
Zwitterionic Polymer Coated and Aptamer Functionalized Flexible Micro-Electrode Arrays for In Vivo Cocaine Sensing and Electrophysiology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. MEA Fabrication for In Vitro and In Vivo Use
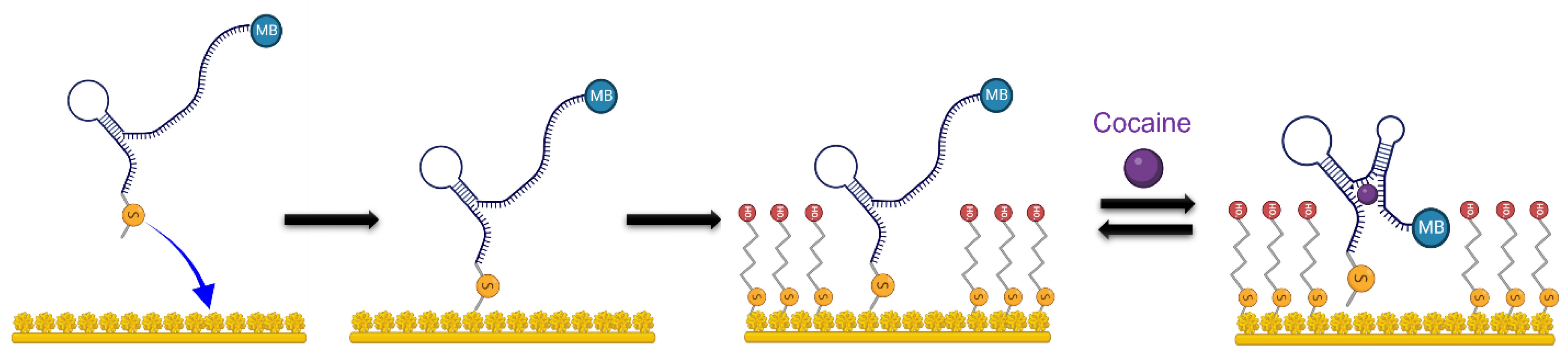
2.3. Aptamer Functionalization of Microelectrode Sites
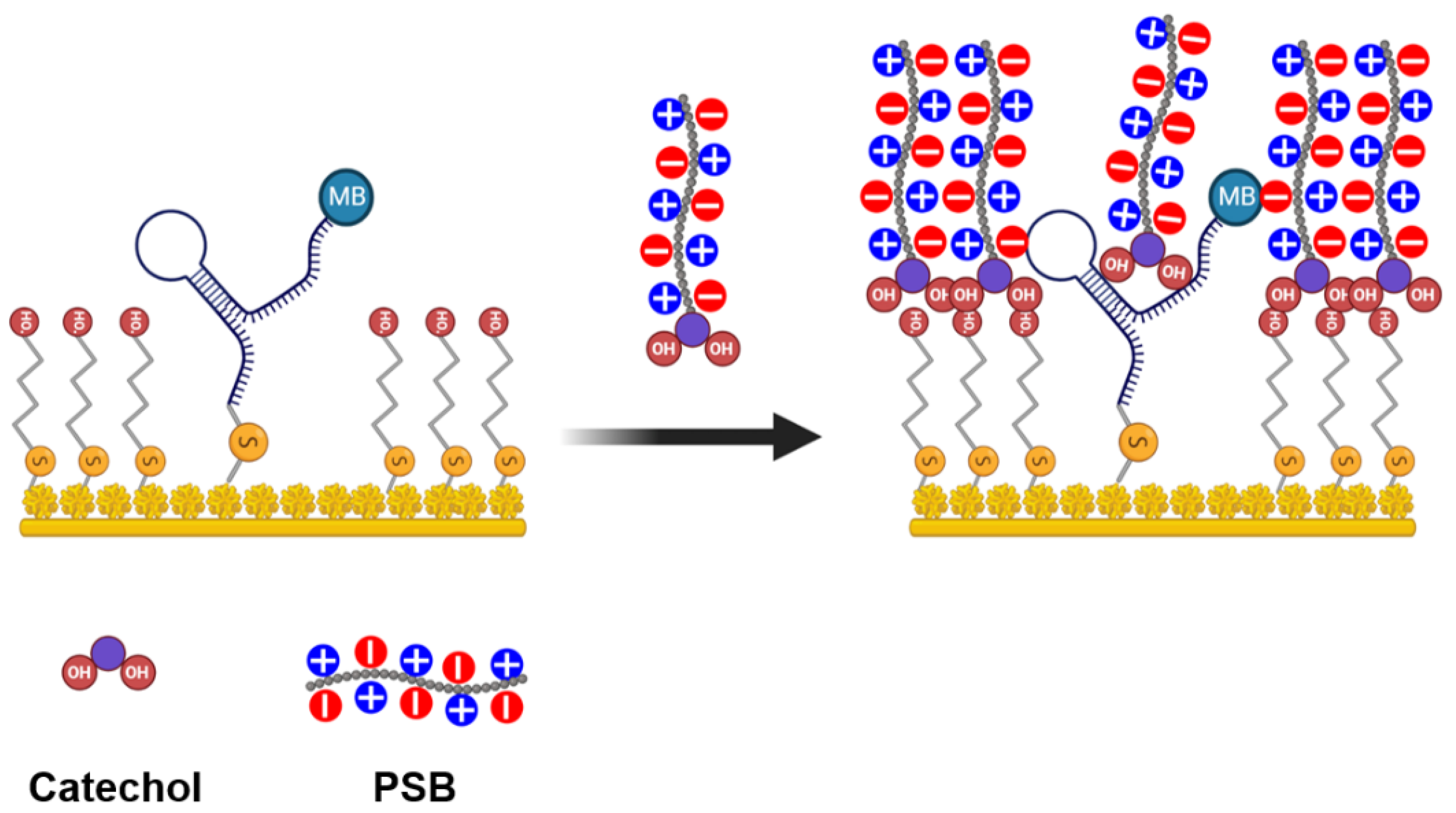
2.4. PSB Synthesis and Coating Procedure
2.5. Electrochemical Methods
2.6. In Vitro Experiment
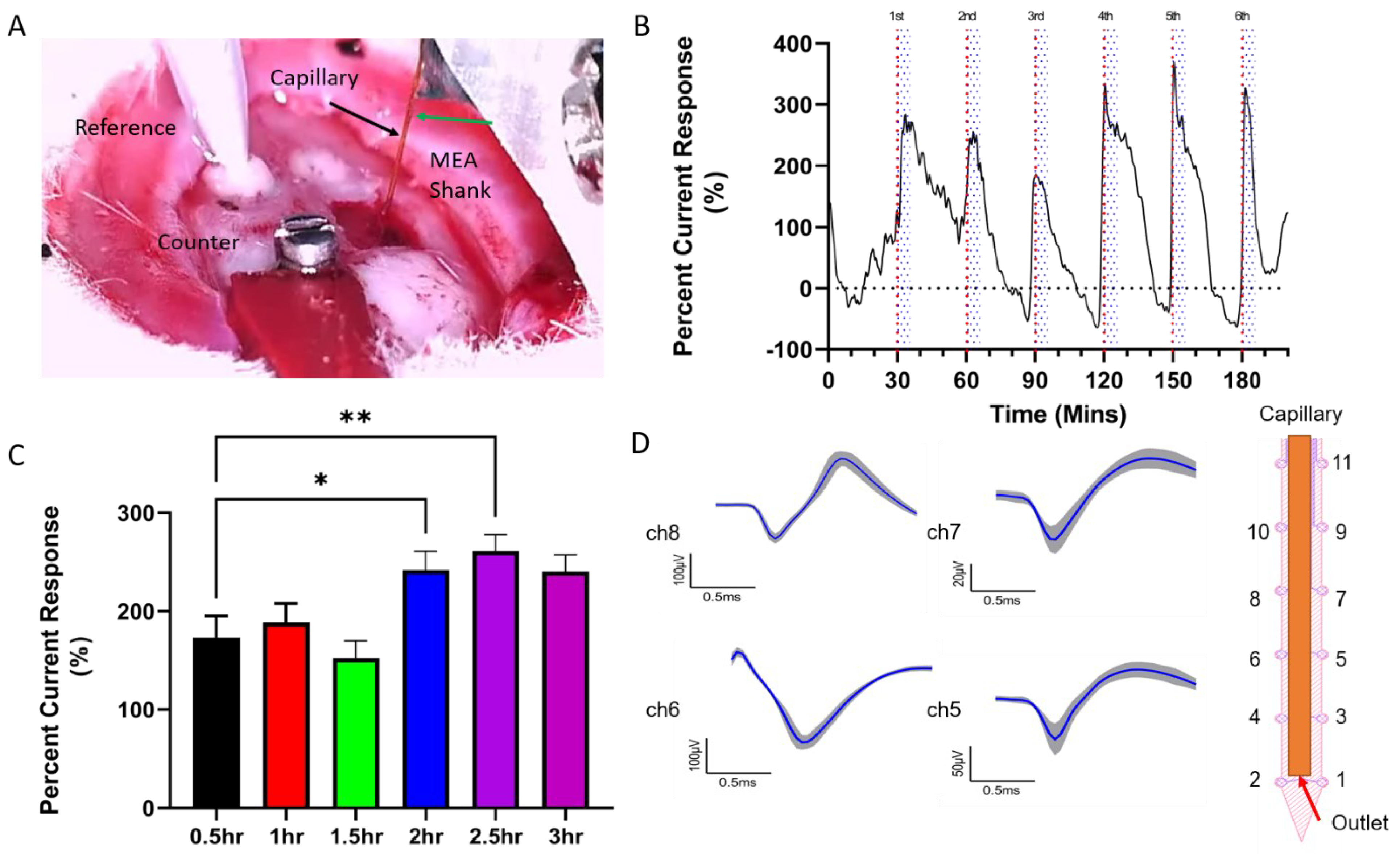
2.7. In Vivo Experiment
2.8. Data Analysis
3. Results
4. Discussion
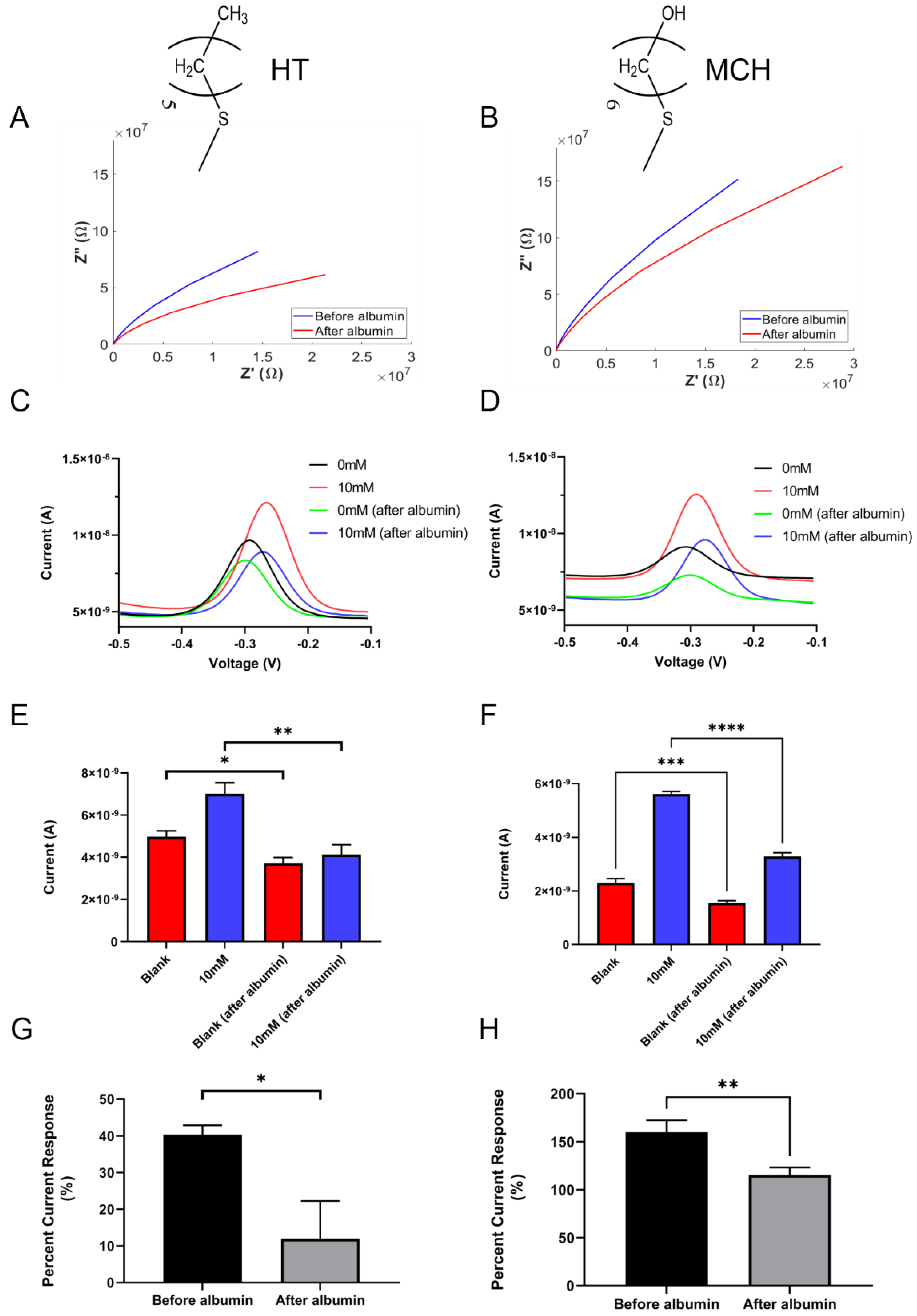
4.1. Effects of Non-Specific Protein Adsorption
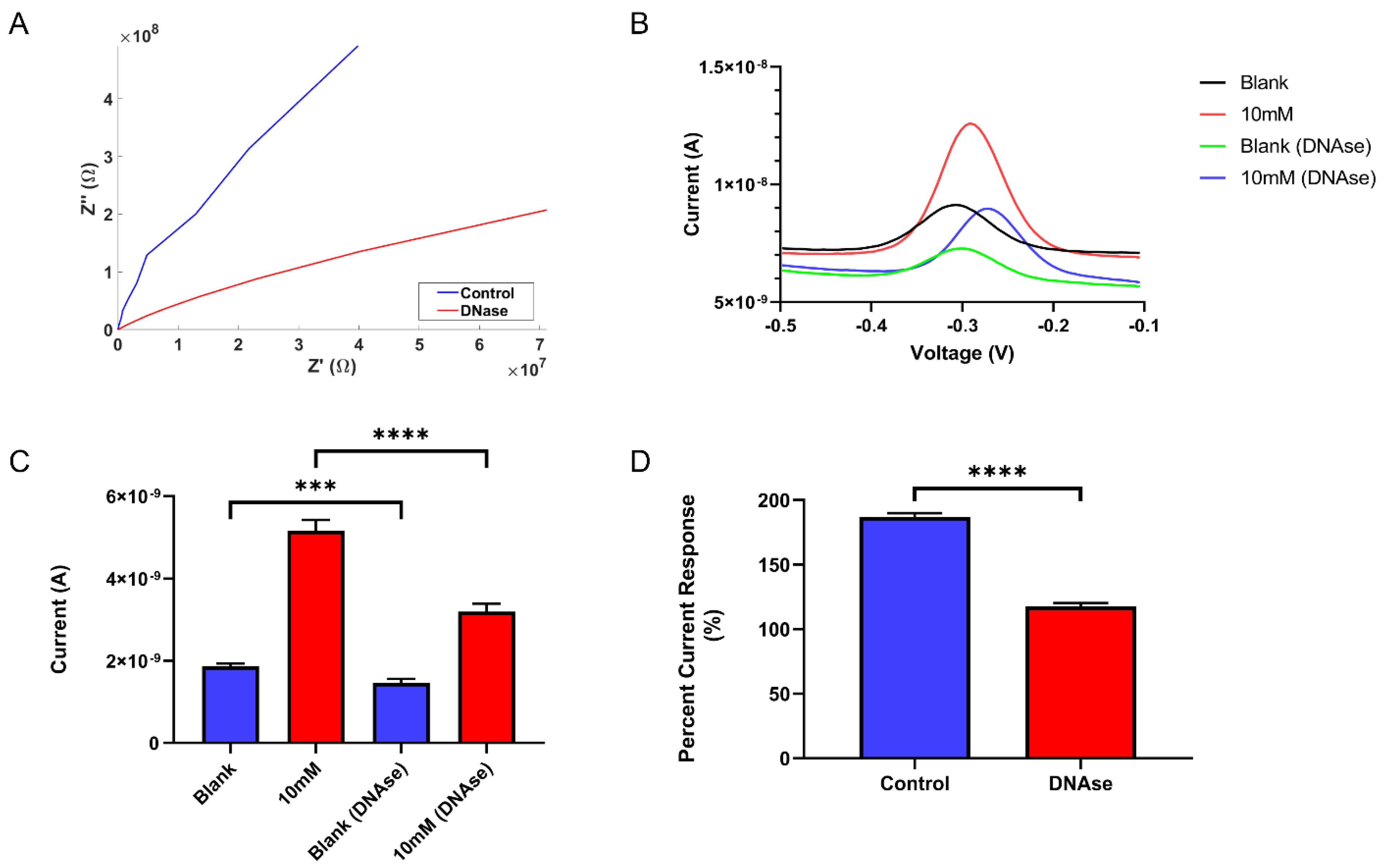
4.2. Effects of DNase-1 Enzyme Digestion
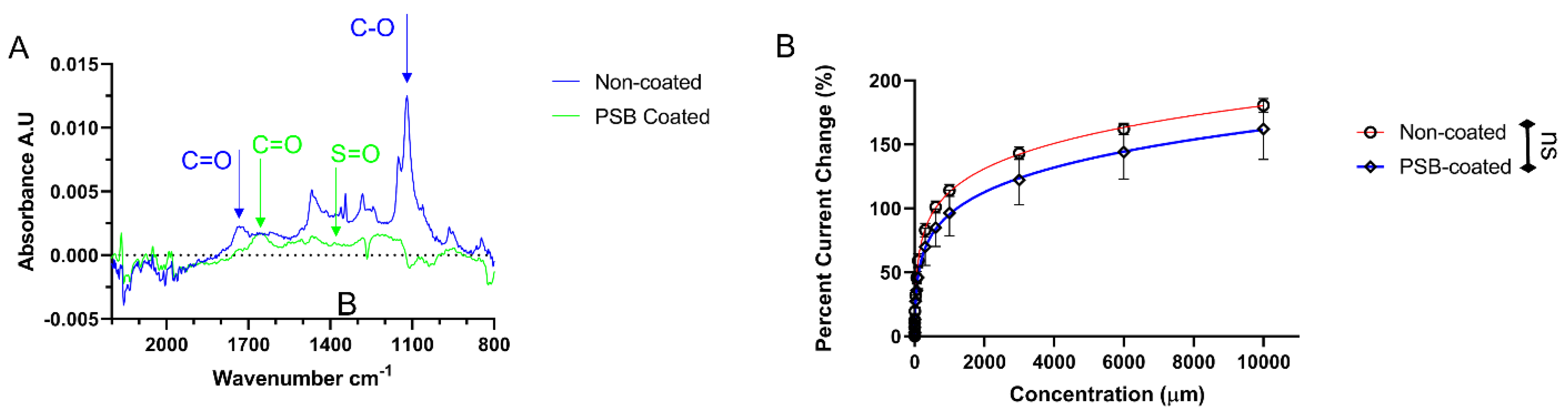
4.3. PSB Coating and Its Protective Effects In Vitro
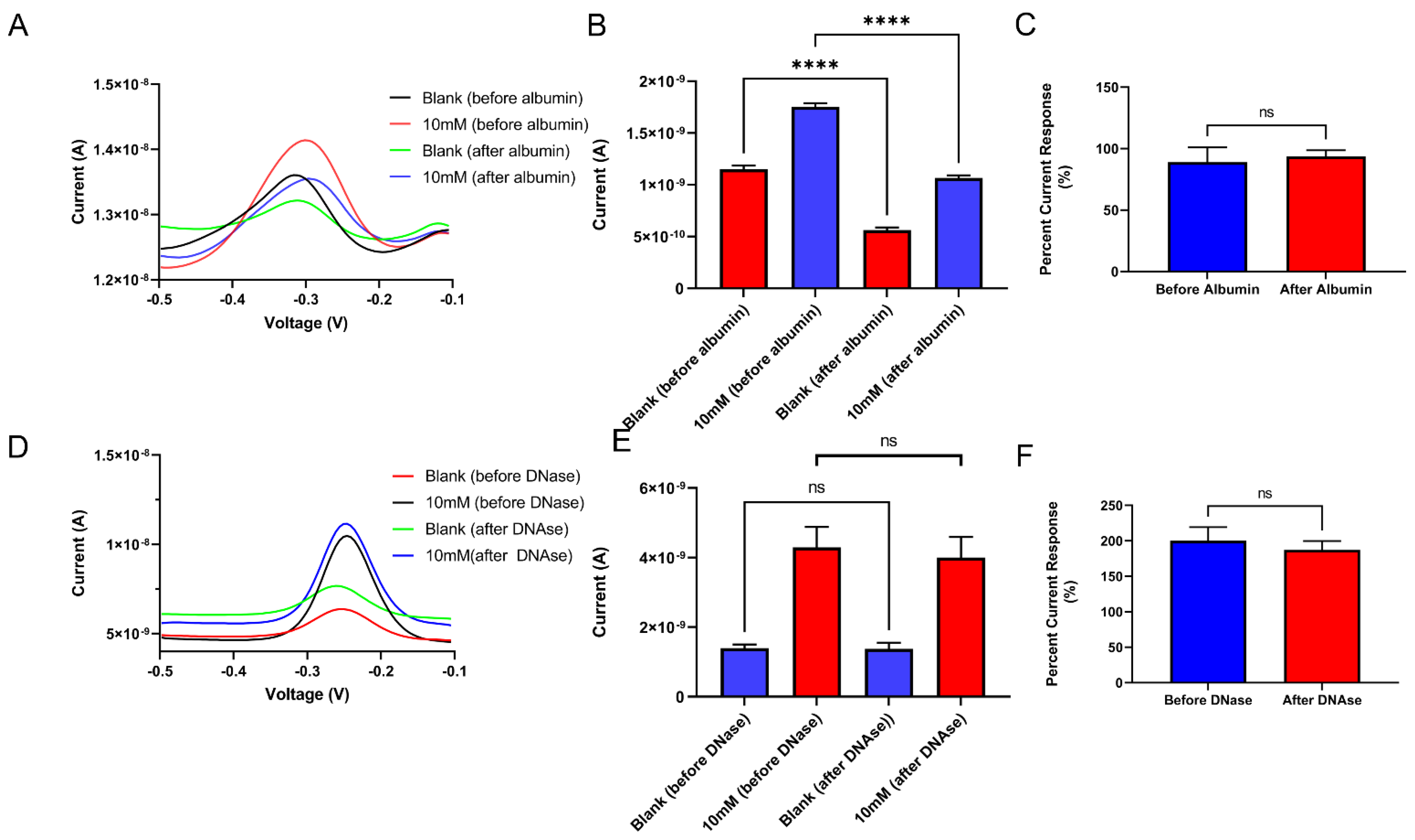
4.4. Stable In Vivo Sensing Performance and Electrophysiology Recording Capability with PSB Coating
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Key Substance Use and Mental Health Indicators in the United States: Results from the 2020 National Survey on Drug Use and Health Center for Behavioral Health Statistics and Quality; Substance Abuse and Mental Health Services Administration: Rockville, MD, USA, 2021.
- Graham, J.; Leonard, J.; Banerji, S.; Wang, G.S. Illicit Drug Exposures in Young Pediatric Patients Reported to the National Poison Data System, 2006–2016. J. Pediatr. 2020, 219, 254–258. [Google Scholar] [CrossRef]
- Nestler, E.J. The neurobiology of cocaine addiction. Sci. Pract. Perspect. 2005, 3, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Carboni, E.; Imperato, A.; Perezzani, L.; Di Chiara, G. Amphetamine, cocaine, phencyclidine and nomifensine increase extracellular dopamine concentrations preferentially in the nucleus accumbens of freely moving rats. Neuroscience 1989, 28, 653–661. [Google Scholar] [CrossRef]
- Hollander, J.A.; Carelli, R.M. Cocaine-associated stimuli increase cocaine seeking and activate accumbens core neurons after abstinence. J. Neurosci. 2007, 27, 3535–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.R.; Garris, P.A.; Wightman, R.M. Different effects of cocaine and nomifensine on dopamine uptake in the caudate-putamen and nucleus accumbens. J. Pharmacol. Exp. Ther. 1995, 274, 396–403. [Google Scholar] [PubMed]
- Hanania, T.; Gulley, J.M.; Salaz, D.O.; Larson, G.A.; Zahniser, N.R. Role of the dopamine transporter in the differential cocaine-induced locomotor activation of inbred long-sleep and short-sleep mice. Neuropsychopharmacology 2004, 29, 1814–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Reith, M.E.; Kuhar, M.J.; Carroll, F.I.; Garris, P.A. Preferential increases in nucleus accumbens dopamine after systemic cocaine administration are caused by unique characteristics of dopamine neurotransmission. J. Neurosci. 2001, 21, 6338–6347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bystrowska, B.; Adamczyk, P.; Moniczewski, A.; Zaniewska, M.; Fuxe, K.; Filip, M. LC/MS/MS evaluation of cocaine and its metabolites in different brain areas, peripheral organs and plasma in cocaine self-administering rats. Pharmacol. Rep. 2012, 64, 1337–1349. [Google Scholar] [CrossRef]
- Giros, B.; Jaber, M.; Jones, S.R.; Wightman, R.M.; Caron, M.G. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 1996, 379, 606–612. [Google Scholar] [CrossRef]
- Greco, P.G.; Garris, P.A. In vivo interaction of cocaine with the dopamine transporter as measured by voltammetry. Eur. J. Pharmacol. 2003, 479, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.M.; Zahniser, N.R. Rapid regulation of dopamine transporter function by substrates, blockers and presynaptic receptor ligands. Eur. J. Pharmacol. 2003, 479, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Rocha, B.A.; Fumagalli, F.; Gainetdinov, R.R.; Jones, S.R.; Ator, R.; Giros, B.; Miller, G.W.; Caron, M.G. Cocaine self-administration in dopamine-transporter knockout mice. Nat. Neurosci. 1998, 1, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.C.; Reith, M.E. Regulation of the dopamine transporter: Aspects relevant to psychostimulant drugs of abuse. Ann. N. Y. Acad. Sci. 2010, 1187, 316–340. [Google Scholar] [CrossRef] [PubMed]
- Maggos, C.E.; Spangler, R.; Zhou, Y.; Schlussman, S.D.; Ho, A.; Kreek, M.J. Quantitation of dopamine transporter mRNA in the rat brain: Mapping, effects of "binge" cocaine administration and withdrawal. Synapse 1997, 26, 55–61. [Google Scholar] [CrossRef]
- Wong, W.C.; Ford, K.A.; Pagels, N.E.; McCutcheon, J.E.; Marinelli, M. Adolescents are more vulnerable to cocaine addiction: Behavioral and electrophysiological evidence. J. Neurosci. 2013, 33, 4913–4922. [Google Scholar] [CrossRef] [Green Version]
- Schramm-Sapyta, N.L.; Walker, Q.D.; Caster, J.M.; Levin, E.D.; Kuhn, C.M. Are adolescents more vulnerable to drug addiction than adults? Evidence from animal models. Psychopharmacology 2009, 206, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Frantz, K.J.; O’Dell, L.E.; Parsons, L.H. Behavioral and neurochemical responses to cocaine in periadolescent and adult rats. Neuropsychopharmacology 2007, 32, 625–637. [Google Scholar] [CrossRef] [Green Version]
- Schramm-Sapyta, N.L.; Cauley, M.C.; Stangl, D.K.; Glowacz, S.; Stepp, K.A.; Levin, E.D.; Kuhn, C.M. Role of individual and developmental differences in voluntary cocaine intake in rats. Psychopharmacology 2011, 215, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Caster, J.M.; Walker, Q.D.; Kuhn, C.M. Enhanced behavioral response to repeated-dose cocaine in adolescent rats. Psychopharmacology 2005, 183, 218–225. [Google Scholar] [CrossRef]
- Bradberry, C.W.; Nobiletti, J.B.; Elsworth, J.D.; Murphy, B.; Jatlow, P.; Roth, R.H. Cocaine and cocaethylene: Microdialysis comparison of brain drug levels and effects on dopamine and serotonin. J. Neurochem. 1993, 60, 1429–1435. [Google Scholar] [CrossRef]
- Benuck, M.; Lajtha, A.; Reith, M.E. Pharmacokinetics of systemically administered cocaine and locomotor stimulation in mice. J. Pharmacol. Exp. Ther. 1987, 243, 144–149. [Google Scholar]
- Borland, L.M.; Shi, G.; Yang, H.; Michael, A.C. Voltammetric study of extracellular dopamine near microdialysis probes acutely implanted in the striatum of the anesthetized rat. J. Neurosci. Methods 2005, 146, 149–158. [Google Scholar] [CrossRef]
- Chefer, V.I.; Thompson, A.C.; Zapata, A.; Shippenberg, T.S. Overview of brain microdialysis. Curr. Protoc. Neurosci. 2009, 47, 7.1.1–7.1.28. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.S.; Michael, A.C. Invasive consequences of using micro-electrodes and microdialysis probes in the brain. TrAC Trends Anal. Chem. 2003, 22, 503–508. [Google Scholar] [CrossRef]
- Jaquins-Gerstl, A.; Michael, A.C. Comparison of the brain penetration injury associated with microdialysis and voltammetry. J. Neurosci. Methods 2009, 183, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Menacherry, S.; Hubert, W.; Justice, J.B., Jr. In vivo calibration of microdialysis probes for exogenous compounds. Anal. Chem. 1992, 64, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, G.S.; Konkol, R.J.; Terry, L.C.; Madden, J.A. Effect of cocaine metabolites on behavior: Possible neuroendocrine mechanisms. Brain Res. Bull. 1996, 39, 43–48. [Google Scholar] [CrossRef]
- Peretti, F.J.; Isenschmid, D.S.; Levine, B.; Caplan, Y.H.; Smialek, J.E. Cocaine fatality: An unexplained blood concentration in a fatal overdose. Forensic Sci. Int. 1990, 48, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Kozai, T.D.; Catt, K.; Du, Z.; Na, K.; Srivannavit, O.; Haque, R.U.; Seymour, J.; Wise, K.D.; Yoon, E.; Cui, X.T. Chronic In Vivo Evaluation of PEDOT/CNT for Stable Neural Recordings. IEEE Trans. Biomed. Eng. 2016, 63, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Boehler, C.; Kleber, C.; Martini, N.; Xie, Y.; Dryg, I.; Stieglitz, T.; Hofmann, U.G.; Asplund, M. Actively controlled release of Dexamethasone from neural microelectrodes in a chronic in vivo study. Biomaterials 2017, 129, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.J.; Luo, X.; Weaver, C.; Cui, X.T. Poly (3, 4-ethylenedioxythiophene)-ionic liquid coating improves neural recording and stimulation functionality of MEAs. J. Mater. Chem. C Mater. Opt. Electron. Devices 2015, 3, 6515–6524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozai, T.D.; Du, Z.; Gugel, Z.V.; Smith, M.A.; Chase, S.M.; Bodily, L.M.; Caparosa, E.M.; Friedlander, R.M.; Cui, X.T. Comprehensive chronic laminar single-unit, multi-unit, and local field potential recording performance with planar single shank electrode arrays. J. Neurosci. Methods. 2015, 242, 15–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castagnola, E.; Robbins, E.M.; Wu, B.; Pwint, M.Y.; Garg, R.; Cohen-Karni, T.; Cui, X.T. Flexible Glassy Carbon Multielectrode Array for In Vivo Multisite Detection of Tonic and Phasic Dopamine Concentrations. Biosensors 2022, 12, 540. [Google Scholar] [CrossRef]
- Taylor, I.M.; Du, Z.; Bigelow, E.T.; Eles, J.R.; Horner, A.R.; Catt, K.A.; Weber, S.G.; Jamieson, B.G.; Cui, X.T. Aptamer-functionalized neural recording electrodes for the direct measurement of cocaine in vivo. J. Mater. Chem. B 2017, 5, 2445–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, I.M.; Patel, N.A.; Freedman, N.C.; Castagnola, E.; Cui, X.T. Direct in Vivo Electrochemical Detection of Resting Dopamine Using Poly(3,4-ethylenedioxythiophene)/Carbon Nanotube Functionalized Microelectrodes. Anal. Chem. 2019, 91, 12917–12927. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.R.; Lai, R.Y.; Wood, M.S.; Doctor, E.H.; Heeger, A.J.; Plaxco, K.W. An electronic, aptamer-based small-molecule sensor for the rapid, label-free detection of cocaine in adulterated samples and biological fluids. J. Am. Chem. Soc. 2006, 128, 3138–3139. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.S.; Gifford, R. Biosensors for real-time in vivo measurements. Biosens. Bioelectron. 2005, 20, 2388–2403. [Google Scholar] [CrossRef]
- Zhao, C.; Cheung, K.M.; Huang, I.W.; Yang, H.; Nakatsuka, N.; Liu, W.; Cao, Y.; Man, T.; Weiss, P.S.; Monbouquette, H.G.; et al. Implantable aptamer-field-effect transistor neuroprobes for in vivo neurotransmitter monitoring. Sci. Adv. 2021, 7, eabj7422. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, C.; Wang, Z.; Yang, K.A.; Cheng, X.; Liu, W.; Yu, W.; Lin, S.; Zhao, Y.; Cheung, K.M.; et al. Wearable aptamer-field-effect transistor sensing system for noninvasive cortisol monitoring. Sci. Adv. 2022, 8, eabk0967. [Google Scholar] [CrossRef]
- Kang, D.; White, R.J.; Xia, F.; Zuo, X.; Vallée-Bélisle, A.; Plaxco, K.W. DNA biomolecular-electronic encoder and decoder devices constructed by multiplex biosensors. NPG Asia Mater. 2012, 4, e1. [Google Scholar] [CrossRef]
- Zhen, J.; Liang, G.; Chen, R.; Jia, W. Label-free hairpin-like aptamer and EIS-based practical, biostable sensor for acetamiprid detection. PLoS ONE 2020, 15, e0244297. [Google Scholar] [CrossRef]
- White, R.J.; Phares, N.; Lubin, A.A.; Xiao, Y.; Plaxco, K.W. Optimization of electrochemical aptamer-based sensors via optimization of probe packing density and surface chemistry. Langmuir 2008, 24, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Swensen, J.S.; Xiao, Y.; Ferguson, B.S.; Lubin, A.A.; Lai, R.Y.; Heeger, A.J.; Plaxco, K.W.; Soh, H.T. Continuous, real-time monitoring of cocaine in undiluted blood serum via a microfluidic, electrochemical aptamer-based sensor. J. Am. Chem. Soc. 2009, 131, 4262–4266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, B.S.; Hoggarth, D.A.; Maliniak, D.; Ploense, K.; White, R.J.; Woodward, N.; Hsieh, K.; Bonham, A.J.; Eisenstein, M.; Kippin, T.E.; et al. Real-time, aptamer-based tracking of circulating therapeutic agents in living animals. Sci. Transl. Med. 2013, 5, 213ra165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoukroun-Barnes, L.R.; Wagan, S.; White, R.J. Enhancing the analytical performance of electrochemical RNA aptamer-based sensors for sensitive detection of aminoglycoside antibiotics. Anal. Chem. 2014, 86, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wagan, S.; Davila Morris, M.; Taylor, J.; White, R.J. Achieving reproducible performance of electrochemical, folding aptamer-based sensors on microelectrodes: Challenges and prospects. Anal. Chem. 2014, 86, 11417–11424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azadbakht, A.; Roushani, M.; Abbasi, A.R.; Derikvand, Z. Design and characterization of electrochemical dopamine-aptamer as convenient and integrated sensing platform. Anal. Biochem. 2016, 507, 47–57. [Google Scholar] [CrossRef]
- Mannironi, C.; Di Nardo, A.; Fruscoloni, P.; Tocchini-Valentini, G.P. In vitro selection of dopamine RNA ligands. Biochemistry 1997, 36, 9726–9734. [Google Scholar] [CrossRef]
- Walsh, R.; DeRosa, M.C. Retention of function in the DNA homolog of the RNA dopamine aptamer. Biochem. Biophys. Res. Commun. 2009, 388, 732–735. [Google Scholar] [CrossRef]
- Xiao, Y.; Lubin, A.A.; Heeger, A.J.; Plaxco, K.W. Label-Free Electronic Detection of Thrombin in Blood Serum by Using an Aptamer-Based Sensor. Angew. Chem. 2005, 117, 5592–5595. [Google Scholar] [CrossRef]
- Xiao, Y.; Piorek, B.D.; Plaxco, K.W.; Heeger, A.J. A reagentless signal-on architecture for electronic, aptamer-based sensors via target-induced strand displacement. J. Am. Chem. Soc. 2005, 127, 17990–17991. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.Y.; Plaxco, K.W.; Heeger, A.J. Aptamer-based electrochemical detection of picomolar platelet-derived growth factor directly in blood serum. Anal. Chem. 2007, 79, 229–233. [Google Scholar] [CrossRef]
- Macazo, F.C.; Karpel, R.L.; White, R.J. Monitoring cooperative binding using electrochemical DNA-based sensors. Langmuir 2015, 31, 868–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Cui, X.T. Reagentless aptamer based impedance biosensor for monitoring a neuro-inflammatory cytokine PDGF. Biosens. Bioelectron. 2007, 23, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Randall, B.A.; Alba, N.A.; Cui, X.T. Conducting polymer-based impedimetric aptamer biosensor for in situ detection. Anal. Bioanal. Chem. 2008, 392, 861–864. [Google Scholar] [CrossRef]
- Luo, X.; Lee, I.; Huang, J.; Yun, M.; Cui, X.T. Ultrasensitive protein detection using an aptamer-functionalized single polyaniline nanowire. Chem. Commun. 2011, 47, 6368–6370. [Google Scholar] [CrossRef]
- Kozai, T.D.; Jaquins-Gerstl, A.S.; Vazquez, A.L.; Michael, A.C.; Cui, X.T. Brain tissue responses to neural implants impact signal sensitivity and intervention strategies. ACS Chem. Neurosci. 2015, 6, 48–67. [Google Scholar] [CrossRef] [Green Version]
- Polikov, V.S.; Tresco, P.A.; Reichert, W.M. Response of brain tissue to chronically implanted neural electrodes. J. Neurosci. Methods 2005, 148, 1–18. [Google Scholar] [CrossRef]
- Salatino, J.W.; Ludwig, K.A.; Kozai, T.D.Y.; Purcell, E.K. Glial responses to implanted electrodes in the brain. Nat. Biomed. Eng. 2017, 1, 862–877. [Google Scholar] [CrossRef]
- McConnell, G.C.; Rees, H.D.; Levey, A.I.; Gutekunst, C.A.; Gross, R.E.; Bellamkonda, R.V. Implanted neural electrodes cause chronic, local inflammation that is correlated with local neurodegeneration. J. Neural Eng. 2009, 6, 056003. [Google Scholar] [CrossRef]
- Biran, R.; Martin, D.C.; Tresco, P.A. Neuronal cell loss accompanies the brain tissue response to chronically implanted silicon microelectrode arrays. Exp. Neurol 2005, 195, 115–126. [Google Scholar] [CrossRef]
- Weltman, A.; Yoo, J.; Meng, E. Flexible, Penetrating Brain Probes Enabled by Advances in Polymer Microfabrication. Micromachines 2016, 7, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Luan, L.; Zhao, Z.; Li, X.; Zhu, H.; Potnis, O.; Xie, C. Nanofabricated Ultraflexible Electrode Arrays for High-Density Intracortical Recording. Adv. Sci. 2018, 5, 1700625. [Google Scholar] [CrossRef]
- Schuhmann, T.G., Jr.; Zhou, T.; Hong, G.; Lee, J.M.; Fu, T.M.; Park, H.G.; Lieber, C.M. Syringe-injectable Mesh Electronics for Stable Chronic Rodent Electrophysiology. J. Vis. Exp. 2018, 137, e58003. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Terayama, Y.; Yamaguchi, H.; Terada, M.; Murakami, D.; Ishihara, K.; Takahara, A. Wettability and antifouling behavior on the surfaces of superhydrophilic polymer brushes. Langmuir 2012, 28, 7212–7222. [Google Scholar] [CrossRef] [PubMed]
- Ladd, J.; Zhang, Z.; Chen, S.; Hower, J.C.; Jiang, S. Zwitterionic polymers exhibiting high resistance to nonspecific protein adsorption from human serum and plasma. Biomacromolecules 2008, 9, 1357–1361. [Google Scholar] [CrossRef]
- Li, G.; Cheng, G.; Xue, H.; Chen, S.; Zhang, F.; Jiang, S. Ultra low fouling zwitterionic polymers with a biomimetic adhesive group. Biomaterials 2008, 29, 4592–4597. [Google Scholar] [CrossRef]
- Li, G.; Xue, H.; Cheng, G.; Chen, S.; Zhang, F.; Jiang, S. Ultralow fouling zwitterionic polymers grafted from surfaces covered with an initiator via an adhesive mussel mimetic linkage. J. Phys. Chem. B 2008, 112, 15269–15274. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, Z.; Bai, T.; Carr, L.; Ella-Menye, J.R.; Irvin, C.; Ratner, B.D.; Jiang, S. Zwitterionic hydrogels implanted in mice resist the foreign-body reaction. Nat. Biotechnol. 2013, 31, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Golabchi, A.; Wu, B.; Cao, B.; Bettinger, C.J.; Cui, X.T. Zwitterionic polymer/polydopamine coating reduce acute inflammatory tissue responses to neural implants. Biomaterials 2019, 225, 119519. [Google Scholar] [CrossRef]
- Yang, Q.; Wu, B.; Eles, J.R.; Vazquez, A.L.; Kozai, T.D.Y.; Cui, X.T. Zwitterionic Polymer Coating Suppresses Microglial Encapsulation to Neural Implants In Vitro and In Vivo. Adv. Biosyst. 2020, 4, e1900287. [Google Scholar] [CrossRef]
- Li, S.C.; Chu, L.N.; Gong, X.Q.; Diebold, U. Hydrogen bonding controls the dynamics of catechol adsorbed on a TiO2(110) surface. Science 2010, 328, 882–884. [Google Scholar] [CrossRef]
- Koizumi, T. Tissue distribution of deoxyribonuclease I (DNase I) activity level in mice and its sexual dimorphism. Exp. Anim. 1995, 44, 181–185. [Google Scholar] [CrossRef] [Green Version]
- Margarson, M.P.; Soni, N. Serum albumin: Touchstone or totem? Anaesthesia 1998, 53, 789–803. [Google Scholar] [CrossRef]
- Shukla, G.S.; Chandra, S.V.; Seth, P.K. Effect of manganese on the levels of DNA, RNA, DNase and RNase in cerebrum, cerebellum and rest of brain regions of rat. Acta Pharmacol. Toxicol. 1976, 39, 562–569. [Google Scholar] [CrossRef]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar] [CrossRef]
- Yang, W.; Sundaram, H.S.; Ella, J.R.; He, N.; Jiang, S. Low-fouling electrospun PLLA films modified with zwitterionic poly(sulfobetaine methacrylate)-catechol conjugates. Acta Biomater. 2016, 40, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Xu, Z.; Xu, H.; Gu, Y.; Xing, Y.; Yan, X.; Liu, H.; Lu, N.; Song, Y.; Zhang, S.; et al. Catechol and zwitterion-bifunctionalized poly(ethylene glycol) based ultrasensitive antifouling electrochemical aptasensor for the quantification of adenosine triphosphate in biological media. Sens. Actuators B Chem. 2019, 288, 469–475. [Google Scholar] [CrossRef]
- Inaba, T.; Stewart, D.J.; Kalow, W. Metabolism of cocaine in man. Clin. Pharmacol. Ther. 1978, 23, 547–552. [Google Scholar] [CrossRef]
- Booze, R.M.; Lehner, A.F.; Wallace, D.R.; Welch, M.A.; Mactutus, C.F. Dose-response cocaine pharmacokinetics and metabolite profile following intravenous administration and arterial sampling in unanesthetized, freely moving male rats. Neurotoxicol. Teratol. 1997, 19, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Hetke, J.F.; Wiler, J.A.; Anderson, D.J.; Martin, D.C. Electrochemical deposition and characterization of conducting polymer polypyrrole/PSS on multichannel neural probes. Sens. Actuators A Phys. 2001, 93, 8–18. [Google Scholar] [CrossRef]
- Neto, J.P.; Baiao, P.; Lopes, G.; Frazao, J.; Nogueira, J.; Fortunato, E.; Barquinha, P.; Kampff, A.R. Does Impedance Matter When Recording Spikes With Polytrodes? Front. Neurosci. 2018, 12, 715. [Google Scholar] [CrossRef] [PubMed]
- Sachan, A.; Ilgu, M.; Kempema, A.; Kraus, G.A.; Nilsen-Hamilton, M. Specificity and Ligand Affinities of the Cocaine Aptamer: Impact of Structural Features and Physiological NaCl. Anal. Chem. 2016, 88, 7715–7723. [Google Scholar] [CrossRef] [Green Version]
- Shoara, A.A.; Reinstein, O.; Borhani, O.A.; Martin, T.R.; Slavkovic, S.; Churcher, Z.R.; Johnson, P.E. Development of a thermal-stable structure-switching cocaine-binding aptamer. Biochimie 2018, 145, 137–144. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, B.; Castagnola, E.; Cui, X.T. Zwitterionic Polymer Coated and Aptamer Functionalized Flexible Micro-Electrode Arrays for In Vivo Cocaine Sensing and Electrophysiology. Micromachines 2023, 14, 323. https://0-doi-org.brum.beds.ac.uk/10.3390/mi14020323
Wu B, Castagnola E, Cui XT. Zwitterionic Polymer Coated and Aptamer Functionalized Flexible Micro-Electrode Arrays for In Vivo Cocaine Sensing and Electrophysiology. Micromachines. 2023; 14(2):323. https://0-doi-org.brum.beds.ac.uk/10.3390/mi14020323
Chicago/Turabian StyleWu, Bingchen, Elisa Castagnola, and Xinyan Tracy Cui. 2023. "Zwitterionic Polymer Coated and Aptamer Functionalized Flexible Micro-Electrode Arrays for In Vivo Cocaine Sensing and Electrophysiology" Micromachines 14, no. 2: 323. https://0-doi-org.brum.beds.ac.uk/10.3390/mi14020323