
Sphingomonas Relies on Chemotaxis to Degrade Polycyclic Aromatic Hydrocarbons and Maintain Dominance in Coking Sites


Abstract
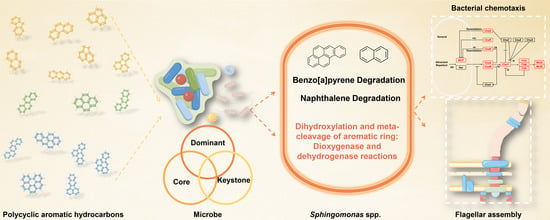
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Collection of Soil Samples
2.2. Determination of 16 PAHs
2.3. DNA Extraction, 16S rRNA Gene Amplicon Sequencing, and Analysis
2.4. Screening for Keystone Species, Core Species, and Dominant Species
2.5. Metagenomic Sequencing
2.6. Genome Assembly
2.7. Data Analysis and Picture Drawing
3. Results and Discussion
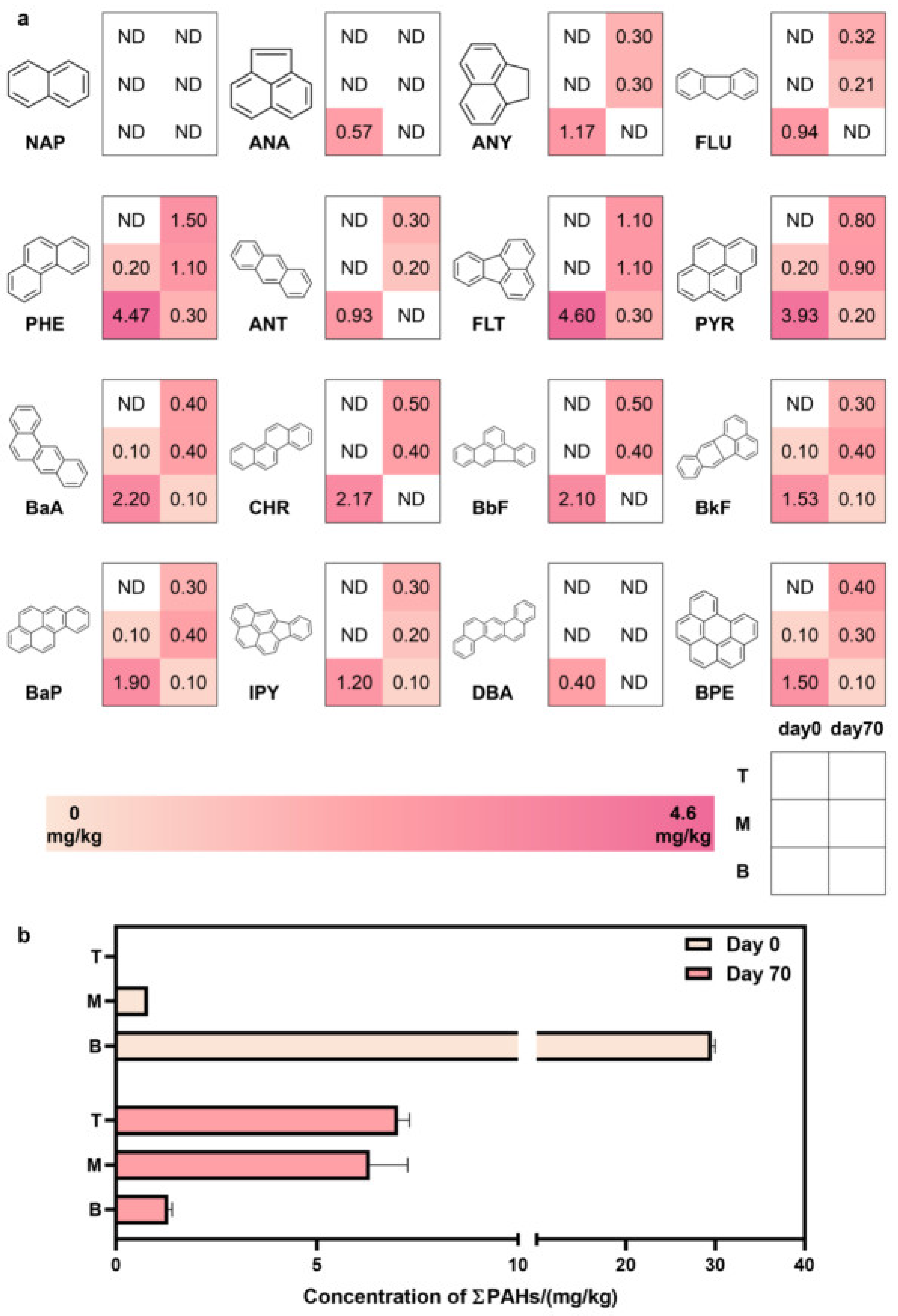
3.1. Occurrence Patterns, Migration, and Transformation Dynamics of PAHs in Soil at Different Depths in Coking Sites
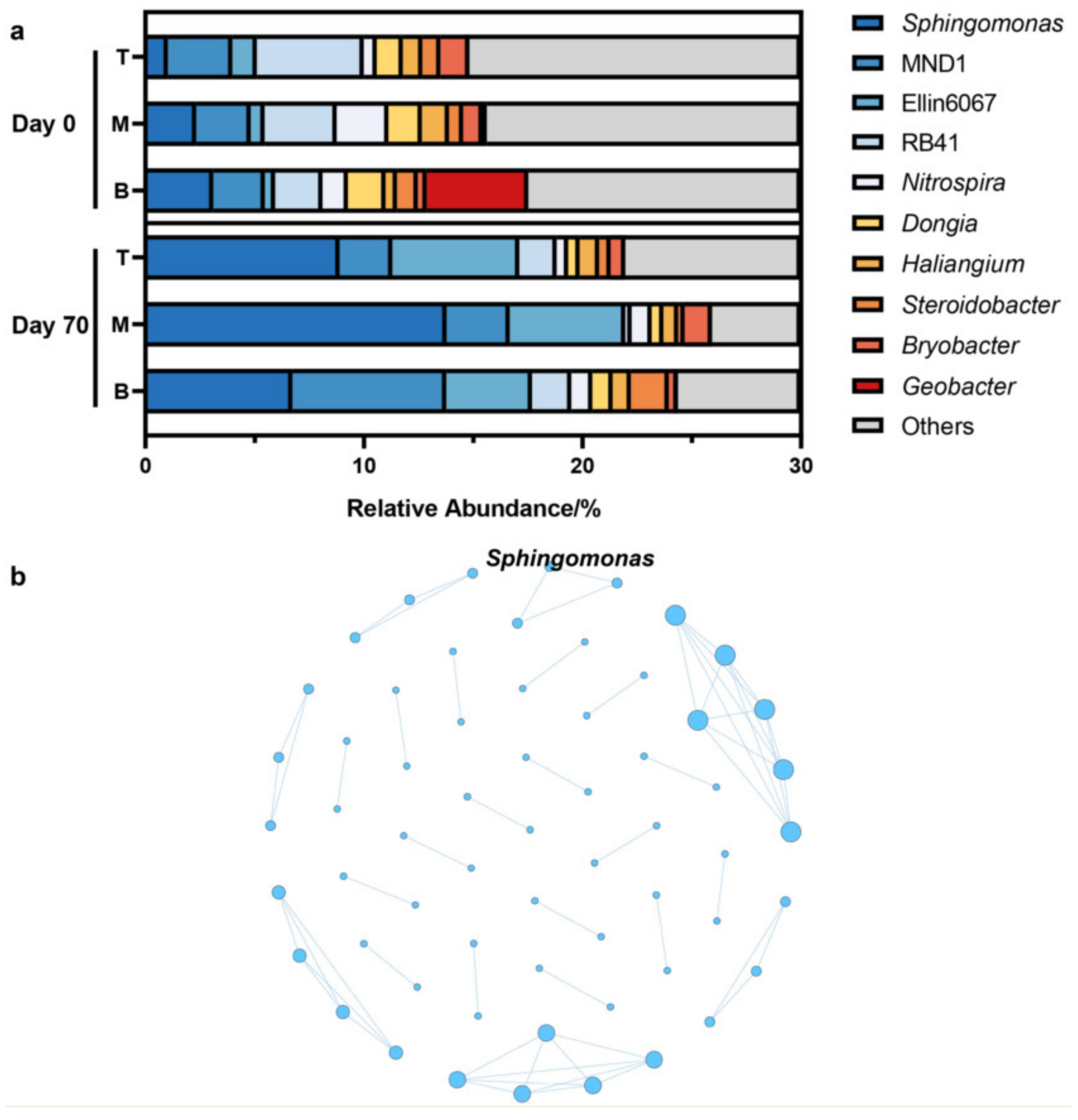
3.2. Dominant, Keystone, and Core Species of Coking Site Soil
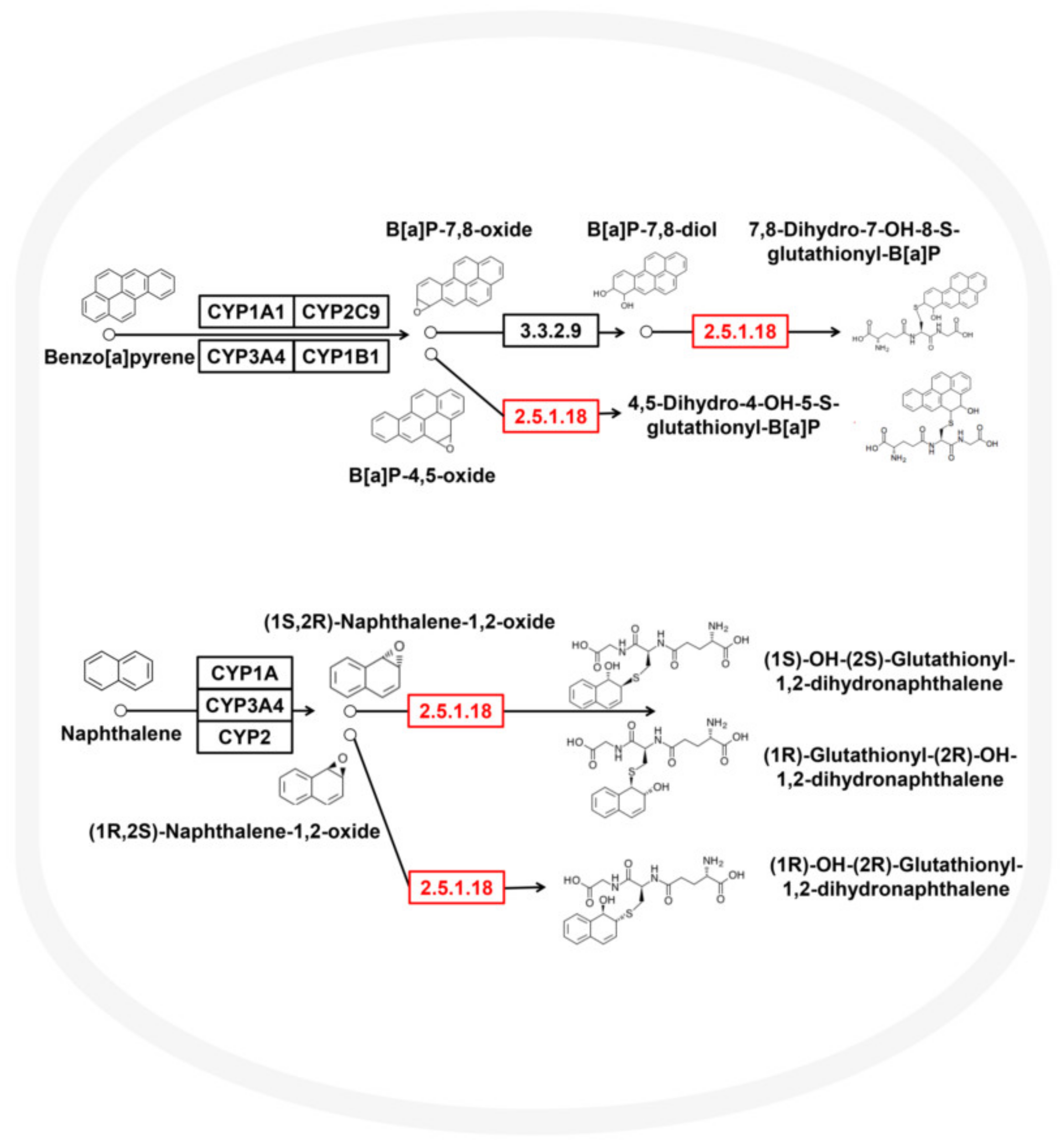
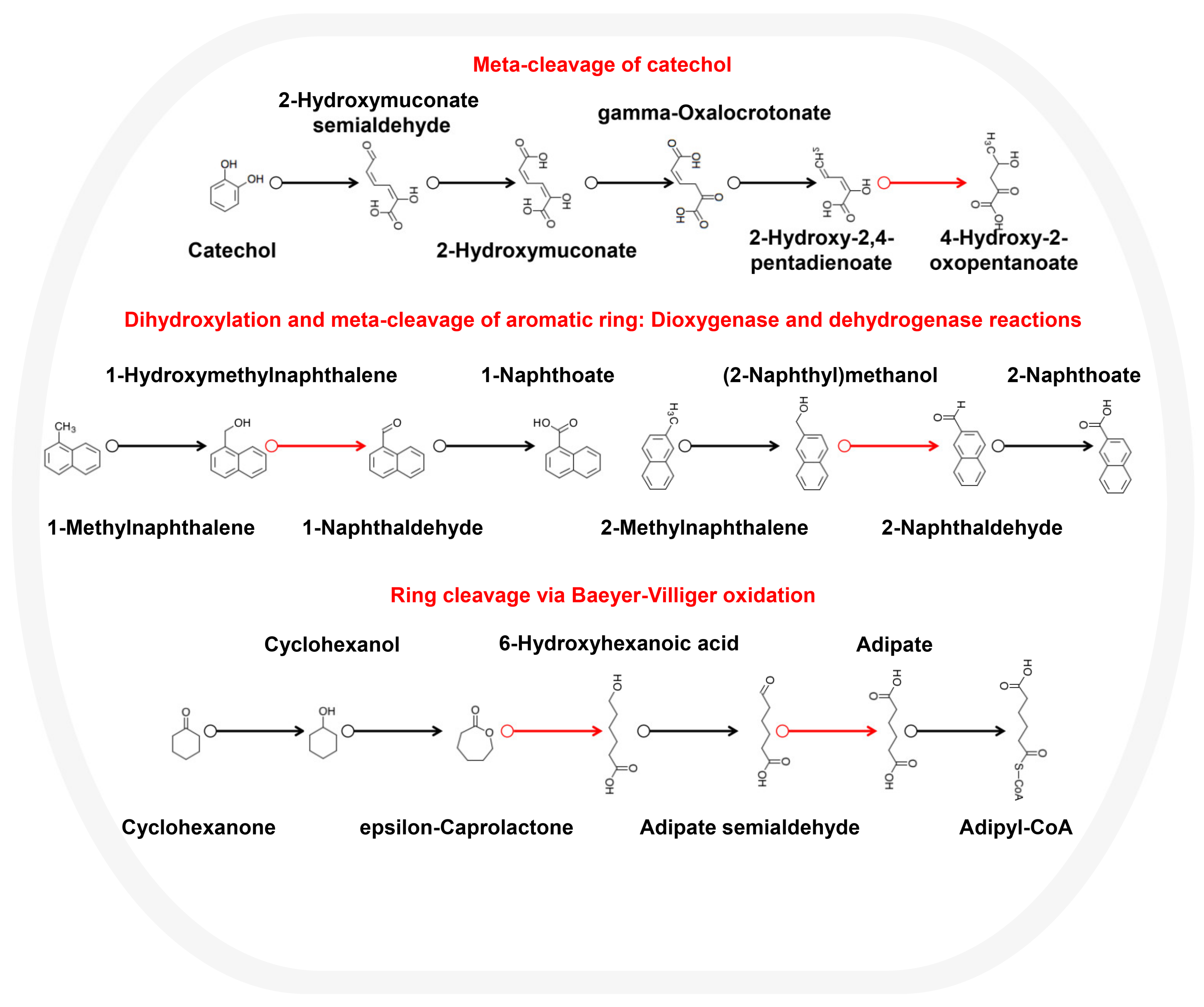
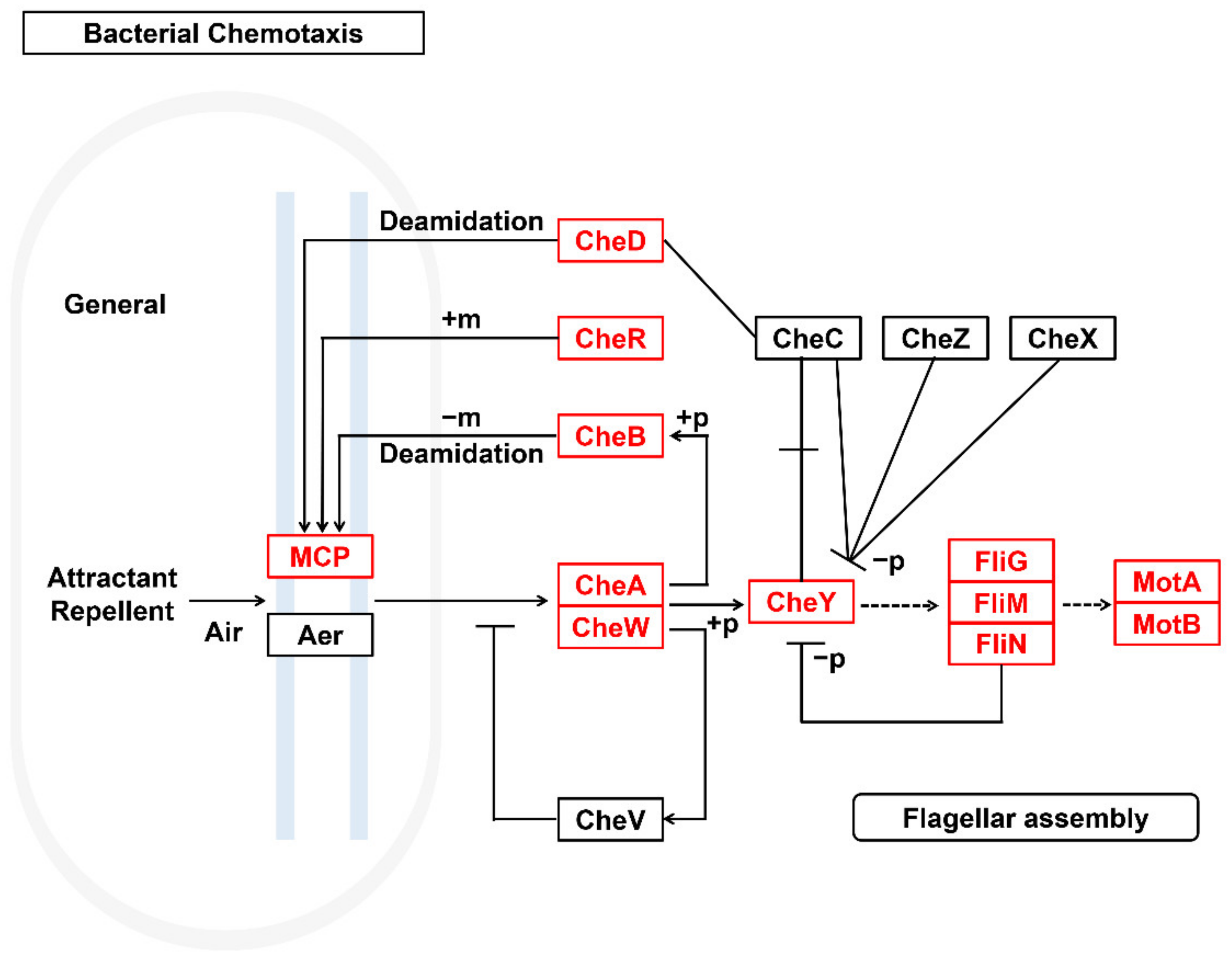
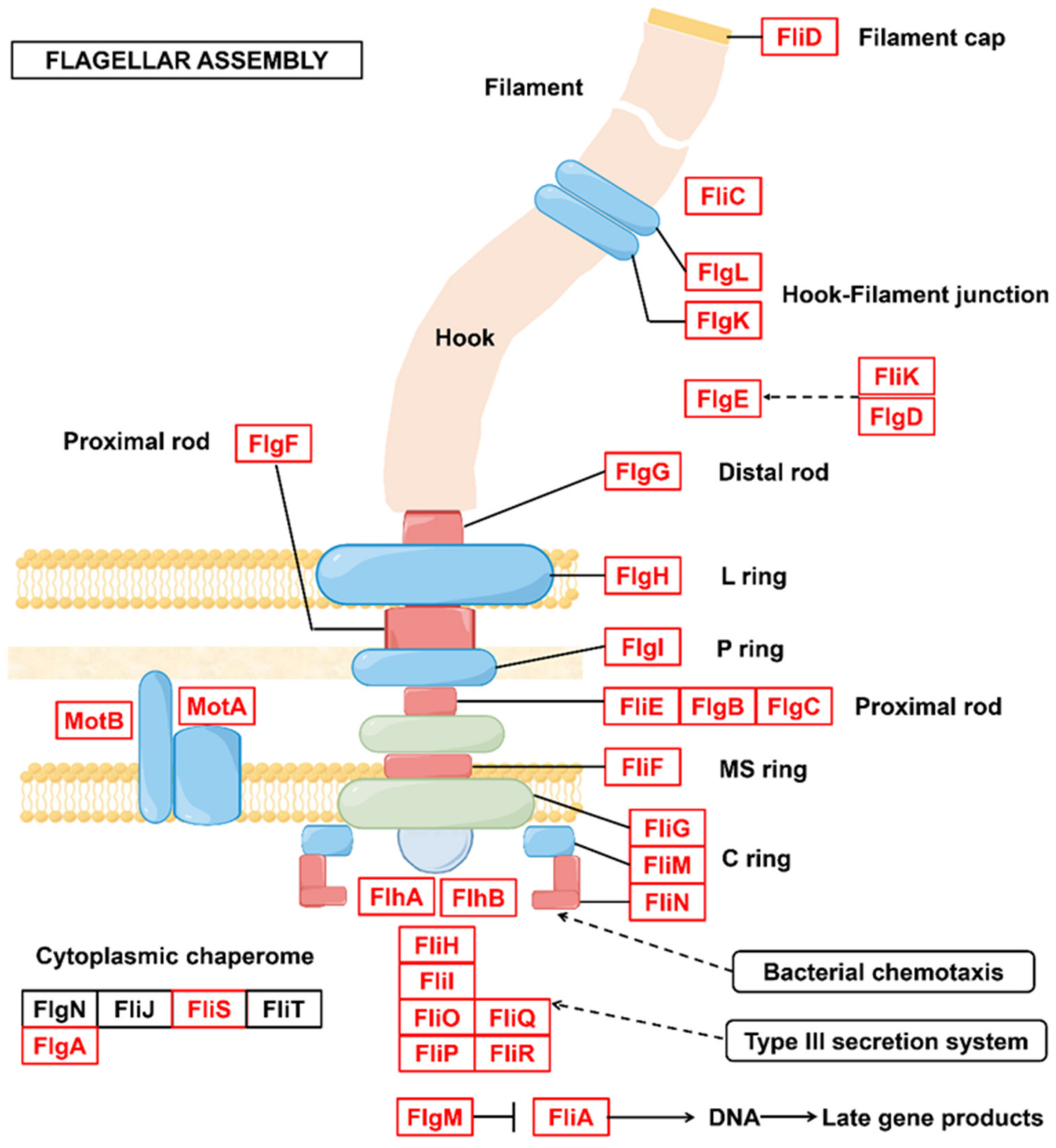
3.3. Metabolic Potential and Survival Strategies of Sphingomonas sp.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Keith, L.H.; Telliard, W.A. Priority pollutants I—A perspective view. Environ. Sci. Technol. 1979, 13, 416–423. [Google Scholar] [CrossRef]
- Mortelmans, K.; Haworth, S.; Lawlor, T.; Speck, W.; Tainer, B.; Zeiger, E. Salmonella mutagenicity tests: 2. Results from the testing of 270 chemicals. Environ. Mutagen. 1986, 8, 1–119. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, S.; Thavamani, P.; Venkateswarlu, K.; Lee, Y.B.; Naidu, R.; Megharaj, M. Remediation approaches for polycyclic aromatic hydrocarbons (PAHs) contaminated soils: Technological constraints, emerging trends and future directions. Chemosphere 2017, 168, 944–968. [Google Scholar] [CrossRef] [PubMed]
- Wild, S.R.; Jones, K.C. polynuclear aromatic-hydrocarbons in the united-kingdom environment—A preliminary source inventory and budget. Environ. Pollut. 1995, 88, 91–108. [Google Scholar] [CrossRef]
- Ghosal, D.; Ghosh, S.; Dutta, T.K.; Ahn, Y. Current State of Knowledge in Microbial Degradation of Polycyclic Aromatic Hydrocarbons (PAHs): A Review. Front. Microbiol. 2016, 7, 1837. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Pathak, B.; Fulekar, M.H. Molecular approaches for biodegradation of polycyclic aromatic hydrocarbon compounds: A review. Rev. Environ. Sci. Bio/Technol. 2015, 14, 241–269. [Google Scholar] [CrossRef]
- Zhang, L.; Qiu, X.; Huang, L.; Xu, J.; Wang, W.; Li, Z.; Xu, P.; Tang, H. Microbial degradation of multiple PAHs by a microbial consortium and its application on contaminated wastewater. J. Hazard. Mater. 2021, 419, 126524. [Google Scholar] [CrossRef]
- Keegstra, J.M.; Carrara, F.; Stocker, R. The ecological roles of bacterial chemotaxis. Nat. Rev. Microbiol. 2022. [Google Scholar] [CrossRef]
- Wadhwa, N.; Berg, H.C. Bacterial motility: Machinery and mechanisms. Nat. Rev. Microbiol. 2022, 20, 161–173. [Google Scholar] [CrossRef]
- Cunliffe, M.; Kertesz, M.A. Autecological properties of soil sphingomonads involved in the degradation of polycyclic aromatic hydrocarbons. Appl. Microbiol. Biotechnol. 2006, 72, 1083–1089. [Google Scholar] [CrossRef]
- Ortega-Calvo, J.J.; Marchenko, A.I.; Vorobyov, A.V.; Borovick, R.V. Chemotaxis in polycyclic aromatic hydrocarbon-degrading bacteria isolated from coal-tar- and oil-polluted rhizospheres. FEMS Microbiol. Ecol. 2003, 44, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, D.; Jakobsen, H.H.; Winding, A.; Mayer, P. Co-Transport of Polycyclic Aromatic Hydrocarbons by Motile Microorganisms Leads to Enhanced Mass Transfer under Diffusive Conditions. Environ. Sci. Technol. 2014, 48, 4368–4375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, F.; Corre, E.; Cebron, A. Stable isotope probing and metagenomics highlight the effect of plants on uncultured phenanthrene-degrading bacterial consortium in polluted soil. ISME J. 2019, 13, 1814–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Li, Q.; Zhang, L.; Cui, J.; Yu, H.; Wang, X.; Ouyang, X.; Tao, F.; Xu, P.; Tang, H. Genetic mapping of highly versatile and solvent-tolerant Pseudomonas putida B6-2 (ATCC BAA-2545) as a ‘superstar’ for mineralization of PAHs and dioxin-like compounds. Environ. Microbiol. 2021, 23, 4309–4325. [Google Scholar] [CrossRef]
- Desta, M.; Wang, W.; Zhang, L.; Xu, P.; Tang, H. Isolation, Characterization, and Genomic Analysis of Pseudomonas sp. Strain SMT-1, an Efficient Fluorene-Degrading Bacterium. Evol. Bioinform. 2019, 15, 1176934319843518. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Luo, C.; Jiang, L.; Peng, K.; Zhang, D.; Zhang, R.; Li, Y.; Zhang, G. The presence of in situ sulphamethoxazole degraders and their interactions with other microbes in activated sludge as revealed by DNA stable isotope probing and molecular ecological network analysis. Environ. Int. 2019, 124, 121–129. [Google Scholar] [CrossRef]
- Li, J.; Luo, C.; Zhang, D.; Zhao, X.; Dai, Y.; Cai, X.; Zhang, G. The catabolic pathways of in situ rhizosphere PAH degraders and the main factors driving PAH rhizoremediation in oil-contaminated soil. Environ. Microbiol. 2021, 23, 7042–7055. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- LaPara, T.M.; Nakatsu, C.H.; Pantea, L.; Alleman, J.E. Phylogenetic analysis of bacterial communities in mesophilic and thermophilic bioreactors treating pharmaceutical wastewater. Appl. Environ. Microbiol. 2000, 66, 3951–3959. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.R.; Hurlbert, A.H.; White, E.P. Opposing Mechanisms Drive Richness Patterns of Core and Transient Bird Species. Am. Nat. 2013, 181, E83–E90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umana, M.N.; Zhang, C.; Cao, M.; Lin, L.; Swenson, N.G. A core-transient framework for trait-based community ecology: An example from a tropical tree seedling community. Ecol. Lett. 2017, 20, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.J.S.; Evans, B.S.; White, E.P.; Hurlbert, A.H. The prevalence and impact of transient species in ecological communities. Ecology 2018, 99, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the Third International Conference on Weblogs and Social Media (ICWSM 2009), San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Szekely, A.J.; Langenheder, S. The importance of species sorting differs between habitat generalists and specialists in bacterial communities. FEMS Microbiol. Ecol. 2014, 87, 102–112. [Google Scholar] [CrossRef]
- Cheng, J.; Ringel-Kulka, T.; Heikamp-de Jong, I.; Ringel, Y.; Carroll, I.; de Vos, W.M.; Salojarvi, J.; Satokari, R. Discordant temporal development of bacterial phyla and the emergence of core in the fecal microbiota of young children. ISME J. 2016, 10, 1002–1014. [Google Scholar] [CrossRef] [Green Version]
- Ju, F.; Zhang, T.J. Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant. ISME J. 2015, 9, 683–695. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Steinegger, M.; Soeding, J. Clustering huge protein sequence sets in linear time. Nat. Commun. 2018, 9, 2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Naidu, R.; Thavamani, P.; Meaklim, J.; Megharaj, M. Managing long-term polycyclic aromatic hydrocarbon contaminated soils: A risk-based approach. Environ. Sci. Pollut. Res. 2015, 22, 8927–8941. [Google Scholar] [CrossRef] [PubMed]
- Bending, G.D.; Lincoln, S.D.; Sorensen, S.R.; Morgan, J.A.W.; Aamand, J.; Walker, A. In-field spatial variability in the degradation of the phenyl-urea herbicide isoproturon is the result of interactions between degradative Sphingomonas spp. and soil pH. Appl. Environ. Microbiol. 2003, 69, 827–834. [Google Scholar] [CrossRef] [Green Version]
- Peng, R.-H.; Xiong, A.-S.; Xue, Y.; Fu, X.-Y.; Gao, F.; Zhao, W.; Tian, Y.-S.; Yao, Q.-H. Microbial biodegradation of polyaromatic hydrocarbons. FEMS Microbiol. Rev. 2008, 32, 927–955. [Google Scholar] [CrossRef] [Green Version]
- Asaf, S.; Numan, M.; Khan, A.L.; Al-Harrasi, A. Sphingomonas: From diversity and genomics to functional role in environmental remediation and plant growth. Crit. Rev. Biotechnol. 2020, 40, 138–152. [Google Scholar] [CrossRef]
- Sampedro, I.; Parales, R.E.; Krell, T.; Hill, J.E. Pseudomonas chemotaxis. FEMS Microbiol. Rev. 2015, 39, 17–46. [Google Scholar] [CrossRef] [Green Version]
- Yun-Hao, W.; Zhou, H.; Shuang-Jiang, L. Chemotaxis towards aromatic compounds: Insights from Comamonas testosteroni. Int. J. Mol. Sci. 2019, 20, 2701. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Liu, S.-J.; Du, W. Chemotactic screening of imidazolinone-degrading bacteria by microfluidic SlipChip. J. Hazard. Mater. 2019, 366, 512–519. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, M.; Liu, Z.; Wang, J.; Zhao, Y.; Hu, B. Sphingomonas Relies on Chemotaxis to Degrade Polycyclic Aromatic Hydrocarbons and Maintain Dominance in Coking Sites. Microorganisms 2022, 10, 1109. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10061109
Zhou M, Liu Z, Wang J, Zhao Y, Hu B. Sphingomonas Relies on Chemotaxis to Degrade Polycyclic Aromatic Hydrocarbons and Maintain Dominance in Coking Sites. Microorganisms. 2022; 10(6):1109. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10061109
Chicago/Turabian StyleZhou, Meng, Zishu Liu, Jiaqi Wang, Yuxiang Zhao, and Baolan Hu. 2022. "Sphingomonas Relies on Chemotaxis to Degrade Polycyclic Aromatic Hydrocarbons and Maintain Dominance in Coking Sites" Microorganisms 10, no. 6: 1109. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10061109