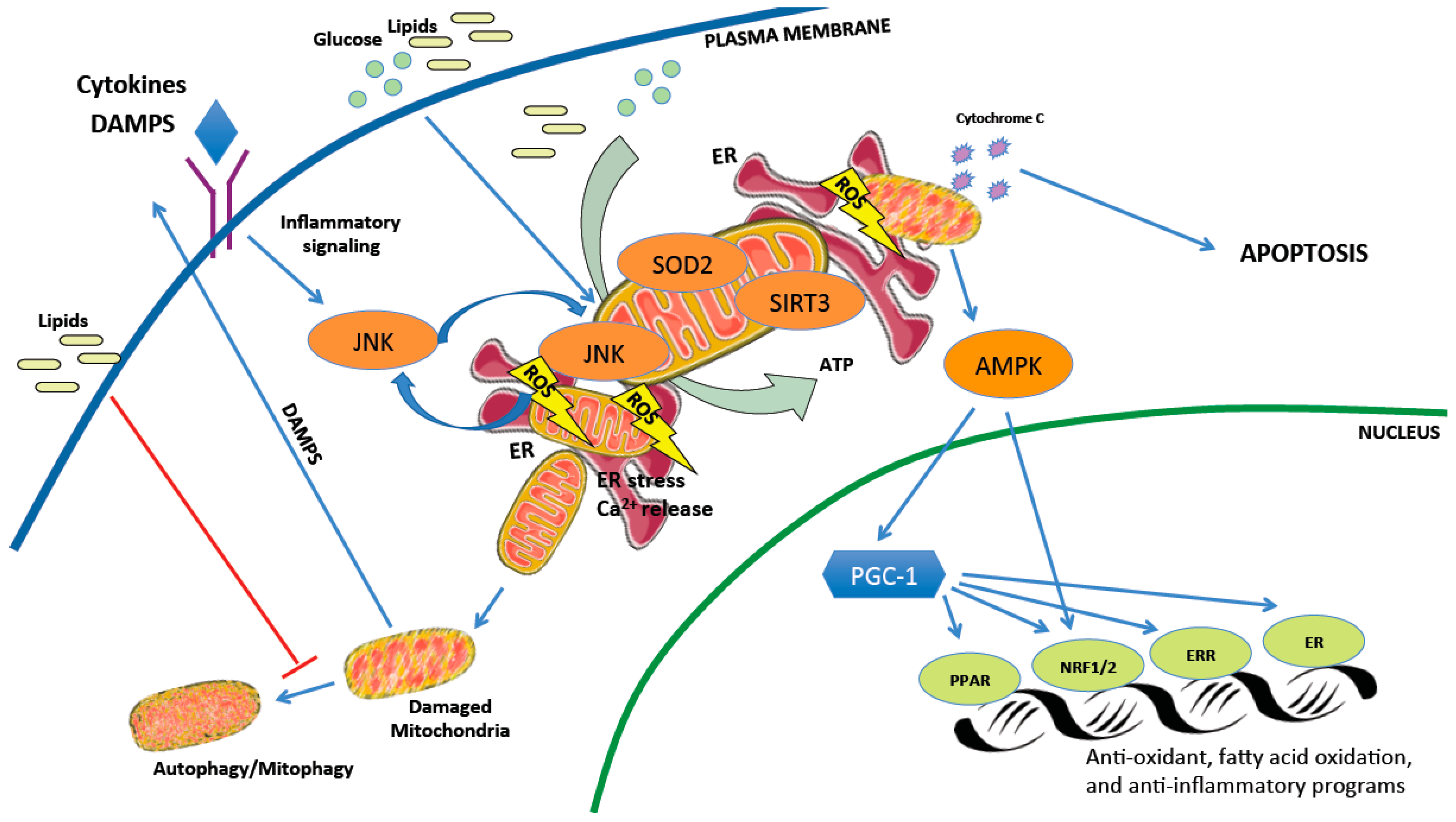
The imbalance of respiratory complex activity during the development of NAFLD and its progression to NASH leads to increased mitochondrial reactive oxygen species (ROS), causing oxidative mitochondrial DNA (mtDNA) damage, mitochondria structural abnormalities, and lipid peroxidation [37,44,45]. In line with this, increased serum markers of oxidation such as malondialdehyde are detected in NAFLD subjects, while antioxidants coenzyme Q10 and CuZn-superoxide dismutase are reduced [46]. Mitochondrial ROS (mtROS) and lipid peroxidation also trigger proinflammatory cytokines such as interleukin 6 (IL-6), tumor necrosis factor α (TNF-α), interleukin 1 beta (IL-1β), which are critical mediators of inflammation in NASH [47,48,49,50]. This is observed in rodent models of NASH, as substantial amounts of lipid peroxidation and proinflammatory cytokines are detected in mice and rats with steatohepatitis caused by NASH-promoting diets [51,52]. Exogenous lipid delivery or high-fat feeding in mice also upregulates mitochondrial oxidative metabolism, resulting in increased oxidative stress and inflammation [53].
Mitochondrial-derived ROS activate mitogen-activated protein kinases (MAPKs) including c-Jun N-terminal kinase (JNK) [54]. Phosphorylated JNK translocates to the mitochondria, where it binds to scaffold proteins inhibiting the mitochondrial respiratory chain and further increasing ROS production [54,55,56]. In animal models, sustained JNK activation mediates liver injury, but blocking translocation of phosphorylated JNK to the mitochondria can prevent the development of fatty liver in mice fed a high-fat diet [55]. Mitochondrial-derived ROS also activate AMP-activated protein kinase (AMPK) secondary to redox changes and mitochondrial ATP production [57]. H2O2 produced by mitochondria induces antioxidant enzyme expression through AMPK-mediated activation of nuclear factor 2 (NRF2), and consistently, NRF2 knockout mice on a high-fat diet develop more severe NASH associated with decreased antioxidant response, reduced β-oxidation genes, and increased lipogenic genes [58]. AMPK activators partially restore fatty acid oxidation in primary hepatocytes, consistent with a study showing that liver-specific activation of AMPK in mice protects against liver steatosis [59]. Taken together, mitochondrial functional status and ROS generation directly impact AMPK and JNK signaling, which both have important roles in maintaining liver metabolic function and health.
AMPK also activates the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), which is required for mitochondrial adaptive responses to oxidative stress [60]. PGC-1α is a transcriptional coactivator coordinating activity of several transcription factors important for mitochondrial biogenesis and function including, but not limited to, peroxisome proliferator-activated receptors (PPARs), nuclear respiratory factors (NRF-1 and -2), and estrogen-related receptors (ERRs). Decreased mitochondrial biogenesis is associated with a reduction of PGC-1α, transcription factor A, mitochondrial (TFAM), or PPARs in fatty livers [61,62,63,64]. Interestingly, PGC-1α coactivates estrogen receptor-alpha (ERα) in a ligand-dependent manner to increase superoxide dismutase 2 (SOD2) and glutathione peroxidase 1 (GPX1), enhancing ROS scavenging and reducing oxidative damage [52]. Besse-Patin et al. showed that female mice express higher levels of PGC-1α in liver tissue compared to male mice and are more susceptible to reduced PGC-1α in obesity, in line with evidence suggesting dependence of estrogen’s antioxidative properties on coactivator activity [52]. It has not yet been investigated whether protection of liver cells from mitochondria-induced ROS via PGC-1α:ERα contributes to sex-dependent differences in human NASH pathogenesis, where pre-menopausal women are more resistant. Although, it is intriguing to note that PGC-1α and mitochondrial mass/function are known to decrease with age [65,66,67,68,69,70,71], and combined with the drop in estrogen during menopause, this may explain why the incidence of NAFLD and NASH in women approaches (and may surpass) that of men after menopause [7,72].
In line with increased liver PGC-1α being beneficial in NAFLD, liver-specific overexpression of PGC-1α in Sprague Dawley male rats increases fatty acid oxidation and tricarboxylic acid (TCA) cycle activity in isolated liver mitochondria, and this was coupled with reduced hepatic and plasma triglycerides [73]. While hepatic PGC-1α is known to increase in a fasting liver to promote lipid catabolism, it is interesting to note that it also increases following acute exercise training in rodents [74,75,76,77]. McCoin et al. investigated whether liver-specific hemizygous disruption of PGC-1α had an impact on hepatic mitochondrial adaptation (respiratory capacity, H2O2 production, mitophagy) in male and female mice fed a high-fat diet (HFD) and whether exercise (voluntary wheel running) affected outcomes [77]. They note wild-type female mice have an inherent ability to increase hepatic mitochondrial function in response to the obesogenic challenge, while males need exercise to equally adapt their respiratory capacity. In striking contrast to male mice, HFD-induced liver pathology is exacerbated by voluntary wheel running in females. Consistent with previous findings [52], reduced hepatic PGC-1α has a greater impact in female mice and led to higher liver triglycerides, markers of liver fibrosis and serum alanine aminotransferase (ALT) after high-fat diet feeding, yet reduction of the coactivator remarkably restored the benefits of exercise on the female liver. These data suggest that exercise has differential effects on hepatic metabolism in male and female livers, and while low hepatic PGC-1α worsens NAFLD in sedentary females, it may also improve their hepatic responses to exercise. Of note, PGC-1α-dependent effects on NAFLD in female mice within this study appeared independent of changes in mitochondrial respiration, implying that detrimental effects of reduced PGC-1α in the liver may be more influential on antioxidant capacity [52] or responses to inflammation [78,79,80]. PGC-1α expression is also negatively correlated with NAFLD severity in humans [81], but it remains to be seen whether this reflects a cause or a consequence of the mitochondrial abnormalities found in humans or whether targeting PGC-1α could be a viable option to treat the disease.
In addition to antioxidant enzymes, mitochondria also contain metabolic sensors such as the nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase sirtuins that protect from the deleterious consequences of oxygen radicals on mtDNA, proteins, and lipids [82]. Sirtuin enzymes regulate biosynthesis, transport, and catabolism of NAD+, a dinucleotide cofactor with the potential to accept electrons in redox reactions [83]. NAD/NADH ratio is essential for mitochondrial ATP production and membrane potential and it appears that mitochondria have their own NAD biosynthetic machinery [84]. Three NAD-dependent deacetylase sirtuins (SIRTs) are localized in mitochondria: SIRT3, SIRT4, and SIRT5. SIRT4 is increased in NAFLD subjects, while the other sirtuins are decreased correlating with increased lipogenic genes including sterol regulatory element binding protein-1 (SREBP-1), fatty acid synthase, and acetyl-CoA carboxylase [85]. SIRT3 is the most investigated mitochondrial sirtuin and deficient SIRT3 activity predisposes mice to NASH [25,86]. SIRT3 is important for mitochondria biogenesis, carbohydrate metabolism, ketogenesis, amino acid metabolism, and stress-related pathways [87]. Deacetylation of the mitochondrial ROS scavenger superoxide dismutase 2 (SOD2) by SIRT3 in mice given streptozotocin (an ROS-inducer) points toward a protective role of this sirtuin in oxidative stress [88]. Moreover, reduced expression of SIRT3 and TFAM in diabetic hearts correlates with decreased transcription of mitochondrial DNA-encoded genes [89] and SIRT3 knockout mice have reduced fatty acid oxidation and low basal levels of ATP in the heart and liver [90,91]. Mice fed high-fat diets for a long time exhibit low SIRT3 activity, impaired mitochondrial function, and hyperacetylation of proteins in their livers [90]. Taken together, SIRT3 appears to play a protective role against NAFLD, possibly by improving mitochondrial function [92]. However, muscle- or liver-specific deletion of SIRT3 in mice subjected to a high-fat diet shows no difference in oxidative stress, glucose tolerance, or insulin sensitivity [93], and little is known about whether a gain-of-function in SIRT3 activity would have any beneficial effect.
2.3. Mitochondrial Networks, Housekeeping, and Cell Death
Chronic liver injury in individuals with NASH is associated with hepatocyte apoptosis [94,95]. Lipid peroxidation, formation of cytotoxic aldehyde, and proinflammatory cytokines activate stellate cells and hepatocyte cell death receptors (Fas, tumor necrosis factor receptor 1 (TNFR1), TNF-related apoptosis-inducing ligand (TRAIL)-receptor), resulting in fibrosis and apoptosis [96,97]. Activation of the mitochondrial intrinsic pathway of apoptosis (or programmed cell death) plays an important role in the progression of NASH [95]. Pro-apoptotic proteins of the B-cell lymphoma 2 (Bcl-2) family (e.g., bcl-2-associated X (Bax), bcl-2 homologous antagonist/killer protein (Bak), BH3 interacting-domain death agonist (Bid)) promote mitochondrial membrane permeabilization (MMP) by translocating to the mitochondrial outer membrane and forming mega channels. The anti-apoptotic proteins (e.g., Bcl-2, B-cell lymphoma-extra large (Bcl-xL), induced myeloid cell leukemia apoptosis regulator (Mcl-1)) prevent these processes [98]. Once triggered, MMP leads to the release of proteins such as apoptosis inducing factor (AIF) and cytochrome c which coordinate both mitochondrial dysfunction and trigger cell death pathways [99]. MMP can be influenced by lipid accumulation, ions (Ca2+), pH, ROS, and ATP levels [99]. In line with this, mice maintained on a high-fat diet and injected with glucose creating glucolipotoxic conditions displayed marked lipid peroxidation, hepatocyte apoptosis, and inflammation [100]. Moreover, mice maintained on a high-fat diet for eight weeks exhibited a disrupted mitochondrial respiratory chain and decreased ATP levels coupled with activated mitochondrial caspase 3-dependent cell death pathway [101]. Treatment with cyclosporin A, which inhibits the mitochondrial permeability transition via enhanced matrix Ca2+ buffering [102], can prevent mitochondrial dysfunction, oxidative stress, and hepatocyte apoptosis [100]. Taken together, there is a strong correlation between impaired mitochondrial function and cell death, and evidence suggests that their dysfunction amplifies the apoptotic signal in liver cells in response to high glucose and lipids.
Although there can be more mitochondria in a metabolically unhealthy liver, NASH is also characterized by the accumulation of abnormal mitochondria, which might be secondary to defects in mitophagy. Excess lipids, as well as insulin resistance and hyperinsulinemia, suppress autophagy by altering vesicular fusion, autophagy protein expression, and autophagy maturation [103]. Mitophagy is thought to protect against NAFLD, and its reduction in liver tissues from subjects with NASH correlates with disease severity [104]. Reduced mitophagy results in the accumulation of severely damaged mitochondria, leading to cell necrosis releasing mitochondrial damage-associated molecular patterns (DAMPs) that can promote liver inflammation and NASH development. Mitochondrial DAMPs (MTDs) activate pattern recognition receptors (PRRs), such as Toll-like receptor (TLR)9 [105,106]. After binding to TLR9, mitochondrial DAMPs trigger numerous downstream pathways, including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), the nucleotide binding oligomerization domain (NOD)-like receptor family, pyrin domain containing 3 (NLRP3), and interferon regulatory factor-dependent type 1 [107] to promote inflammation. In line with this, a decline in mitophagy is associated with activation of hepatic NLRP3 inflammasome in a murine mouse model of NASH and palmitic acid-treated primary hepatocytes [108], suggesting that decreased mitophagy might promote inflammation and NASH development.
While mitochondrial dysfunction is considered a critical component in NASH development, other potential mechanisms such as endoplasmic reticulum (ER) stress may contribute to disease progression. Both organelles physically interact through mitochondrial-associated membranes (MAMs) to dynamically adjust metabolic demand and responses to stress stimuli [109]. These contact sites normally allow the exchange of lipids, calcium (Ca2+), and ROS [110], and evidence suggests a relationship between MAM formation and the unfolded protein response (UPR), as ER stress sensors are found at the MAM interface and loss of MAM proteins induces UPR signaling [111,112]. ER stress markers are elevated in NASH-affected livers [113] and mitochondrial dysfunction correlates with excessive endoplasmic reticulum stress [30]. Mitochondrial ROS and oxidative stress can disrupt ER function leading to inappropriate release of calcium in liver parenchymal cells [114]. Recent studies also show that MAMs are important for hepatic insulin signaling, nutrient sensing, and glucose homeostasis [111,112]. Therefore, disruption of ER-mitochondria contact sites could exacerbate hepatic lipid accumulation and miscommunication between organelles appears to be related to the pathology of hepatic metabolic diseases. The complex interplay of cellular mechanisms impacting mitochondrial health and how this can lead to hepatocyte cell damage is illustrated in Figure 2.
2.4. We Are What We Eat
NAFLD is highly correlated with obesity and calorically high, nutritionally poor diets. Overconsumption of a diet rich in fat is also linked to the development of fatty liver disease [115]. Excessive storage of triacylglycerols in liver is a key feature of fatty liver disease and interestingly individuals with NASH have significantly more saturated fatty acids in their triacylglycerols [116]. Saturated fats promote fatty liver and accumulation of lipotoxic byproducts including ceramides and diacylglycerols [117,118] is associated with hepatic inflammation and mitochondrial ROS production resulting in liver cell death [119]. Moreover, in vitro treatment of liver cells with saturated fatty acids (palmitic and stearic acids) reproduces mitochondrial dysfunction found in NASH, including decreasing cellular ATP content and mtDNA-encoded oxidative phosphorylation (OXPHOS) subunit expression, coupled with increased oxidative stress [120].
In addition to fat, excessive glucose consumption (or hyperglycemia associated with conditions such as diabetes) could be a primary cause of mitochondria respiratory chain dysfunction by increasing the burden on oxidative phosphorylation and causing oxidative stress [121]. Vanhorebeek et al. reported that normoglycemia or strict control of blood glucose in humans prevents or reverses ultrastructural or functional abnormalities of hepatocyte mitochondria [121]. Consistent with this, high glucose induces mitochondrial fragmentation, ROS production, loss of mitochondrial membrane potential, and ATP depletion in several rodent and human cell models [122,123,124,125,126,127]. In addition to glucose, fructose appears to be particularly harmful to liver mitochondria [128]. In addition to its ability to promote de novo lipogenesis and block β-oxidation of fatty acids, fructose consumption seems to cause a drop in ATP and an elevation of uric acid, which can further induce mitochondrial oxidative stress [129]. Moreover, a diet rich in fructose is associated with increased oxidative mtDNA lesions in rat liver coupled with reduced mitochondrial repair capacity [130] and rats consuming a diet high in fat and rich in fructose have increased hepatocyte damage, inflammation, and lipid peroxidation coupled with impaired mitochondrial respiration and activity [131]. Studies show that dietary fructose induces fatty liver [132,133,134] and inflammation in mice after 8–24 weeks of exposure [52,135,136], as well as hepatic fibrosis in rhesus monkeys after seven years [137]. While these data in animals and humans implicate fructose as a risk factor for fatty liver [138], further studies are needed to investigate the direct relationship between mitochondrial dysfunction and fructose consumption in humans.
Dietary cholesterol is also implicated in the development of NASH in mouse models and humans [139]. More precisely, dietary-induced hypercholesterolemia causes oxidative stress, loss of mitochondrial membrane potential, reduction in ATP content, loss of mitochondrial cristae, and hepatic steatosis in mice [140]. It is also suggested that overload of free cholesterol disrupts mitochondrial and ER membrane integrity, triggering mitochondrial oxidative injury and ER stress [141]. Bellanti et al. showed through targeted lipidomic analysis that rats fed diets rich in cholesterol exhibited increased toxic hepatic triol, which induces apoptosis and impaired mitochondrial respiration in vitro [142]. Gan et al. showed that mitochondrial-free cholesterol deposition causes hepatocyte apoptosis and necrosis through c-Jun N-terminal kinase (JNK) activation, associated with increased high-mobility group box 1 (HMGB1), and cytolytic effects on neighboring hepatocytes driven by Toll-like receptor 4 (TLR4) [143]. Moreover, mitochondrial-free cholesterol sensitizes liver cells to TNFα- and Fas-mediated steatohepatitis and causes mitochondrial-reduced glutathione (mGSH) depletion [144], which is also reported in animal models and patients with NASH [145]. Thus, accumulating evidences suggest that cholesterol overload in mitochondria induces redox imbalances leading to oxidative stress and cell death associated with steatohepatitis [146,147,148]. Consumption of high calorie diets rich in sugar, saturated fat, and cholesterol contributes to obesity, a major risk factor of fatty liver disease. While disease in the liver may simply be due to accumulating lipids, evidence suggests that the direct effect of these nutrients and their metabolism on mitochondrial health, independent of weight gain, likely plays a significant role in pathogenesis.
3. Mitochondrial Dysfunction in NASH-Associated HCC
3.1. From Cell Death to Proliferation
Prolonged exposure of hepatocytes to lipids and inflammation can result in the exhaustion of liver defense mechanisms, leading to chronic liver disease that promotes the development of hepatocellular carcinoma (HCC). Multiple mechanisms are proposed for the pathogenesis of obesity-associated HCC [149], and they can be different from cancers stemming from alcohol or viral origins. Among these, mitochondrial dysfunction is linked to cancer progression through increased ROS production, impaired mitochondrial respiration, ER stress, and alteration of nutrient metabolism [150]. Overproduction of ROS impairs the mitochondrial respiratory chain, leading to cytochrome c release, and apoptotic death signals in cancer cells as well [151]. Moreover, oxidative stress and ROS lead to the release of calcium by the endoplasmic reticulum, further increasing mitochondria ROS production and potentiating proapoptotic pathways [151,152]. While apoptosis impedes the growth of hepatocellular carcinoma cells [153], it is also believed to promote cancer initiation through compensatory proliferation of progenitor cells in the liver [154,155]. This increased proliferation within an environment of higher ROS production and DNA damage causes gene mutations to accumulate in mature and newly formed hepatocytes. As time passes, cells that can now resist apoptotic pressure and escape cell cycle checkpoints persist and contribute to malignant transformation and cancer development [50]. Furthermore, an environment where mitochondrial function is already reduced by metabolic stress could favor survival of cells that do not depend on mitochondria for energy production, a feature of many cancer cell types [156]. Lastly, damage or mutation of mtDNA itself may potentiate HCC. A low mtDNA copy number is associated with liver cancer and this may confer resistance to chemotherapy [157,158]. Thus, whether it be ROS-induced accumulation of DNA mutations (both in mtDNA and ncDNA), increased mitochondrial-mediated apoptosis due to metabolic exhaustion, or a combination of the two, mitochondrial abnormalities can contribute in many ways to cancer development in obesity and metabolic disease.
Just like in NASH, metabolic stress, mitochondrial dysfunction, and oxidized mtDNA also indirectly contribute to the progression of HCC through influences on inflammation and immunologic pathways, including activation of the inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ)/NF-κB signaling pathway, a key node that plays a crucial role in hepatocyte survival and inflammatory responses [154,158]. ROS and DNA/lipid peroxidation increase the release of cytokines including TNF-α and IL-6, which activate pro-oncogenic pathways via JNK, signal transducer and activator of transcription 3 (STAT3), Janus kinase 2 (JAK2), MAPK, and phosphoinositide 3-kinase (PI3K) [159,160,161]. TNF-α and IL-6 promote the development of obesity-induced HCC development in mice exposed to the liver carcinogen diethylnitrosamine when combined with a high-fat diet [159]. Moreover, TNF-α and IL-6 promote iron accumulation, which further fuels oxidative stress-driven cell toxicity and can activate fibrogenesis and carcinogenesis in metabolic syndrome [162]. Indeed, detrimental effects of hepatic iron overload seem to impact HCC development in NASH patients [163,164]. Thus, mitochondrial dysfunction, in combination with or driving cytokine imbalances and iron accumulation, are part of a damaging cycle that potentiate hepatocyte injury, NAFLD progression, and malignant transformation of hepatocytes.
The production of ROS also stabilizes hypoxia inducible factor alpha (HIFα) subunits by inactivating prolyl hydroxylase domain-containing protein (PHD) enzymatic activity [165]. Since α-ketoglutarate is needed for PHD function, conditions promoting low concentrations of this metabolite (another consequence of mitochondrial dysfunction) will lead to an overall increase in HIF transcriptional activity [165]. HIFα subunits are stable under hypoxia, while they are rapidly degraded under normal oxygen tension. Changes in oxygen tension occur during steatohepatitis, and when combined with inflammation, may be sufficient to promote a hypoxic response that increases HIF-regulated transcription of genes involved in cellular metabolism, angiogenesis, and proliferation, all of which promote tumor development [165]. Moreover, a hypoxic environment further compromises ATP generation, leading to heavier reliance on glycolysis [166]. In agreement with this, mice with hepatocyte-specific deletion of HIF-2α have reduced liver tumor numbers and size on a choline-deficient L-amino acid refined diet following diethylnitrosamine administration [167]. Additionally, increases in hepatic HIF-1α accelerate the transition from NASH to HCC in a model of NASH-associated liver cancer involving a high-fat–high-cholesterol–high-sugar diet combined with diethylnitrosamine [168]. Thus, mitochondrial dysfunction could promote the transition from NASH toward HCC by potentiating pathways designed to protect cells from hypoxic damage that unfortunately also promote tumor development within permissive environments.
3.2. Cancer Cells: It is Not Only What You Burn, But When
Metabolic reprogramming is a key event in HCC development and progression [28,29]. Metabolites generated during this process can be cancer-promoting and favorable metabolite signatures seem to be selected for tumor cells [28]. Well-characterized metabolic changes thought to promote cancer include the Warburg effect (a switch toward anaerobic glycolysis) [169], increased glutamine metabolism, and defects in one-carbon metabolism [170]. Early studies in malignant tumors describe a fundamental reprogramming of gene expression resulting in a highly glycolytic “Warburg” phenotype and suppression of mitochondrial biogenesis. However, new evidence suggests that because of sustained proliferation and nutrient depletion in malignancies, a second “wave” of cancer cell adaptation is required to restore the more efficient process of oxidative phosphorylation to accommodate increasing energy demands [171]. Interestingly, increased lactate production is observed in NASH patients, suggesting there may be a shift toward a glycolytic phenotype in the first steps of liver carcinogenesis [172]. In the second steps of malignant transformation, increased cellular dependency on glutamine can occur to support the high bioenergetic demand, generating high levels of α-ketoglutarate and citrate to support the TCA cycle to provide carbon cycles for amino acid biosynthesis [173].
Proliferative tissues are highly sensitive to changes in one-carbon (folate) metabolism and dysregulation of this pathway is reported in NASH and HCC [174,175]. Mitochondrial one-carbon metabolism is required for several biological reactions including nucleotide synthesis, amino acid metabolism, NADPH production, and epigenetic methylation [176]. Alterations in major sources of one-carbon units such as glutathione, choline, carnitine, serine, and glycine compromise mitochondrial performance and metabolic control [174]. For instance, serine is the most important donor of one-carbon units through the mitochondrial serine hydroxymethyltransferase (SHMT2), which catalyzes the reversible conversion of serine to glycine [174]. Although the conversion rate of serine has not been reported in obesity and associated metabolic disorders, decreased plasma level of serine is seen in NASH patients [177,178,179,180]. In contrast, serum levels of serine are increased in humans with HCC compared with healthy subjects [181,182] and high SHMT2 expression is associated with negative prognosis in human hepatocellular carcinoma [183]. Downregulation of SHMT2 suppresses human HCC cell proliferation and liver tumor incidence and growth in a xenograft model [184]. It is also suggested that cancer cells benefit from enhanced serine-dependent one-carbon metabolism for the production of NADPH [185] and this may be exacerbated by obesity. Disturbances in liver one-carbon metabolism, including increases in transcript level of mitochondrial SHMT2, are associated with hepatic lipid accumulation in mice fed a high-fat diet [186].
Altered or enhanced lipid metabolism is also implicated in metabolic reprogramming of cancer cells [187]. A lipid-rich condition characterized by large droplet steatosis and ballooning, and associated with pericellular fibrosis and inflammation, is observed in tumor cells of steatohepatitic variants of HCC [188]. Metabolic profiling by liquid chromatography-mass spectrometry (LC-MS) reveals extensive accumulation of long-chain acylcarnitine species in HFD-fed HCC tissues of mice correlated with the increased expression of the mitochondrial transporter carnitine palmitoyltransferase-1 (CPT1) and decreased expression of CPT2, an important enzyme of the mitochondrial long-chain fatty acid oxidation [28]. Lipid metabolic reprogramming mediated by CPT2 downregulation in HCC cells can promote liver cancer through accumulation of acylcarnitine as an oncometabolite [28]. Indeed, decreases in CPT2 are reported in HCC and in subjects with chemoresistance to cisplatin [189]. These findings are in line with previous studies showing that the expression of acylcarnitine metabolism-related genes are altered in NASH-driven HCC mouse models, including HFD-fed major urinary protein (MUP)- urokinase-type plasminogen activator (uPA) [190] mice and phosphatidylinositol 3-kinase catalytic subunit alpha PI3KCA transgenic mice [191]. Overall, mitochondria are involved in several biochemical pathways that generate metabolic precursors needed to support the sustained bioenergetic demands of NASH and HCC pathogenesis.
3.3. Transcriptional Regulation of Mitochondrial Adaptation in HCC
Given the role of PGC-1α as a key regulator of mitochondrial metabolism, adaptation, and antioxidant defense, several studies have investigated its role in cancer development, including hepatocellular carcinoma. Whether it sustains or interferes with tumorigenesis remains contradictory. Reduced hepatic PGC-1α expression is reported in human HCC associated with increased glycolysis and reduction of blood glucose [192]. Decreased PGC-1α is also associated with dedifferentiation of human hepatoma cell lines through impairment of hepatocyte nuclear factor 4 alpha (HNF4α), a transcription factor critical for liver development [193]. Consistent with the coactivator having tumor-suppressive functions, Lee et al. showed that overexpression of PGC-1α increases the epithelial marker E-cadherin and reduces motility of HepG2 cells possibly through PPARγ [194] and PGC-1α can bind p53 and enhance transcription of proarrest genes including cyclin-dependent kinase inhibitor 1 (p21), growth arrest and DNA damage-inducible 45 (GADD45), TP53-inducible glycolysis and apoptosis regulator (TIGAR), SCO cytochrome C oxidase assembly Protein 2 (SCO2), and sestrin2 in human hepatoma cells [195]. On the other hand, Ballah et al. showed that loss of PGC-1α in whole-body knockout mice prevented diethylnitrosamine (DEN)-induced liver cancer development and overexpression of the coactivator promotes tumor development in a xenograft model by increasing lipogenesis [196]. Interestingly, mice with liver-specific overexpression of the related coactivator PGC-1β also exhibited increased chemically-induced (DEN) and genetically-induced (Abcb4−/−) liver cancer resulting from increased ROS scavenging and tumor anabolism [197]. Interestingly, the authors’ report reduced PGC-1α in liver tumors of these mice and, vice versa, mice with liver-specific ablation of PGC-1α had a compensatory increase in PGC-1β [52]. Although PGC-1α and PGC-1β are often viewed as redundant in function, there is evidence in liver that they may regulate different gene programs related to nutrient metabolism, namely PGC-1β is the primary isoform-enhancing lipogenesis [52,81,197,198,199]. This may suggest that PGC-1β is a driver of tumor formation, while PGC-1α confers resistance. However, although there is strong evidence linking PGC-1 coactivators to HCC pathogenesis, it still remains unclear how this family of coactivators directly affects cancer cell biology and tumor development, particularly when metabolism is altered as in obesity or metabolic disease.
Answering this question is complicated by the multiple ways by which PGC-1 activity is controlled in cells. In addition to changes in mRNA and protein level, post-translational modification of PGC-1α has been implicated in hepatocarcinogenesis, including its phosphorylation by AMPK and deacetylation by sirtuins [29]. Zhang et al. showed that activation of the AMPK-PGC-1α pathway inhibits proliferation and induces apoptosis of hepatocellular carcinoma cell lines [200]. Li et al. showed that SIRT1/PGC-1α axis facilitates hepatocellular carcinoma metastasis through increased mitochondrial biogenesis [201]. Apart from the deacetylase activity of SIRT1 on PGC-1α, less is known about the impact of PGC-1α on sirtuins themselves. Kong et al. showed that SIRT3 is a downstream target gene of PGC-1α in hepatocytes and mediates the suppressive effects of PGC-1α on ROS production [202]. SIRT3 can also inhibit growth and proliferation of the human hepatoma cells HepG2 and induce apoptosis [203] and is a tumor suppressor in the human hepatoma cells Huh7, reducing phosphorylation of PI3K/Akt [204]. Of note, SIRT3 expression is downregulated in human HCC [203,204] and low SIRT3 expression is associated with poor differentiation and unfavorable prognosis [205]. Thus, mitochondrial SIRT3 appears to play a protective role in HCC. Given the potential link between PGC-1α and SIRT3, it could be interesting to investigate the role of this partnership in NAFLD-associated liver cancer. Taken together, studies suggest that PGC-1α may have different roles in tumor development depending on the cell type, environment, and current metabolic state. Further studies are needed to clarify whether PGC-1α plays a significant role in hepatic cancer development and whether this role changes in different metabolic environments based on the need or dependence of cancer cells on mitochondrial metabolism.
4. Challenges in Targeting Mitochondria for NASH-Related HCC Treatment
Currently, the tyrosine kinase inhibitor sorafenib is the only approved systemic medication for the treatment of HCC, increasing patient survival by months [206]. Chemotherapy and radiotherapy are generally ineffective, as HCC cells are highly chemoresistant. There is critical need for new effective and more targeted therapies for this disease. As evidence in animal models supports mitochondrial dysfunction as a key player in metabolic liver disease pathogenesis, targeting mitochondria represents an attractive strategy to stop or slow down NASH-related HCC progression. However, what has been revealed from these efforts is at times paradoxical. Mitochondrial-based therapies do indeed have varying efficacy to treat liver disease and cancer, though is not clear whether the correct strategy should be to boost or inhibit their function. There is currently no available treatment specifically targeting mitochondria in NASH-related HCC. We will discuss this as a potential treatment strategy, its associated challenges, and paradoxical findings, and avenues that remain to be investigated.
4.1. Lifestyle Intervention
Regular exercise and caloric restriction are the only physiological interventions proven to reduce steatosis, inflammation, and fibrosis in NAFLD patients [207]. Mechanistic studies in mice show that exercise improves liver mitochondrial morphology, and increases mitochondria biogenesis (PGC-1α and TFAM), autophagy-related proteins, and SIRT3 [208,209]. However, whether these outcomes on mitochondrial health help to prevent HCC in humans is extremely difficult to determine. A large prospective study suggests that increased physical activity reduces liver cancer risk in men; however, the etiology of liver cancer in this study was not restricted to NASH [210]. On the other hand, rapid weight loss with severe dieting or malnutrition increases liver inflammation and fibrosis [211]. Unfortunately, despite the known benefits of caloric restriction to reduce obesity and lengthen lifespan, lifestyle modification is very difficult to implement, and even more difficult to maintain, in the general population. Notably, since the diagnosis of liver cancer is often made late in its progression when overall health and energy of the patient are already failing, diet and exercise may not always represent a recommendable or feasible course of action.
4.2. Antioxidants
Since both increased mitochondrial function and dysfunction causes ROS production, antioxidant supplementation is predicted to prevent steatohepatitis and possibly have anti-cancer effects. However, broad specificity of this therapy may also have detrimental effects on cellular pathways that require ROS, such as redox signaling [212], a caveat not often considered during evaluation of these approaches. Indeed, the reduced glutathione (GSH) precursor N-acetylcysteine (NAC) prevents development of steatosis in methionine-choline deficient (MCD)-diet fed rats and ob/ob mice [213,214]. It also attenuates hepatocarcinogenesis in TLR2-deficient mice by inhibiting ROS and ER stress, and induces apoptosis in human liver cancer cells [215,216]. Vitamin E (α-tocopherol) can act as a peroxyl radical scavenger [217,218]. Deficiencies in vitamin E can be found in NASH patients [219] and the PIVENS (Pioglitazone, Vitamin E or Placebo for Nonalcoholic Steatohepatitis) trial in NASH patients showed a reduction in hepatic steatosis, lobular inflammation, and transaminases [220]. Interestingly, vitamin E treatment in children with NAFLD may provide no improvement of liver function [221]. Based on these results, the American Association for the Study of Liver Disease (AASLD) recommended in 2012 that vitamin E should be considered for non-diabetic adults with biopsy-proven NASH [222]. Vitamin E prevents HCC in mice via downregulation of inducible nitric oxide synthase (iNOS) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [223] and protects against oxidative DNA damage in human HCC cell lines [224]. Zhang et al. also reported that dietary vitamin E decreases the risk of developing hepatitis B virus (HBV)-related HCC in humans [225]. Finally, while antioxidants may help prevent cancer initiation by reducing ROS, they may also lessen ROS to levels that support cancer cell proliferation and metastasis by minimizing some of the negative effects (i.e., DNA damage) of ROS on malignant cells, thus cancer stage and whether ROS is a major driver influence outcomes. Interestingly, a very effective method to reduce cellular ROS is to inhibit mitochondrial oxidative phosphorylation with mitochondrial toxins (e.g., metformin) or mild chemical uncouplers. Inhibition of mitochondrial function with these chemicals also effectively reverses NAFLD and prevents HCC in mice [226] and zebrafish [227], arguing that it might not be reduced mitochondrial function or oxidative capacity that is pathogenic per se, but rather an inability to effectively deal with the bioproducts of their activity.
4.3. Pioglitazone
The antihyperglycemic drug pioglitazone is an interesting candidate for the treatment of NASH [228]. It belongs to the thiazolidinedione family and acts as a potent ligand for the nuclear factor peroxisome proliferator-activated receptor gamma (PPARy). Cusi et al. showed that 18 and 36 months of pioglitazone treatment in prediabetic or type 2 diabetic patients with biopsy-proven NASH improved fibrosis score, hepatic triglyceride content, as well as hepatic and muscle insulin sensitivity [229]. It also ameliorates steatosis and necroinflammation in humans and diabetic mice with NASH [230,231] and improves hepatic mitochondrial oxidative capacity in a mouse model of NASH [232]. It effectively delays liver fibrosis and hepatocarcinogenesis in two rodent models of HCC induced by diethylnitrosamine alone or diethylnitrosamine combined with a choline deficient, L-amino acid defined, high-fat diet [233]. Interestingly, thiazolidinediones including pioglitazone lower the risk of liver cancer incidence and HCC recurrence in diabetic patients [234,235]. Whether the beneficial effects of this drug are due to improved hepatic mitochondria function, or the many other known benefits of PPARγ activation such as improved insulin sensitivity, remains to be determined. However, concerns over possible hepatotoxicity and other undesirable side effects make PPARγ agonists unlikely to be used widely for NASH or HCC, or at least not in their current form.
4.4. NAD+ Precursors
Boosting mitochondrial redox homeostasis using NAD+ precursors may also be attractive. For example, resveratrol increases the NAD+/NADH ratio by activating AMPK in combination with SIRT1 or SIRT3, resulting in increased fatty acid β-oxidation and tricarboxylic acid cycle in liver cells [236]. Resveratrol also reduces oxidative stress, inflammation, fibrosis, hepatic lipogenesis, and steatosis in animal models of NASH and NAFLD [237], and in obese persons [238]. On the other hand, resveratrol impairs F0/F1 ATP synthase activity in mitochondria isolated from rat brain and liver, which is suspected to result in cell death by apoptosis or necrosis [239]. While the pro-apoptotic effect of resveratrol could be useful for cancer cell eradication [240], this could also be harmful for healthy cells. In humans, its effectiveness as a treatment for NAFLD remains inconclusive, as resveratrol has only been tested in a small number of clinical trials [241]. Instead, it is suggested that it might be more effective as supplementation to additional lifestyle changes. Finally, it is important to note that the bioavailability of resveratrol is very limited in humans. Other approaches to boost NAD+ levels include inhibition of NAD+-consuming enzymes (such as poly ADP-ribose polymerase; PARPs and CD38) and supplementation with NAD+ precursors [242,243]. Inhibition of the glycohydrolase CD38 in mice protects from HFD-induced obesity and liver steatosis via NAD-dependent activation of the SIRT1-PGC-1α pathway [244]. Importantly, PARP1 is robustly activated in the liver of NAFLD patients and positively correlates with hepatic steatosis (i.e., triglycerides and free fatty acids) [245]. Mechanistic studies in mice show that inhibition of PARP protects from NAFLD and NASH [246,247]. Gariani et al. demonstrated that it replenishes NAD+ levels and promotes mitochondrial biogenesis and hepatic β-oxidation [246]. Supplementation with the NAD precursor pyridine nucleoside form of vitamin B3, nicotinamide riboside, protects and reverts NAFLD through SIRT1- and SIRT3-dependent mitochondrial unfolded protein response, increasing TCA cycle, OXPHOS, and hepatic β-oxidation [248]. Nicotinamide riboside supplementation also reduces body weight and attenuates liver fibrosis in a diet-induced mouse model of NAFLD [249]. Since NAD+ boosters have shown efficacy in rodents and appear to raise NAD+ levels in mouse and humans [248,250,251,252,253], research interest into the effects of NAD+ precursors in humans is growing considerably. However, efficacy in subjects with metabolic disorders remains to be determined. Boosting NAD+ for the prevention and treatment of liver cancer has also been proposed [254,255]. However, the effects of nicotinamide ribosides on mitochondria in the context of HCC remains to be investigated.
4.5. Master Regulators of Metabolism
Another potential therapeutic strategy could be to use AMPK activators such as the 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), which can increase insulin sensitivity, improve glucose uptake, and reverse hepatocyte injury [58,256]. AICAR inhibits hepatosteatosis and liver tumor development in a mouse model of HFD-related HCC [257] and AICAR reduces human HCC cell adhesion and proliferation via cell cycle arrest in G1/S [258,259]. However, similar to resveratrol, AICAR is poorly bioavailable when administered orally [260] and can also induce mitochondrial-mediated apoptosis [261,262]. Given the diverse and numerous effects of AMPK activation on cells in general, in addition to their low bioavailability, the use of these molecules requires a more in-depth understanding of the downstream molecular mechanisms implicated in their anti-NAFLD effects and whether these would be different in cancer cells versus differentiated liver cells. PGC-1α may also represent an interesting target, as it regulates a vast transcriptional network of pathways centered on enhancement and maintenance of mitochondrial biogenesis, oxidative capacity, fatty acid oxidation, and ROS detoxification. However, targeting PGC-1α is not easy, as in contrast to nuclear receptors, enzymes, or cell-surface receptors, transcriptional coactivators lack highly specific ligand-binding domains [81]. Interestingly, many treatments acting through SIRT1 and AMPK, such as the AICAR, metformin, and resveratrol have been shown to induce or activate PGC-1α in different organs [263,264,265,266,267]. However, PGC-1α-dependent effects of these molecules remain to be determined.
An important final consideration for all NASH and HCC therapies is whether the duration of treatment reverses and/or maintains improved liver health. Clinical studies reporting improvements in NAFLD and prevention of HCC are generally performed over short periods of time (i.e., over two years) [268]. Given that the progress to HCC from NAFLD/NASH occurs over a relatively long period (10–20 years), long-term safety and efficacy for any treatment for NASH and associated liver cancer would need to be established. In addition, lifestyle changes including weight loss, exercise, and diet modification are likely needed to prevent reoccurrence of the disease. More research is needed to understand how these environmental and lifestyle modifications affect treatment efficacy and safety, as level of compliance (ranging anywhere from intense intervention to minimal participation) is hard to control in humans.
5. Conclusions
Mitochondria play a key role in liver metabolism and likely lie at the core of NASH. They are important for adaptation to nutrient overload through their ability to increase β-oxidation of fatty acids and oxidative phosphorylation. However, this comes at a considerable cost, as increases in activity increases ROS production that can lead to apoptosis and compensatory cell proliferation contributing to malignant transformation. Emerging evidence clearly suggests that mitochondrial dysfunction is central to NASH and HCC. Recent advances in animal models shed some light on the complex network involved in the pathogenesis with many pointing toward the significance of inappropriate ROS regulation, but replicating the full spectrum of human NASH-HCC in rodents remains challenging, as we still do not yet know the precise diet, lifestyle, and environmental variables that trigger NASH in humans, let alone other species. Determining whether the mitochondrial defects observed in the NASH-HCC transition translate into causative explanations for the disease is a major priority, as it is at this stage of the disease where targeting will have the most benefit. It is difficult to dismiss the abundance of evidence linking mitochondrial bioenergetics, ROS imbalance, ER stress, inflammation, and cell death to metabolic disease and carcinogenesis. Thus, the therapeutic potential of agents targeting mitochondrial function in NASH-HCC transition remains a very interesting avenue to pursue, but it appears that there is still much more we can learn about these important organelles, particularly within the setting of human disease.
Author Contributions
M.L. and J.L.E. co-wrote the manuscript.
Funding
This study was supported by the IDRC grant (108591-001) and an FRQS doctoral fellowship to M.L. and Chercheur-Boursier Jr2 award to J.L.E.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yang, K.C.; Hung, H.-F.; Lu, C.-W.; Chang, H.-H.; Lee, L.-T.; Huang, K.-C. Association of Non-alcoholic Fatty Liver Disease with Metabolic Syndrome Independently of Central Obesity and Insulin Resistance. Sci. Rep. 2016, 6, 27034. [Google Scholar] [CrossRef] [Green Version]
- Asrih, M.; Jornayvaz, F.R. Metabolic syndrome and nonalcoholic fatty liver disease: Is insulin resistance the link? Mol. Cell. Endocrinol. 2015, 418, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef]
- Portillo-Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High Prevalence of Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Mellitus and Normal Plasma Aminotransferase Levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef]
- Maximos, M.; Bril, F.; Sanchez, P.P.; Lomonaco, R.; Orsak, B.; Biernacki, D.; Suman, A.; Weber, M.; Cusi, K. The role of liver fat and insulin resistance as determinants of plasma aminotransferase elevation in nonalcoholic fatty liver disease. Hepatology 2015, 61, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.-J.; Fallon, M.B. Gender and racial differences in nonalcoholic fatty liver disease. World J. Hepatol. 2014, 6, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in NAFLD: State of the Art and Identification of Research Gaps. Hepatology 2019. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 451–496. [Google Scholar] [CrossRef]
- Calzadilla Bertot, L.; Adams, L. The Natural Course of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359.e2. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Otgonsuren, M.; Henry, L.; Venkatesan, C.; Mishra, A.; Erario, M.; Hunt, S. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015, 62, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Poklepovic, A.; Moyneur, E.; Barghout, V. Population-based risk factors and resource utilization for HCC: US perspective. Curr. Med. Res. Opin. 2010, 26, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, A.; Sood, G.K. Hepatocellular carcinoma review: Current treatment, and evidence-based medicine. World J. Gastroenterol. 2014, 20, 4115–4127. [Google Scholar] [CrossRef]
- Edeline, J.; Raoul, J.-L.; Vauleon, E.; Guillygomac’h, A.; Boudjema, K.; Boucher, E. Systemic chemotherapy for hepatocellular carcinoma in non-cirrhotic liver: A retrospective study. World J. Gastroenterol. 2009, 15, 713–716. [Google Scholar] [CrossRef]
- Medavaram, S.; Zhang, Y. Emerging therapies in advanced hepatocellular carcinoma. Exp. Hematol. Oncol. 2018, 7, 17. [Google Scholar] [CrossRef]
- Balogh, J.; Victor, D.; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P. Hepatocellular carcinoma: A review. J. Hepatocell. Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef]
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World J. Hepatol. 2017, 9, 533–543. [Google Scholar] [CrossRef]
- Kawada, N.; Imanaka, K.; Kawaguchi, T.; Tamai, C.; Ishihara, R.; Matsunaga, T.; Gotoh, K.; Yamada, T.; Tomita, Y. Hepatocellular carcinoma arising from non-cirrhotic nonalcoholic steatohepatitis. J. Gastroenterol. 2009, 44, 1190–1194. [Google Scholar] [CrossRef]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: A pathological analysis. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Said, A.; Ghufran, A. Epidemic of non-alcoholic fatty liver disease and hepatocellular carcinoma. World J. Clin. Oncol. 2017, 8, 429–436. [Google Scholar] [CrossRef]
- Shaker, M.; Tabbaa, A.; Albeldawi, M.; Alkhouri, N. Liver transplantation for nonalcoholic fatty liver disease: New challenges and new opportunities. World J. Gastroenterol. 2014, 20, 5320. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, D.; Hamelin, J.; Bosselut, N.; Saffroy, R.; Sebagh, M.; Pommier, A.; Martel, C.; Lemoine, A. Mitochondrial roles and cytoprotection in chronic liver injury. Biochem. Res. Int. 2012, 2012, 387626. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of Mitochondria in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers 2018, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Mello, T.; Simeone, I.; Galli, A. Mito-Nuclear Communication in Hepatocellular Carcinoma Metabolic Rewiring. Cells 2019, 8, 417. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.E.; Kalavalapalli, S.; Williams, C.M.; Nautiyal, M.; Mathew, J.T.; Martinez, J.; Reinhard, M.K.; McDougall, D.J.; Rocca, J.R.; Yost, R.A.; et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol.-Endocrinol. Metab. 2016, 310, E484–E494. [Google Scholar] [CrossRef]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, L.; Grieco, A.; Armuzzi, A.; Candelli, M.; Forgione, A.; Gasbarrini, A.; Gasbarrini, G. Hepatic mitochondrial beta-oxidation in patients with nonalcoholic steatohepatitis assessed by 13C-octanoate breath test. Am. J. Gastroenterol. 2003, 98, 2335–2336. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Satapati, S.; Fu, X.; He, T.; Mehdibeigi, R.; Spring-Robinson, C.; Duarte, J.; Potthoff, M.J.; Browning, J.D.; Burgess, S.C. Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet. Am. J. Physiol.-Endocrinol. Metab. 2010, 298, E1226–E1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; d’Avignon, D.A.; Graham, M.J.; Dietzen, D.J.; Brunt, E.M.; Patti, G.J.; Crawford, P.A. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef] [Green Version]
- Männistö, V.T.; Simonen, M.; Hyysalo, J.; Soininen, P.; Kangas, A.J.; Kaminska, D.; Matte, A.K.; Venesmaa, S.; Käkelä, P.; Kärjä, V.; et al. Ketone body production is differentially altered in steatosis and non-alcoholic steatohepatitis in obese humans. Liver Int. 2015, 35, 1853–1861. [Google Scholar] [CrossRef]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Chavin, K.D.; Yang, S.; Lin, H.Z.; Chatham, J.; Chacko, V.P.; Hoek, J.B.; Walajtys-Rode, E.; Rashid, A.; Chen, C.H.; Huang, C.C.; et al. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J. Biol. Chem. 1999, 274, 5692–5700. [Google Scholar] [CrossRef]
- Nair, S.; Chacko, V.P.; Arnold, C.; Diehl, A.M. Hepatic ATP reserve and efficiency of replenishing: Comparison between obese and nonobese normal individuals. Am. J. Gastroenterol. 2003, 98, 466–470. [Google Scholar]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in Liver ATP Homeostasis in Human Nonalcoholic Steatohepatitis: A Pilot Study. JAMA 1999, 282, 1659–1664. [Google Scholar] [CrossRef]
- Schmid, A.I.; Szendroedi, J.; Chmelik, M.; Krššák, M.; Moser, E.; Roden, M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 2011, 34, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Chmelik, M.; Schmid, A.I.; Nowotny, P.; Brehm, A.; Krššák, M.; Moser, E.; Roden, M. Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology 2009, 50, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, M.; Koliaki, C.; Livingstone, R.; Phielix, E.; Bierwagen, A.; Meisinger, M.; Jelenik, T.; Strassburger, K.; Zimmermann, S.; Brockmann, K.; et al. Time course of postprandial hepatic phosphorus metabolites in lean, obese, and type 2 diabetes patients. Am. J. Clin. Nutr. 2015, 102, 1051–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, S.H.; Crespo, D.M.; Kang, H.S.; Al-Osaimi, A.M.S. Obesity and hepatocellular carcinoma. Gastroenterology 2004, 127, S97–S103. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef]
- Niederreiter, L.; Tilg, H. Cytokines and fatty liver diseases. Liver Res. 2018, 2, 14–20. [Google Scholar] [CrossRef]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef]
- Fromenty, B.; Robin, M.A.; Igoudjil, A.; Mansouri, A.; Pessayre, D. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 2004, 30, 121–138. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef]
- Leclercq, I.A.; Farrell, G.C.; Field, J.; Bell, D.R.; Gonzalez, F.J.; Robertson, G.R. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J. Clin. Investig. 2000, 105, 1067–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besse-Patin, A.; Léveillé, M.; Oropeza, D.; Nguyen, B.N.; Prat, A.; Estall, J.L. Estrogen Signals Through Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α to Reduce Oxidative Damage Associated With Diet-Induced Fatty Liver Disease. Gastroenterology 2017, 152, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Kucejova, B.; Duarte, J.A.G.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Livingston, K.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Zhang, J.; Oo, C.; Min, R.W.M.; Kaplowitz, N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 2018, 67, 2013–2024. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Min, R.W.M.; Aghajan, M.; Kaplowitz, N. JNK mediates mouse liver injury through a novel Sab (SH3BP5) dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Le, B.H.A.; García-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [Green Version]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L.J. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar]
- Garcia, D.; Hellberg, K.; Chaix, A.; Wallace, M.; Herzig, S.; Badur, M.G.; Lin, T.; Shokhirev, M.N.; Pinto, A.F.M.; Ross, D.S.; et al. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Rep. 2019, 26, 192–208.e6. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.-P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; Pierre, J.S.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Mello, T.; Materozzi, M.; Galli, A. PPARs and Mitochondrial Metabolism: From NAFLD to HCC. PPAR Res. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Casellas, A.; Amengual-Cladera, E.; Proenza, A.M.; Lladó, I.; Gianotti, M. Long-term high-fat-diet feeding impairs mitochondrial biogenesis in liver of male and female rats. Cell. Physiol. Biochem. 2010, 26, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kamat, A.; Pergola, P.; Swamy, A.; Tio, F.; Cusi, K. Metabolic factors in the development of hepatic steatosis and altered mitochondrial gene expression in vivo. Metabolism 2011, 60, 1090–1099. [Google Scholar] [CrossRef]
- Sczelecki, S.; Besse-Patin, A.; Abboud, A.; Kleiner, S.; Laznik-Bogoslavski, D.; Wrann, C.D.; Ruas, J.L.; Haibe-Kains, B.; Estall, J.L. Loss of Pgc-1α expression in aging mouse muscle potentiates glucose intolerance and systemic inflammation. Am. J. Physiol.-Endocrinol. Metab. 2013, 306, E157–E167. [Google Scholar] [CrossRef] [PubMed]
- Derbré, F.; Gomez-Cabrera, M.C.; Nascimento, A.L.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Tresguerres, J.A.F.; Fuentes, T.; Gratas-Delamarche, A.; Monsalve, M.; Viña, J. Age associated low mitochondrial biogenesis may be explained by lack of response of PGC-1α to exercise training. Age 2012, 34, 669–679. [Google Scholar] [CrossRef]
- Seo, A.Y.; Joseph, A.-M.; Dutta, D.; Hwang, J.C.Y.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: Mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial Aging and Age-Related Dysfunction of Mitochondria. Available online: https://www.hindawi.com/journals/bmri/2014/238463/ (accessed on 25 July 2019).
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef]
- Suzuki, A.; Abdelmalek, M.F. Nonalcoholic fatty liver disease in women. Womens Health 2009, 5, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.M.; Meers, G.M.E.; Booth, F.W.; Fritsche, K.L.; Hardin, C.D.; Thyfault, J.P.; Ibdah, J.A. PGC-1α overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 303, G979–G992. [Google Scholar] [CrossRef] [PubMed]
- Laye, M.J.; Rector, R.S.; Borengasser, S.J.; Naples, S.P.; Uptergrove, G.M.; Ibdah, J.A.; Booth, F.W.; Thyfault, J.P. Cessation of daily wheel running differentially alters fat oxidation capacity in liver, muscle, and adipose tissue. J. Appl. Physiol. 2009, 106, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, T.N.; Ringholm, S.; Leick, L.; Biensø, R.S.; Kiilerich, K.; Johansen, S.; Nielsen, M.M.; Wojtaszewski, J.F.; Hidalgo, J.; Pedersen, P.A.; et al. Role of PGC-1α in exercise and fasting-induced adaptations in mouse liver. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1501–R1509. [Google Scholar] [CrossRef]
- Lezi, E.; Lu, J.; Selfridge, J.E.; Burns, J.M.; Swerdlow, R.H. Lactate administration reproduces specific brain and liver exercise-related changes. J. Neurochem. 2013, 127, 91–100. [Google Scholar] [Green Version]
- McCoin, C.S.; Von Schulze, A.; Allen, J.; Fuller, K.N.; Xia, Q.; Koestler, D.C.; Houchen, C.J.; Maurer, A.; Dorn, G.W., II; Shankar, K.; et al. Sex modulates hepatic mitochondrial adaptations to high fat diet and physical activity. Am. J. Physiol.-Endocrinol. Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Buler, M.; Aatsinki, S.-M.; Skoumal, R.; Komka, Z.; Tóth, M.; Kerkelä, R.; Georgiadi, A.; Kersten, S.; Hakkola, J. Energy-sensing factors coactivator peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) and AMP-activated protein kinase control expression of inflammatory mediators in liver: Induction of interleukin 1 receptor antagonist. J. Biol. Chem. 2012, 287, 1847–1860. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Fuente-Martin, E.; Finan, B.; Kim, M.; Frank, A.; Garcia-Caceres, C.; Navas, C.R.; Gordillo, R.; Neinast, M.; Kalainayakan, S.P.; et al. Hypothalamic PGC-1α protects against high-fat diet exposure by regulating ERα. Cell Rep. 2014, 9, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Barroso, W.A.; Victorino, V.J.; Jeremias, I.C.; Petroni, R.C.; Ariga, S.K.K.; Salles, T.A.; Barbeiro, D.F.; de Lima, T.M.; de Souza, H.P. High-fat diet inhibits PGC-1α suppressive effect on NFκB signaling in hepatocytes. Eur. J. Nutr. 2018, 57, 1891–1900. [Google Scholar] [CrossRef]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol Hepatol. 2018, 16, 160–174. [Google Scholar] [CrossRef]
- Ricci, C.; Pastukh, V.; Leonard, J.; Turrens, J.; Wilson, G.; Schaffer, D.; Schaffer, S.W. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am. J. Physiol.-Cell Physiol. 2008, 294, C413–C422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. TEM 2012, 23, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-Sensitive Mitochondrial NAD+ Levels Dictate Cell Survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Liu, Y.; Fu, Y.; Liu, X.; Zhou, X. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418. [Google Scholar] [PubMed]
- He, J.; Hu, B.; Shi, X.; Weidert, E.R.; Lu, P.; Xu, M.; Huang, M.; Kelley, E.E.; Xie, W. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol. Cell. Biol. 2013, 33, 2047–2055. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Sirtuins and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 10084–10092. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, L.; Wang, P.; Li, X.; Qiu, D.; Li, L.; Zhang, J.; Hou, X.; Han, L.; Ge, J.; et al. Sirt3-dependent deacetylation of SOD2 plays a protective role against oxidative stress in oocytes from diabetic mice. Cell Cycle 2017, 16, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Bagul, P.K.; Katare, P.B.; Bugga, P.; Dinda, A.K.; Banerjee, S.K. SIRT-3 Modulation by Resveratrol Improves Mitochondrial Oxidative Phosphorylation in Diabetic Heart through Deacetylation of TFAM. Cells 2018, 7, 235. [Google Scholar] [CrossRef]
- Choudhury, M.; Jonscher, K.R.; Friedman, J.E. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging 2011, 3, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.-H. SIRT3 as a Regulator of Non-alcoholic Fatty Liver Disease. J. Lifestyle Med. 2014, 4, 80–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Marcos, P.J.; Jeninga, E.H.; Canto, C.; Harach, T.; de Boer, V.C.J.; Andreux, P.; Moullan, N.; Pirinen, E.; Yamamoto, H.; Houten, S.M.; et al. Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Sci. Rep. 2012, 2, 425. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Quan, X.-B.; Zeng, W.-J.; Yang, X.-O.; Wang, M.-J. Mechanism of Hepatocyte Apoptosis. J. Cell Death 2016, 9, 19–29. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Kroemer, G. Mitochondrial control of apoptosis: An overview. Biochem. Soc. Symp. 1999, 66, 1–15. [Google Scholar] [CrossRef]
- Gupta, S.; Kass, G.E.; Szegezdi, E.; Joseph, B. The mitochondrial death pathway: A promising therapeutic target in diseases. J. Cell. Mol. Med. 2009, 13, 1004–1033. [Google Scholar] [CrossRef]
- Yin, X.; Zheng, F.; Pan, Q.; Zhang, S.; Yu, D.; Xu, Z.; Li, H. Glucose fluctuation increased hepatocyte apoptosis under lipotoxicity and the involvement of mitochondrial permeability transition opening. J. Mol. Endocrinol. 2015, 55, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.; Johnson, C.D.; Hennebelle, M.; Holtmann, T.; Taha, A.Y.; Kirpich, I.A.; Eguchi, A.; Ramsden, C.E.; Papouchado, B.G.; McClain, C.J.; et al. Oxidized linoleic acid metabolites induce liver mitochondrial dysfunction, apoptosis, and NLRP3 activation in mice. J. Lipid Res. 2018, 59, 1597–1609. [Google Scholar] [CrossRef] [Green Version]
- Mishra, J.; Davani, A.J.; Stowe, D.F.; Kwok, W.-M.; Camara, A.K.S. Cyclosporin A: New Insights into its Potential Role in Mitochondrial Calcium Buffering. Biophys. J. 2018, 114, 659a. [Google Scholar] [CrossRef]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010, 24, 3052–3065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Sasai, M.; Linehan, M.M.; Iwasaki, A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science 2010, 329, 1530–1534. [Google Scholar] [CrossRef]
- Zhang, N.-P.; Liu, X.-J.; Xie, L.; Shen, X.-Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Investig. 2019, 199, 749–763. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Pihán, P.; Hetz, C. The Unfolded Protein Response: At the Intersection between Endoplasmic Reticulum Function and Mitochondrial Bioenergetics. Front. Oncol. 2017, 7, 1. [Google Scholar] [CrossRef]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.-A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef]
- Theurey, P.; Tubbs, E.; Vial, G.; Jacquemetton, J.; Bendridi, N.; Chauvin, M.-A.; Alam, M.R.; Le Romancer, M.; Vidal, H.; Rieusset, J. Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 2016, 8, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Brumbaugh, D.E.; Friedman, J.E. Developmental origins of nonalcoholic fatty liver disease. Pediatric Res. 2014, 75, 140–147. [Google Scholar] [CrossRef]
- Peng, K.-Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res. 2018, 59, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Rosqvist, F.; Kullberg, J.; Ståhlman, M.; Cedernaes, J.; Heurling, K.; Johansson, H.-E.; Iggman, D.; Wilking, H.; Larsson, A.; Eriksson, O.; et al. Overeating saturated fat promotes fatty liver and ceramides compared to polyunsaturated fat: A randomized trial. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New Aspects of Lipotoxicity in Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2034. [Google Scholar] [CrossRef]
- García-Ruiz, I.; Solís-Muñoz, P.; Fernández-Moreira, D.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. In vitro treatment of HepG2 cells with saturated fatty acids reproduces mitochondrial dysfunction found in nonalcoholic steatohepatitis. Dis. Model. Mech. 2015, 8, 183–191. [Google Scholar] [CrossRef]
- Vanhorebeek, I.; De Vos, R.; Mesotten, D.; Wouters, P.J.; De Wolf-Peeters, C.; Van den Berghe, G. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 2005, 365, 53–59. [Google Scholar] [CrossRef]
- Sun, L.; Xie, P.; Wada, J.; Kashihara, N.; Liu, F.; Zhao, Y.; Kumar, D.; Chugh, S.S.; Danesh, F.R.; Kanwar, Y.S. Rap1b GTPase Ameliorates Glucose-Induced Mitochondrial Dysfunction. J. Am. Soc. Nephrol. 2008, 19, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Dassanayaka, S.; Readnower, R.D.; Salabei, J.K.; Long, B.W.; Aird, A.L.; Zheng, Y.-T.; Muthusamy, S.; Facundo, H.T.; Hill, B.G.; Jones, S.P. High glucose induces mitochondrial dysfunction independently of protein O-GlcNAcylation. Biochem. J. 2015, 467, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tien, T.; Zhang, J.; Muto, T.; Kim, D.; Sarthy, V.P.; Roy, S. High Glucose Induces Mitochondrial Dysfunction in Retinal Müller Cells: Implications for Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2915–2921. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.-L.; Zhu, C.; Zhao, Y.-P.; Chen, X.-H.; Ji, C.-B.; Zhang, C.-M.; Zhu, J.-G.; Xia, Z.-K.; Tong, M.-L.; Guo, X.-R. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 2010, 320, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.W.; Golovoy, D.; Vincent, A.M.; Mahendru, P.; Olzmann, J.A.; Mentzer, A.; Feldman, E.L. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 2002, 16, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, K.; Molina, A.J.A.; Roy, S. High glucose induces mitochondrial morphology and metabolic changes in retinal pericytes. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8657–8664. [Google Scholar] [CrossRef] [PubMed]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.-J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef]
- Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Liverini, G.; Lanni, A.; Goglia, F.; Iossa, S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 2017, 9, 323. [Google Scholar] [CrossRef]
- García-Berumen, C.I.; Ortiz-Avila, O.; Vargas-Vargas, M.A.; del Rosario-Tamayo, B.A.; Guajardo-López, C.; Saavedra-Molina, A.; Rodríguez-Orozco, A.R.; Cortés-Rojo, C. The severity of rat liver injury by fructose and high fat depends on the degree of respiratory dysfunction and oxidative stress induced in mitochondria. Lipids Health Dis. 2019, 18, 78. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, Z.; Oron-Herman, M.; Grozovski, M.; Rosenthal, T.; Pappo, O.; Link, G.; Sela, B.-A. Fructose-induced fatty liver disease: Hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension 2005, 45, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Collison, K.S.; Maqbool, Z.M.; Inglis, A.L.; Makhoul, N.J.; Saleh, S.M.; Bakheet, R.H.; Al-Johi, M.A.; Al-Rabiah, R.K.; Zaidi, M.Z.; Al-Mohanna, F.A.; et al. Effect of dietary monosodium glutamate on HFCS-induced hepatic steatosis: Expression profiles in the liver and visceral fat. Obesity 2010, 18, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Schuschke, D.A.; Zhou, Z.; Chen, T.; Shi, X.; Zhang, J.; Zhang, X.; Pierce, W.M.; Johnson, W.T.; Vos, M.B.; et al. Modest Fructose Beverage Intake Causes Liver Injury and Fat Accumulation in Marginal Copper Deficient Rats. Obesity 2013, 21, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.-K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, A.; Neil, D.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Hepatic adverse effects of fructose consumption independent of overweight/obesity. Int. J. Mol. Sci. 2013, 14, 21873–21886. [Google Scholar] [CrossRef] [PubMed]
- Bremer, A.A.; Stanhope, K.L.; Graham, J.L.; Cummings, B.P.; Wang, W.; Saville, B.R.; Havel, P.J. Fructose-fed rhesus monkeys: A nonhuman primate model of insulin resistance, metabolic syndrome, and type 2 diabetes. Clin. Transl. Sci. 2011, 4, 243–252. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.Q.; Teoh, N.; Xu, L.; Pok, S.; Li, X.; Chu, E.S.H.; Chiu, J.; Dong, L.; Arfianti, E.; Haigh, W.G.; et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 2018, 9, 4490. [Google Scholar] [CrossRef]
- Domínguez-Pérez, M.; Simoni-Nieves, A.; Rosales, P.; Nuño-Lámbarri, N.; Rosas-Lemus, M.; Souza, V.; Miranda, R.U.; Bucio, L.; Uribe Carvajal, S.; Marquardt, J.U.; et al. Cholesterol burden in the liver induces mitochondrial dynamic changes and resistance to apoptosis. J. Cell. Physiol. 2019, 234, 7213–7223. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef] [Green Version]
- Bellanti, F.; Mitarotonda, D.; Tamborra, R.; Blonda, M.; Iannelli, G.; Petrella, A.; Sanginario, V.; Iuliano, L.; Vendemiale, G.; Serviddio, G. OP2–6—Oxysterols induce mitochondrial impairment and hepatocellular toxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2014, 75, S16–S17. [Google Scholar] [CrossRef]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernández, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Capitanio, N.; Romano, A.D.; Sastre, J.; Vendemiale, G.; Altomare, E. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 2008, 57, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, C.; Mari, M.; Colell, A.; Morales, A.; Caballero, F.; Montero, J.; Terrones, O.; Basañez, G.; Fernández-Checa, J.C. Mitochondrial cholesterol in health and disease. Histol. Histopathol. 2009, 24, 117–132. [Google Scholar] [PubMed]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enríquez-Cortina, C.; Bello-Monroy, O.; Rosales-Cruz, P.; Souza, V.; Miranda, R.U.; Toledo-Pérez, R.; Luna-López, A.; Simoni-Nieves, A.; Hernández-Pando, R.; Gutiérrez-Ruiz, M.C.; et al. Cholesterol overload in the liver aggravates oxidative stress-mediated DNA damage and accelerates hepatocarcinogenesis. Oncotarget 2017, 8, 104136–104148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X. NAFLD Related-HCC: The Relationship with Metabolic Disorders. In Obesity, Fatty Liver and Liver Cancer; Yu, J., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2018; pp. 55–62. ISBN 978-981-10-8684-7. [Google Scholar]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin. Transl. Med. 2016, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis- (NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xie, Q.; Zhou, X.; Yao, J.; Zhu, X.; Huang, P.; Zhang, L.; Wei, J.; Xie, H.; Zhou, L.; et al. Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 2015, 358, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Mezale, D.; Strumfa, I.; Vanags, A.; Mezals, M.; Fridrihsone, I.; Strumfs, B.; Balodis, D. Non-Alcoholic Steatohepatitis, Liver Cirrhosis and Hepatocellular Carcinoma: The Molecular Pathways. Liver Cirrhosis-Update Curr. Chall. 2017. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, H.D.; Bergmann, A. The Role of Apoptosis-Induced Proliferation for Regeneration and Cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008797. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R. Mitochondria and Cancer: A Growing Role in Apoptosis, Cancer Cell Metabolism and Dedifferentiation. In Advances in Mitochondrial Medicine; Scatena, R., Bottoni, P., Giardina, B., Eds.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2012; pp. 287–308. ISBN 978-94-007-2869-1. [Google Scholar]
- Yu, C.; Wang, X.; Huang, L.; Tong, Y.; Chen, L.; Wu, H.; Xia, Q.; Kong, X. Deciphering the Spectrum of Mitochondrial DNA Mutations in Hepatocellular Carcinoma Using High-Throughput Sequencing. Gene Expr. J. Liver Res. 2018, 18, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.-C.; Lee, H.-C.; Wei, Y.-H. Mitochondrial DNA alterations and mitochondrial dysfunction in the progression of hepatocellular carcinoma. World J. Gastroenterol. 2013, 19, 8880–8886. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Min, H.-K.; Mirshahi, F.; Verdianelli, A.; Pacana, T.; Patel, V.; Park, C.-G.; Choi, A.; Lee, J.-H.; Park, C.-B.; Ren, S.; et al. Activation of the GP130-STAT3 axis and its potential implications in nonalcoholic fatty liver disease. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 308, G794–G803. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Sun, R.; Sun, Q. Leptin signaling molecular actions and drug target in hepatocellular carcinoma. Drug Des. Dev. Ther. 2014, 8, 2295–2302. [Google Scholar]
- Fernández-Real, J.M.; López-Bermejo, A.; Ricart, W. Cross-talk between iron metabolism and diabetes. Diabetes 2002, 51, 2348–2354. [Google Scholar] [CrossRef]
- Uysal, S.; Armutcu, F.; Aydogan, T.; Akin, K.; Ikizek, M.; Yigitoglu, M.R. Some inflammatory cytokine levels, iron metabolism and oxidan stress markers in subjects with nonalcoholic steatohepatitis. Clin. Biochem. 2011, 44, 1375–1379. [Google Scholar] [CrossRef]
- Pietrangelo, A. Iron in NASH, chronic liver diseases and HCC: How much iron is too much? J. Hepatol. 2009, 50, 249–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, G.K.; Tennant, D.A.; McKeating, J.A. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: Current understanding and future directions. J. Hepatol. 2014, 61, 1397–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weljie, A.M.; Jirik, F.R. Hypoxia-induced metabolic shifts in cancer cells: Moving beyond the Warburg effect. Int. J. Biochem. Cell Biol. 2011, 43, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Foglia, B.; Sutti, S.; Morello, E.; Cannito, S.; Novo, E.; Bocca, C.; Maggiora, M.; Bruzzì, S.; Ramavath, N.N.; Rosso, C.; et al. NASH-related liver carcinogenesis is critically affected by hypoxia-inducible factor 2α. Dig. Liver Dis. 2019, 51, e8. [Google Scholar] [CrossRef]
- Ambade, A.; Satishchandran, A.; Saha, B.; Gyongyosi, B.; Lowe, P.; Kodys, K.; Catalano, D.; Szabo, G. Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1αinduced IL-10. Oncoimmunology 2016, 5, e1221557. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Smolková, K.; Plecitá-Hlavatá, L.; Bellance, N.; Benard, G.; Rossignol, R.; Ježek, P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol. 2011, 43, 950–968. [Google Scholar] [CrossRef]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011, 60, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Cabré, N.; Camps, J.; Joven, J. Inflammation, mitochondrial metabolism and nutrition: The multi-faceted progression of non-alcoholic fatty liver disease to hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2016, 5, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Dreval, K.; Kindrat, I.; Melnyk, S.; Jimenez, L.; de Conti, A.; Tryndyak, V.; Pogribna, M.; Ortega, J.F.; James, S.J.; et al. Epigenetically mediated inhibition of S-adenosylhomocysteine hydrolase and the associated dysregulation of 1-carbon metabolism in nonalcoholic steatohepatitis and hepatocellular carcinoma. FASEB J. 2018, 32, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Konno, M.; Koseki, J.; Nishida, N.; Kawamoto, K.; Yamada, D.; Asaoka, T.; Noda, T.; Wada, H.; Gotoh, K.; et al. The mitochondrial one-carbon metabolic pathway is associated with patient survival in pancreatic cancer. Oncol. Lett. 2018, 16, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Söderlund, S.; Ståhlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kilicarslan, M.; et al. Personal model-assisted identification of NAD+ and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [Green Version]
- Gaggini, M.; Carli, F.; Rosso, C.; Buzzigoli, E.; Marietti, M.; Latta, V.D.; Ciociaro, D.; Abate, M.L.; Gambino, R.; Cassader, M.; et al. Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance. Hepatology 2018, 67, 145–158. [Google Scholar] [CrossRef]
- Yamakado, M.; Tanaka, T.; Nagao, K.; Imaizumi, A.; Komatsu, M.; Daimon, T.; Miyano, H.; Tani, M.; Toda, A.; Yamamoto, H.; et al. Plasma amino acid profile associated with fatty liver disease and co-occurrence of metabolic risk factors. Sci. Rep. 2017, 7, 14485. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Cheng, J.; Fan, C.; Shi, X.; Cao, Y.; Sun, B.; Ding, H.; Hu, C.; Dong, F.; Yan, X. Serum Metabolomics to Identify the Liver Disease-Specific Biomarkers for the Progression of Hepatitis to Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 18175. [Google Scholar] [CrossRef]
- Stepien, M.; Duarte-Salles, T.; Fedirko, V.; Floegel, A.; Barupal, D.K.; Rinaldi, S.; Achaintre, D.; Assi, N.; Tjønneland, A.; Overvad, K.; et al. Alteration of amino acid and biogenic amine metabolism in hepatobiliary cancers: Findings from a prospective cohort study. Int. J. Cancer 2016, 138, 348–360. [Google Scholar] [CrossRef]
- Ji, L.; Tang, Y.; Pang, X.; Zhang, Y. Increased Expression of Serine Hydroxymethyltransferase 2 (SHMT2) is a Negative Prognostic Marker in Patients with Hepatocellular Carcinoma and is Associated with Proliferation of HepG2 Cells. Med. Sci. Monit. 2019, 25, 5823–5832. [Google Scholar] [CrossRef]
- Woo, C.C.; Chen, W.C.; Teo, X.Q.; Radda, G.K.; Lee, P.T.H. Downregulating serine hydroxymethyltransferase 2 (SHMT2) suppresses tumorigenesis in human hepatocellular carcinoma. Oncotarget 2016, 7, 53005–53017. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial one-carbon metabolism maintains redox balance during hypoxia. Cancer Discov. 2014, 4, 1371–1373. [Google Scholar] [CrossRef]
- Rubio-Aliaga, I.; Roos, B.D.; Sailer, M.; McLoughlin, G.A.; Boekschoten, M.; van Erk, M.J.; Bachmair, E.M.; van Schothorst, E.M.; Keijer, J.; Coort, S.L.M.; et al. Alterations in hepatic one-carbon metabolism and related pathways following a high-fat dietary intervention. Physiol. Genom. 2011, 43, 408–416. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Salomao, M.; Remotti, H.; Vaughan, R.; Siegel, A.B.; Lefkowitch, J.H.; Moreira, R.K. The steatohepatitic variant of hepatocellular carcinoma and its association with underlying steatohepatitis. Hum. Pathol. 2012, 43, 737–746. [Google Scholar] [CrossRef]
- Lin, M.; Lv, D.; Zheng, Y.; Wu, M.; Xu, C.; Zhang, Q.; Wu, L. Downregulation of CPT2 promotes tumorigenesis and chemoresistance to cisplatin in hepatocellular carcinoma. OncoTargets Ther. 2018, 11, 3101–3110. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef]
- Kudo, Y.; Tanaka, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Mohri, D.; Isomura, Y.; Seto, M.; Nakagawa, H.; Asaoka, Y.; et al. Altered composition of fatty acids exacerbates hepatotumorigenesis during activation of the phosphatidylinositol 3-kinase pathway. J. Hepatol. 2011, 55, 1400–1408. [Google Scholar] [CrossRef]
- Wang, B.; Hsu, S.-H.; Frankel, W.; Ghoshal, K.; Jacob, S.T. Stat3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha. Hepatology 2012, 56, 186–197. [Google Scholar] [CrossRef]
- Martínez-Jiménez, C.P.; Gómez-Lechón, M.J.; Castell, J.V.; Jover, R. Underexpressed coactivators PGC1alpha and SRC1 impair hepatocyte nuclear factor 4 alpha function and promote dedifferentiation in human hepatoma cells. J. Biol. Chem. 2006, 281, 29840–29849. [Google Scholar] [CrossRef]
- Lee, H.-J.; Su, Y.; Yin, P.-H.; Lee, H.-C.; Chi, C.-W. PPARγ/PGC-1α Pathway in E-Cadherin Expression and Motility of HepG2 Cells. Anticancer Res. 2009, 29, 5057–5063. [Google Scholar]
- Sen, N.; Satija, Y.K.; Das, S. PGC-1α, a Key Modulator of p53, Promotes Cell Survival upon Metabolic Stress. Mol. Cell 2011, 44, 621–634. [Google Scholar] [CrossRef]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L.; et al. PGC1α promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011, 71, 6888–6898. [Google Scholar] [CrossRef]
- Piccinin, E.; Peres, C.; Bellafante, E.; Ducheix, S.; Pinto, C.; Villani, G.; Moschetta, A. Hepatic peroxisome proliferator-activated receptor γ coactivator 1β drives mitochondrial and anabolic signatures that contribute to hepatocellular carcinoma progression in mice. Hepatology 2018, 67, 884–898. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Chambers, K.T.; Chen, Z.; Lai, L.; Leone, T.C.; Towle, H.C.; Kralli, A.; Crawford, P.A.; Finck, B.N. PGC-1β and ChREBP partner to cooperatively regulate hepatic lipogenesis in a glucose concentration-dependent manner. Mol. Metab. 2013, 2, 194–204. [Google Scholar] [CrossRef]
- Zhang, X.; Han, K.; Yuan, D.-H.; Meng, C.-Y. Overexpression of NAD(P)H: Quinone Oxidoreductase 1 Inhibits Hepatocellular Carcinoma Cell Proliferation and Induced Apoptosis by Activating AMPK/PGC-1α Pathway. DNA Cell Biol. 2017, 36, 256–263. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Li, J.; Zheng, L.; Feng, M.; Wang, X.; Han, K.; Pi, H.; Li, M.; Huang, X.; et al. SIRT1 facilitates hepatocellular carcinoma metastasis by promoting PGC-1α-mediated mitochondrial biogenesis. Oncotarget 2016, 7, 29255–29274. [Google Scholar]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a New Target of PGC-1α, Plays an Important Role in the Suppression of ROS and Mitochondrial Biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.-L.; Cheng, W.; Yin, X.-M.; Jiang, B. The expression of SIRT3 in primary hepatocellular carcinoma and the mechanism of its tumor suppressing effects. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 978–998. [Google Scholar]
- Zeng, X.; Wang, N.; Zhai, H.; Wang, R.; Wu, J.; Pu, W. SIRT3 functions as a tumor suppressor in hepatocellular carcinoma. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.Z.; Liu, L.; Cai, M.; Pan, Y.; Fu, J.; Cao, Y.; Yun, J. Low SIRT3 Expression Correlates with Poor Differentiation and Unfavorable Prognosis in Primary Hepatocellular Carcinoma. PLoS ONE 2012, 7, e51703. [Google Scholar] [CrossRef]
- Daher, S.; Massarwa, M.; Benson, A.A.; Khoury, T. Current and Future Treatment of Hepatocellular Carcinoma: An Updated Comprehensive Review. J. Clin. Transl. Hepatol. 2018, 6, 69–78. [Google Scholar] [CrossRef]
- Hannah, W.N.; Harrison, S.A. Lifestyle and Dietary Interventions in the Management of Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1365–1374. [Google Scholar] [CrossRef]
- Dethlefsen, M.M.; Kristensen, C.M.; Tøndering, A.S.; Lassen, S.B.; Ringholm, S.; Pilegaard, H. Impact of liver PGC-1α on exercise and exercise training-induced regulation of hepatic autophagy and mitophagy in mice on HFF. Physiol. Rep. 2018, 6, e13731. [Google Scholar] [CrossRef]
- Santos-Alves, E.; Marques-Aleixo, I.; Rizo-Roca, D.; Torrella, J.R.; Oliveira, P.J.; Magalhães, J.; Ascensão, A. Exercise modulates liver cellular and mitochondrial proteins related to quality control signaling. Life Sci. 2015, 135, 124–130. [Google Scholar] [CrossRef]
- Yun, Y.H.; Lim, M.K.; Won, Y.-J.; Park, S.M.; Chang, Y.J.; Oh, S.W.; Shin, S.A. Dietary preference, physical activity, and cancer risk in men: National health insurance corporation study. BMC Cancer 2008, 8, 366. [Google Scholar] [CrossRef]
- Tsai, J.-H.; Ferrell, L.D.; Tan, V.; Yeh, M.M.; Sarkar, M.; Gill, R.M. Aggressive non-alcoholic steatohepatitis following rapid weight loss and/or malnutrition. Mod. Pathol. 2017, 30, 834–842. [Google Scholar] [CrossRef] [Green Version]
- Besse-Patin, A.; Estall, J.L. An Intimate Relationship between ROS and Insulin Signalling: Implications for Antioxidant Treatment of Fatty Liver Disease. Int. J. Cell Biol. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Baumgardner, J.N.; Shankar, K.; Hennings, L.; Albano, E.; Badger, T.M.; Ronis, M.J.J. N-acetylcysteine attenuates progression of liver pathology in a rat model of nonalcoholic steatohepatitis. J. Nutr. 2008, 138, 1872–1879. [Google Scholar] [CrossRef]
- De Oliveira, C.P.M.S.; de Lima, V.M.R.; Simplicio, F.I.; Soriano, F.G.; de Mello, E.S.; de Souza, H.P.; Alves, V.A.F.; Laurindo, F.R.M.; Carrilho, F.J.; de Oliveira, M.G. Prevention and reversion of nonalcoholic steatohepatitis in OB/OB mice by S-nitroso-N-acetylcysteine treatment. J. Am. Coll. Nutr. 2008, 27, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Kretzmann, N.A.; Chiela, E.; Matte, U.; Marroni, N.; Marroni, C.A. N-acetylcysteine improves antitumoural response of Interferon alpha by NF-kB downregulation in liver cancer cells. Comp. Hepatol. 2012, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liu, X.; Yu, J.; Hua, F.; Hu, Z. Antioxidant N-acetylcysteine attenuates hepatocarcinogenesis by inhibiting ROS/ER stress in TLR2 deficient mouse. PLoS ONE 2013, 8, e74130. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Atkinson, J. Vitamin E, Antioxidant and Nothing More. Free Radic. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Draeger, C.L.; Naves, A.; Marques, N.; Baptistella, A.B.; Carnauba, R.A.; Paschoal, V.; Nicastro, H. Controversies of antioxidant vitamins supplementation in exercise: Ergogenic or ergolytic effects in humans? J. Int. Soc. Sports Nutr. 2014, 11, 4. [Google Scholar] [CrossRef]
- Erhardt, A.; Stahl, W.; Sies, H.; Lirussi, F.; Donner, A.; Häussinger, D. Plasma levels of vitamin E and carotenoids are decreased in patients with nonalcoholic steatohepatitis (NASH). Eur. J. Med. Res. 2011, 16, 76–78. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Hironaka, K.; Factor, V.M.; Thorgeirsson, S.S. Vitamin E down-modulates iNOS and NADPH oxidase in c-Myc/TGF-alpha transgenic mouse model of liver cancer. J. Hepatol. 2004, 41, 815–822. [Google Scholar] [CrossRef]
- Fantappiè, O.; Lodovici, M.; Fabrizio, P.; Marchetti, S.; Fabbroni, V.; Solazzo, M.; Lasagna, N.; Pantaleo, P.; Mazzanti, R. Vitamin E Protects DNA from Oxidative Damage in Human Hepatocellular Carcinoma Cell Lines. Free Radic. Res. 2004, 38, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shu, X.-O.; Li, H.; Yang, G.; Cai, H.; Ji, B.-T.; Gao, J.; Gao, Y.-T.; Zheng, W.; Xiang, Y.-B. Vitamin intake and liver cancer risk: A report from two cohort studies in China. J. Natl. Cancer Inst. 2012, 104, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Tajima, K.; Nakamura, A.; Shirakawa, J.; Togashi, Y.; Orime, K.; Sato, K.; Inoue, H.; Kaji, M.; Sakamoto, E.; Ito, Y.; et al. Metformin prevents liver tumorigenesis induced by high-fat diet in C57Bl/6 mice. Am. J. Physiol.-Endocrinol. Metab. 2013, 305, E987–E998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, S.; Houseright, R.A.; Graves, A.L.; Golenberg, N.; Korte, B.G.; Miskolci, V.; Huttenlocher, A. Metformin modulates innate immune-mediated inflammation and early progression of NAFLD-associated hepatocellular carcinoma in zebrafish. J. Hepatol. 2019, 70, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases: Hepatology, Vol. XX, No. X, 2017. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients with Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar] [CrossRef]
- Kalavalapalli, S.; Bril, F.; Koelmel, J.P.; Abdo, K.; Guingab, J.; Andrews, P.; Li, W.-Y.; Jose, D.; Yost, R.A.; Frye, R.F.; et al. Pioglitazone improves hepatic mitochondrial function in a mouse model of nonalcoholic steatohepatitis. Am. J. Physiol.-Endocrinol. Metab. 2018, 315, E163–E173. [Google Scholar] [CrossRef]
- Li, S.; Ghoshal, S.; Sojoodi, M.; Arora, G.; Masia, R.; Erstad, D.J.; Lanuti, M.; Hoshida, Y.; Baumert, T.F.; Tanabe, K.K.; et al. Pioglitazone Reduces Hepatocellular Carcinoma Development in Two Rodent Models of Cirrhosis. J. Gastrointest. Surg. 2019, 23, 101–111. [Google Scholar] [CrossRef]
- Sumie, S.; Kawaguchi, T.; Kawaguchi, A.; Kuromatsu, R.; Nakano, M.; Satani, M.; Yamada, S.; Okamura, S.; Yonezawa, Y.; Kakuma, T.; et al. Effect of pioglitazone on outcome following curative treatment for hepatocellular carcinoma in patients with hepatitis C virus infection: A prospective study. Mol. Clin. Oncol. 2015, 3, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Lin, J.-W.; Wu, L.-C.; Lai, M.-S.; Chuang, L.-M.; Chan, K.A. Association of thiazolidinediones with liver cancer and colorectal cancer in type 2 diabetes mellitus. Hepatology 2012, 55, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Desquiret-Dumas, V.; Gueguen, N.; Leman, G.; Baron, S.; Nivet-Antoine, V.; Chupin, S.; Chevrollier, A.; Vessières, E.; Ayer, A.; Ferré, M.; et al. Resveratrol Induces a Mitochondrial Complex I-dependent Increase in NADH Oxidation Responsible for Sirtuin Activation in Liver Cells. J. Biol. Chem. 2013, 288, 36662–36675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusdon, A.M.; Song, K.-X.; Qu, S. Nonalcoholic Fatty liver disease: Pathogenesis and therapeutics from a mitochondria-centric perspective. Oxid. Med. Cell. Longev. 2014, 2014, 637027. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef]
- De Oliveira, M.R.; Nabavi, S.F.; Manayi, A.; Daglia, M.; Hajheydari, Z.; Nabavi, S.M. Resveratrol and the mitochondria: From triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochim. Biophys. Acta BBA-Gen. Subj. 2016, 1860, 727–745. [Google Scholar] [CrossRef]
- Singh, C.K.; Ndiaye, M.A.; Ahmad, N. Resveratrol and cancer: Challenges for clinical translation. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2015, 1852, 1178–1185. [Google Scholar] [CrossRef]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 35. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The in Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Okabe, K.; Yaku, K.; Tobe, K.; Nakagawa, T. Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci. 2019, 26, 34. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.T.P.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Du, M.; Tan, X.; Yang, L.; Li, X.; Jiang, Y.; Wang, C.; Zhang, F.; Zhu, F.; Cheng, M.; et al. PARP1-mediated PPARα poly(ADP-ribosyl) action suppresses fatty acid oxidation in non-alcoholic fatty liver disease. J. Hepatol. 2017, 66, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Ryu, D.; Menzies, K.J.; Yi, H.-S.; Stein, S.; Zhang, H.; Perino, A.; Lemos, V.; Katsyuba, E.; Jha, P.; et al. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J. Hepatol. 2017, 66, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, P.; Horváth, B.; Rajesh, M.; Varga, Z.V.; Gariani, K.; Ryu, D.; Cao, Z.; Holovac, E.; Park, O.; Zhou, Z.; et al. PARP inhibition protects against alcoholic and non-alcoholic steatohepatitis. J. Hepatol. 2017, 66, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef]
- Pham, T.X.; Bae, M.; Kim, M.-B.; Lee, Y.; Hu, S.; Kang, H.; Park, Y.-K.; Lee, J.-Y. Nicotinamide riboside, an NAD+ precursor, attenuates the development of liver fibrosis in a diet-induced mouse model of liver fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2451–2463. [Google Scholar] [CrossRef]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD (+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Airhart, S.E.; Shireman, L.M.; Risler, L.J.; Anderson, G.D.; Nagana Gowda, G.A.; Raftery, D.; Tian, R.; Shen, D.D.; O’Brien, K.D. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 2017, 12, e0186459. [Google Scholar] [CrossRef]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Djouder, N. Boosting NAD+ for the prevention and treatment of liver cancer. Mol. Cell. Oncol. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Tummala, K.S.; Gomes, A.L.; Yilmaz, M.; Graña, O.; Bakiri, L.; Ruppen, I.; Ximénez-Embún, P.; Sheshappanavar, V.; Rodriguez-Justo, M.; Pisano, D.G.; et al. Inhibition of De Novo NAD + Synthesis by Oncogenic URI Causes Liver Tumorigenesis through DNA Damage. Cancer Cell 2014, 26, 826–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.W.S.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xiong, R.; Xiong, D.; Zhao, W.; Zhang, S.; Yin, T.; Zhang, X.; Jiang, G.; Yin, Z. The Adenosine Monophosphate (AMP) Analog, 5-Aminoimidazole-4-Carboxamide Ribonucleotide (AICAR) Inhibits Hepatosteatosis and Liver Tumorigenesis in a High-Fat Diet Murine Model Treated with Diethylnitrosamine (DEN). Med. Sci. Monit. 2018, 24, 8533–8543. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Huang, T.; Li, Y.; Guo, Y.; Zhu, Y.; Wang, Q.; Tan, X.; Chen, W.; Zhang, Y.; Cheng, W.; et al. AMP-Activated Protein Kinase Suppresses the in Vitro and in Vivo Proliferation of Hepatocellular Carcinoma. PLoS ONE 2014, 9, e93256. [Google Scholar] [CrossRef]
- Yang, C.-C.; Chang, S.-F.; Chao, J.-K.; Lai, Y.-L.; Chang, W.-E.; Hsu, W.-H.; Kuo, W.-H. Activation of AMP-activated protein kinase attenuates hepatocellular carcinoma cell adhesion stimulated by adipokine resistin. BMC Cancer 2014, 14, 112. [Google Scholar] [CrossRef]
- Pavlov, T.S.; Levchenko, V.; Ilatovskaya, D.V.; Li, H.; Palygin, O.; Pastor-Soler, N.M.; Hallows, K.R.; Staruschenko, A. Lack of Effects of Metformin and AICAR Chronic Infusion on the Development of Hypertension in Dahl Salt-Sensitive Rats. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Morishita, M.; Kawamoto, T.; Hara, H.; Onishi, Y.; Ueha, T.; Minoda, M.; Katayama, E.; Takemori, T.; Fukase, N.; Kurosaka, M.; et al. AICAR induces mitochondrial apoptosis in human osteosarcoma cells through an AMPK-dependent pathway. Int. J. Oncol. 2016, 50, 23–30. [Google Scholar] [CrossRef]
- Jose, C.; Hébert-Chatelain, E.; Bellance, N.; Larendra, A.; Su, M.; Nouette-Gaulain, K.; Rossignol, R. AICAR inhibits cancer cell growth and triggers cell-type distinct effects on OXPHOS biogenesis, oxidative stress and Akt activation. Biochim. Biophys. Acta 2011, 1807, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Suwa, M.; Egashira, T.; Nakano, H.; Sasaki, H.; Kumagai, S. Metformin increases the PGC-1alpha protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J. Appl. Physiol. 2006, 101, 1685–1692. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Khang, R.; Ham, S.; Jeong, G.R.; Kim, H.; Jo, M.; Lee, B.D.; Lee, Y.I.; Jo, A.; Park, C.; et al. Activation of the ATF2/CREB-PGC-1α pathway by metformin leads to dopaminergic neuroprotection. Oncotarget 2017, 8, 48603–48618. [Google Scholar] [PubMed]
- Higashida, K.; Kim, S.H.; Jung, S.R.; Asaka, M.; Holloszy, J.O.; Han, D.-H. Effects of Resveratrol and SIRT1 on PGC-1α Activity and Mitochondrial Biogenesis: A Reevaluation. PLoS Biol. 2013, 11, e1001603. [Google Scholar] [CrossRef] [PubMed]
- Aatsinki, S.-M.; Buler, M.; Salomäki, H.; Koulu, M.; Pavek, P.; Hakkola, J. Metformin induces PGC-1α expression and selectively affects hepatic PGC-1α functions. Br. J. Pharmacol. 2014, 171, 2351–2363. [Google Scholar] [CrossRef]
- Uchida, D.; Takaki, A.; Adachi, T.; Okada, H. Beneficial and Paradoxical Roles of Anti-Oxidative Nutritional Support for Non-Alcoholic Fatty Liver Disease. Nutrients 2018, 10, 977. [Google Scholar] [CrossRef]
Figure 1.
Roles for mitochondria in the progression of steatohepatitis to hepatocellular carcinoma. Metabolic stress induced by calorically high, nutritionally poor diets leads to metabolic disturbances in hepatic mitochondria. Mitochondrial adaptation and flexibility become compromised once (or possibly just before) the disease progresses to steatohepatitis (NASH). This results in incomplete β-oxidation, impaired ketogenesis, reduced mitochondria respiratory chain activity and ATP production, coupled with overactive TCA cycle potentially to meet the high energy demand. Sustained mitochondrial oxidative flux results in increased ROS production associated with mtDNA damage, ER stress, tissue inflammation and eventual cell death. Just like in NASH, oxidative stress, mitochondrial dysfunction, and oxidized mtDNA also contribute to the progression of HCC through influences on inflammation, cell death, and ER stress. As time passes, changes in lipid metabolism, one-carbon metabolism, and amino acid biosynthesis take place, fueling cancer cells for compensatory proliferation and further enhancing hypoxia, DNA damage, mutations, and escape from cell cycle checkpoints. NASH: nonalcoholic steatohepatitis, ER: endoplasmic reticulum, TCA: tricarboxylic acid cycle, HCC: hepatocellular carcinoma, mtDNA: mitochondrial DNA, ATP: adenosine triphosphate, ROS: reactive oxygen species, FFA: free fatty acids.
Figure 1.
Roles for mitochondria in the progression of steatohepatitis to hepatocellular carcinoma. Metabolic stress induced by calorically high, nutritionally poor diets leads to metabolic disturbances in hepatic mitochondria. Mitochondrial adaptation and flexibility become compromised once (or possibly just before) the disease progresses to steatohepatitis (NASH). This results in incomplete β-oxidation, impaired ketogenesis, reduced mitochondria respiratory chain activity and ATP production, coupled with overactive TCA cycle potentially to meet the high energy demand. Sustained mitochondrial oxidative flux results in increased ROS production associated with mtDNA damage, ER stress, tissue inflammation and eventual cell death. Just like in NASH, oxidative stress, mitochondrial dysfunction, and oxidized mtDNA also contribute to the progression of HCC through influences on inflammation, cell death, and ER stress. As time passes, changes in lipid metabolism, one-carbon metabolism, and amino acid biosynthesis take place, fueling cancer cells for compensatory proliferation and further enhancing hypoxia, DNA damage, mutations, and escape from cell cycle checkpoints. NASH: nonalcoholic steatohepatitis, ER: endoplasmic reticulum, TCA: tricarboxylic acid cycle, HCC: hepatocellular carcinoma, mtDNA: mitochondrial DNA, ATP: adenosine triphosphate, ROS: reactive oxygen species, FFA: free fatty acids.

Figure 2.
Mitochondrial pathways implicated in the pathogenesis of NASH and HCC. Mitochondria are responsible for converting nutrients such as lipids and glucose into energy (ATP). Mitochondrial activity leads to an increase in reactive oxygen species (ROS) production, which are detoxified by anti-oxidant defenses including SIRT3 and SOD2. Decreased mitochondrial efficiency (accumulation of ADP) and ROS activate AMPK and transcriptional machinery, including PGC-1α, to stimulate expression of gene pathways promoting mitochondrial adaptation. Damaged mitochondria are usually eliminated by autophagy/mitophagy which is attenuated by lipid accumulation. Prevention of autophagy further leads to accumulation of damaged mitochondria that can release inflammatory DAMPs and cytochrome C to promote cell death, and/or exacerbate endoplasmic reticulum (ER) stress. SIRT3: NAD+-dependent deacetylase sirtuin-3, SOD2: superoxide dismutase 2, ADP: adenosine diphosphate, AMPK: AMP-activated protein kinase, PPAR: peroxisome proliferator-activated receptor, NRF1/2: nuclear respiratory factor 1/2, ERR: estrogen related receptor, JNK: c-Jun N-terminal kinase, PGC-1: peroxisome proliferator-activated gamma coactivator-1, DAMPs: damage-associated mitochondrial molecular patterns.
Figure 2.
Mitochondrial pathways implicated in the pathogenesis of NASH and HCC. Mitochondria are responsible for converting nutrients such as lipids and glucose into energy (ATP). Mitochondrial activity leads to an increase in reactive oxygen species (ROS) production, which are detoxified by anti-oxidant defenses including SIRT3 and SOD2. Decreased mitochondrial efficiency (accumulation of ADP) and ROS activate AMPK and transcriptional machinery, including PGC-1α, to stimulate expression of gene pathways promoting mitochondrial adaptation. Damaged mitochondria are usually eliminated by autophagy/mitophagy which is attenuated by lipid accumulation. Prevention of autophagy further leads to accumulation of damaged mitochondria that can release inflammatory DAMPs and cytochrome C to promote cell death, and/or exacerbate endoplasmic reticulum (ER) stress. SIRT3: NAD+-dependent deacetylase sirtuin-3, SOD2: superoxide dismutase 2, ADP: adenosine diphosphate, AMPK: AMP-activated protein kinase, PPAR: peroxisome proliferator-activated receptor, NRF1/2: nuclear respiratory factor 1/2, ERR: estrogen related receptor, JNK: c-Jun N-terminal kinase, PGC-1: peroxisome proliferator-activated gamma coactivator-1, DAMPs: damage-associated mitochondrial molecular patterns.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Léveillé, M.; Estall, J.L. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites 2019, 9, 233. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo9100233
AMA Style
Léveillé M, Estall JL. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites. 2019; 9(10):233. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo9100233
Chicago/Turabian StyleLéveillé, Mélissa, and Jennifer L. Estall. 2019. "Mitochondrial Dysfunction in the Transition from NASH to HCC" Metabolites 9, no. 10: 233. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo9100233
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.