
Phylogenetic Diversity of Animal Oral and Gastrointestinal Viromes Useful in Surveillance of Zoonoses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
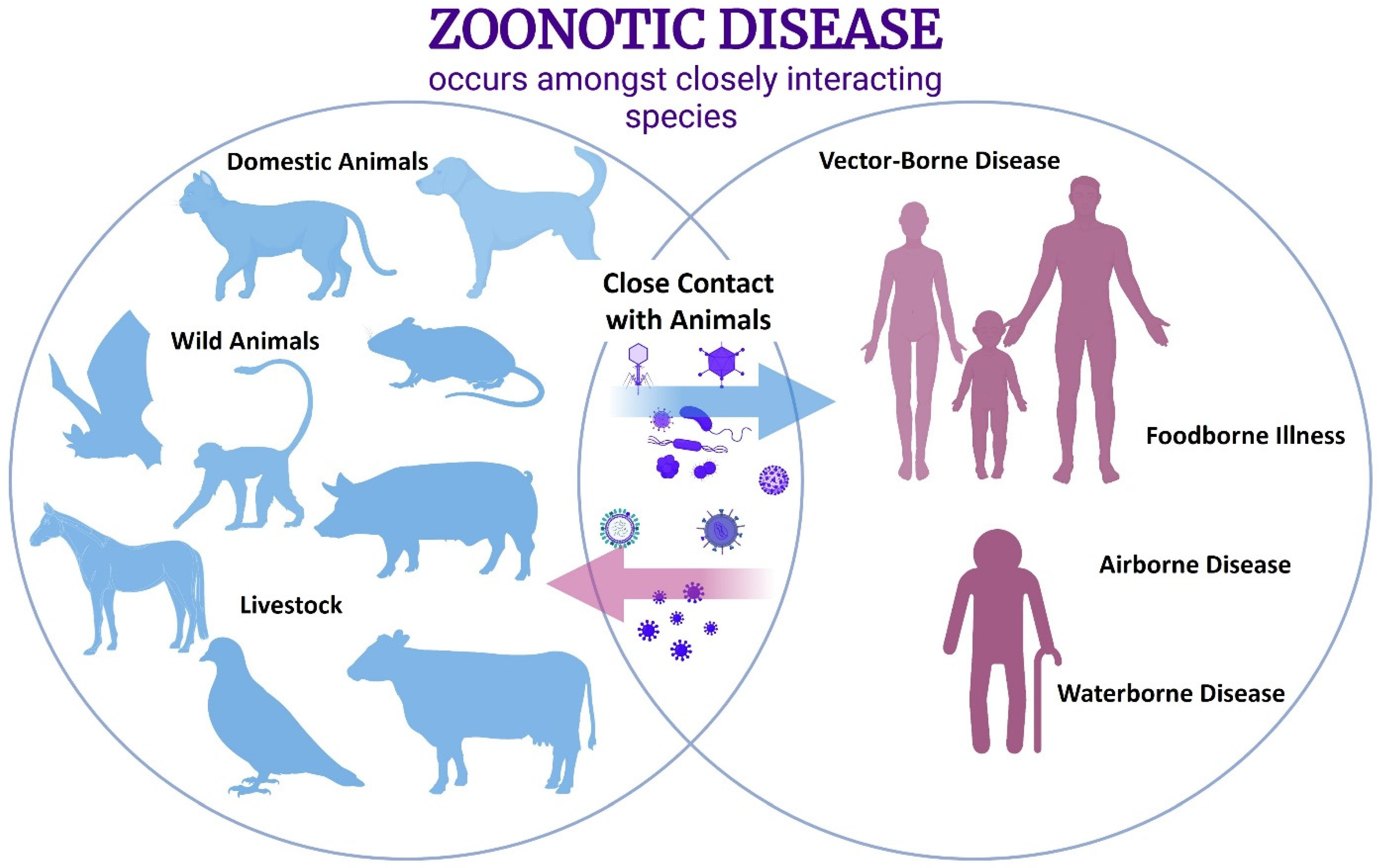
:1. Introduction
2. Virome Analyses of Animals as Critical Tools in the Identification of Novel Viruses and the Surveillance of Known Viral Reservoirs
2.1. Wild Animals
2.2. Domestic Animals
2.3. Livestock Animals
3. Human Virome Metagenomic Analyses Also Provide Valuable Information
3.1. Human Oral Virome Significance
3.2. Human Gut Virome Significance
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Santiago-Rodriguez, T.M.; Hollister, E.B. Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut. Viruses 2019, 11, 656. [Google Scholar] [CrossRef] [PubMed]
- Temmam, S.; Davoust, B.; Berenger, J.-M.; Raoult, D.; Desnues, C. Viral Metagenomics on Animals as a Tool for the Detection of Zoonoses Prior to Human Infection? Int. J. Mol. Sci. 2014, 15, 10377–10397. [Google Scholar] [CrossRef] [PubMed]
- Keesing, F.; Ostfeld, R.S. Impacts of Biodiversity and Biodiversity Loss on Zoonotic Diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2023540118. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Breman, J.G.; Calisher, C.H.; Doherty, P.C.; Hahn, B.H.; Keusch, G.T.; Kramer, L.D.; LeDuc, J.W.; Monath, T.P.; Taubenberger, J.K. The Origin of COVID-19 and Why It Matters. Am. J. Trop. Med. Hyg. 2020, 103, 955–959. [Google Scholar] [CrossRef]
- Nikhra, V. The Trans-Zoonotic Virome Interface: Measures to Balance, Control and Treat Epidemics. Ann. Biomed. Sci. Eng. 2020, 4, 020–027. [Google Scholar]
- Sawyer, A.; Free, T.; Martin, J. Metagenomics: Preventing Future Pandemics. BioTechniques 2021, 70, 1–4. [Google Scholar] [CrossRef]
- Xiao, P.; Han, J.; Zhang, Y.; Li, C.; Guo, X.; Wen, S.; Tian, M.; Li, Y.; Wang, M.; Liu, H.; et al. Metagenomic Analysis of Flaviviridae in Mosquito Viromes Isolated From Yunnan Province in China Reveals Genes From Dengue and Zika Viruses. Front. Cell. Infect. Microbiol. 2018, 8, 359. [Google Scholar] [CrossRef]
- Irving, A.T.; Ahn, M.; Goh, G.; Anderson, D.E.; Wang, L.-F. Lessons from the Host Defences of Bats, a Unique Viral Reservoir. Nature 2021, 589, 363–370. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Curr. Biol. 2020, 30, 2196–2203.e3. [Google Scholar] [CrossRef]
- Li, Y.; Altan, E.; Reyes, G.; Halstead, B.; Deng, X.; Delwart, E. Virome of Bat Guano from Nine Northern California Roosts. J. Virol. 2021, 95, e01713–e01720. [Google Scholar] [CrossRef]
- Zobba, R.; Visco, S.; Sotgiu, F.; Pinna Parpaglia, M.L.; Pittau, M.; Alberti, A. Molecular Survey of Parvovirus, Astrovirus, Coronavirus, and Calicivirus in Symptomatic Dogs. Vet. Res. Commun. 2021, 45, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Uwishema, O.; Adriano, L.F.; Chalhoub, E.; Onyeaka, H.; Mhanna, M.; David, S.C.; Nasrallah, Y.; Ribeiro, L.L.P.A.; Berjaoui, C. Bird Flu Outbreak amidst COVID-19 Pandemic in South Africa: Efforts and Challenges at Hand. J. Med. Virol. 2021, 93, 5676–5679. [Google Scholar] [CrossRef]
- Shan, T.; Yang, S.; Wang, H.; Wang, H.; Zhang, J.; Gong, G.; Xiao, Y.; Yang, J.; Wang, X.; Lu, J.; et al. Virome in the Cloaca of Wild and Breeding Birds Revealed a Diversity of Significant Viruses. Microbiome 2022, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.M.; Silva, F.J.M.; Brito, R.C.; Teixeira, S.D.; Melo, L.F.; Ribeiro, M.B.; Nagata, T.; Campos, S.F. Faecal Virome Analysis of Wild Animals from Brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.-W.; To, K.K.-W.; Chen, H.; Yuen, K.-Y. Cross-Species Transmission and Emergence of Novel Viruses from Birds. Curr. Opin. Virol. 2015, 10, 63–69. [Google Scholar] [CrossRef]
- Díez-Villaseñor, C.; Rodriguez-Valera, F. CRISPR Analysis Suggests That Small Circular Single-Stranded DNA Smacoviruses Infect Archaea Instead of Humans. Nat. Commun. 2019, 10, 294. [Google Scholar] [CrossRef]
- Ng, T.F.F.; Mesquita, J.R.; Nascimento, M.S.J.; Kondov, N.O.; Wong, W.; Reuter, G.; Knowles, N.J.; Vega, E.; Esona, M.D.; Deng, X.; et al. Feline Fecal Virome Reveals Novel and Prevalent Enteric Viruses. Vet. Microbiol. 2014, 171, 102–111. [Google Scholar] [CrossRef]
- Pietsch, C.; Liebert, U.G. Evidence for Presumable Feline Origin of Sporadic G6P[9] Rotaviruses in Humans. Infect. Genet. Evol. 2018, 63, 180–194. [Google Scholar] [CrossRef]
- Zhang, W.; Li, L.; Deng, X.; Kapusinszky, B.; Pesavento, P.A.; Delwart, E. Faecal Virome of Cats in an Animal Shelter. J. Gen. Virol. 2014, 95, 2553–2564. [Google Scholar] [CrossRef]
- Li, Y.; Gordon, E.; Idle, A.; Altan, E.; Seguin, M.A.; Estrada, M.; Deng, X.; Delwart, E. Virome of a Feline Outbreak of Diarrhea and Vomiting Includes Bocaviruses and a Novel Chapparvovirus. Viruses 2020, 12, 506. [Google Scholar] [CrossRef]
- Shi, Y.; Tao, J.; Li, B.; Shen, X.; Cheng, J.; Liu, H. The Gut Viral Metagenome Analysis of Domestic Dogs Captures Snapshot of Viral Diversity and Potential Risk of Coronavirus. Front. Vet. Sci. 2021, 8, 695088. [Google Scholar] [CrossRef] [PubMed]
- Fahsbender, E.; Altan, E.; Seguin, M.A.; Young, P.; Estrada, M.; Leutenegger, C.; Delwart, E. Chapparvovirus DNA Found in 4% of Dogs with Diarrhea. Viruses 2019, 11, 398. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.S.; Wagner, J.; Mansfield, C.S.; Stevens, M.; Gilkerson, J.R.; Kirkwood, C.D. Characterisation of the Canine Faecal Virome in Healthy Dogs and Dogs with Acute Diarrhoea Using Shotgun Metagenomics. PLoS ONE 2017, 12, e0178433. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Kobayashi, T. FAST Proteins: Development and Use of Reverse Genetics Systems for Reoviridae Viruses. Annu. Rev. Virol. 2021, 8, 515–536. [Google Scholar] [CrossRef]
- Hasson, S.S.; Al-Jabri, A.A. Immunized Camels and COVID-19. Asian Pac. J. Trop. Med. 2020, 13, 239. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, B.; Agwanda, B.; Fang, Y.; Wang, J.; Kuria, S.; Yang, J.; Masika, M.; Tang, S.; Lichoti, J.; et al. Viromes and Surveys of RNA Viruses in Camel-Derived Ticks Revealing Transmission Patterns of Novel Tick-Borne Viral Pathogens in Kenya. Emerg. Microbes Infect. 2021, 10, 1975–1987. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Teng, J.L.L.; Tsang, A.K.L.; Joseph, M.; Wong, E.Y.M.; Tang, Y.; Sivakumar, S.; Bai, R.; Wernery, R.; et al. Metagenomic Analysis of Viromes of Dromedary Camel Fecal Samples Reveals Large Number and High Diversity of Circoviruses and Picobirnaviruses. Virology 2014, 471–473, 117–125. [Google Scholar] [CrossRef]
- Amimo, J.O.; El Zowalaty, M.E.; Githae, D.; Wamalwa, M.; Djikeng, A.; Nasrallah, G.K. Metagenomic Analysis Demonstrates the Diversity of the Fecal Virome in Asymptomatic Pigs in East Africa. Arch. Virol. 2016, 161, 887–897. [Google Scholar] [CrossRef]
- Zhirakovskaia, E.; Tikunov, A.; Tymentsev, A.; Sokolov, S.; Sedelnikova, D.; Tikunova, N. Changing Pattern of Prevalence and Genetic Diversity of Rotavirus, Norovirus, Astrovirus, and Bocavirus Associated with Childhood Diarrhea in Asian Russia, 2009–2012. Infect. Genet. Evol. 2019, 67, 167–182. [Google Scholar] [CrossRef]
- Miyata, H.; Tsunoda, H.; Kazi, A.; Yamada, A.; Khan, M.A.; Murakami, J.; Kamahora, T.; Shiraki, K.; Hino, S. Identification of a Novel GC-Rich 113-Nucleotide Region To Complete the Circular, Single-Stranded DNA Genome of TT Virus, the First Human Circovirus. J. Virol. 1999, 73, 3582. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, Á.; Pankovics, P. Kobuviruses—A Comprehensive Review. Rev. Med. Virol. 2011, 21, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, L.; Zheng, Y.; Zhang, J.; Guo, B.; Yoon, K.-J.; Gauger, P.C.; Harmon, K.M.; Main, R.G.; Li, G. Metagenomic Analysis of the RNA Fraction of the Fecal Virome Indicates High Diversity in Pigs Infected by Porcine Endemic Diarrhea Virus in the United States. Virol. J. 2018, 15, 95. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Li, L.; Simmonds, P.; Wang, C.; Moeser, A.; Delwart, E. The Fecal Virome of Pigs on a High-Density Farm. J. Virol. 2011, 85, 11697–11708. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Gong, W.; Yan, X.; Zhao, Z.; Yang, L.; Tan, Z.; Xu, L.; Zhu, A.; Zhang, J.; Rao, J.; et al. Viral Metagenome-Based Precision Surveillance of Pig Population at Large Scale Reveals Viromic Signatures of Sample Types and Influence of Farming Management on Pig Virome. mSystems 2021, 6, e00420–e00421. [Google Scholar] [CrossRef]
- Meng, X.-J.; Halbur, P.G.; Shapiro, M.S.; Govindarajan, S.; Bruna, J.D.; Mushahwar, I.K.; Purcell, R.H.; Emerson, S.U. Genetic and Experimental Evidence for Cross-Species Infection by Swine Hepatitis E Virus. J. Virol. 1998, 72, 9714–9721. [Google Scholar] [CrossRef]
- Chua, K.B.; Bellini, W.J.; Rota, P.A.; Harcourt, B.H.; Tamin, A.; Lam, S.K.; Ksiazek, T.G.; Rollin, P.E.; Zaki, S.R.; Shieh, W. Nipah Virus: A Recently Emergent Deadly Paramyxovirus. Science 2000, 288, 1432–1435. Available online: https://www.science.org/doi/abs/10.1126/science.288.5470.1432 (accessed on 5 July 2022). [CrossRef]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and Pandemic Potential of Swine-Origin H1N1 Influenza Virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef]
- Ricklin, M.E.; García-Nicolás, O.; Brechbühl, D.; Python, S.; Zumkehr, B.; Nougairede, A.; Charrel, R.N.; Posthaus, H.; Oevermann, A.; Summerfield, A. Vector-Free Transmission and Persistence of Japanese Encephalitis Virus in Pigs. Nat. Commun. 2016, 7, 10832. [Google Scholar] [CrossRef]
- Kubacki, J.; Qi, W.; Fraefel, C. Differential Viral Genome Diversity of Healthy and RSS-Affected Broiler Flocks. Microorganisms 2022, 10, 1092. [Google Scholar] [CrossRef]
- Varsani, A.; Krupovic, M. Smacoviridae: A New Family of Animal-Associated Single-Stranded DNA Viruses. Arch. Virol. 2018, 163, 2005–2015. [Google Scholar] [CrossRef]
- Vibin, J.; Chamings, A.; Klaassen, M.; Bhatta, T.R.; Alexandersen, S. Metagenomic Characterisation of Avian Parvoviruses and Picornaviruses from Australian Wild Ducks. Sci. Rep. 2020, 10, 12800. [Google Scholar] [CrossRef] [PubMed]
- Roediger, B.; Lee, Q.; Tikoo, S.; Cobbin, J.C.A.; Henderson, J.M.; Jormakka, M.; O’Rourke, M.B.; Padula, M.P.; Pinello, N.; Henry, M.; et al. An Atypical Parvovirus Drives Chronic Tubulointerstitial Nephropathy and Kidney Fibrosis. Cell 2018, 175, 530–543.e24. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Bilic, I.; Viloux, N.; Palmieri, N.; Albaric, O.; Chatenet, X.; Tvarogová, J.; Dinhopl, N.; Heidl, S.; Liebhart, D.; et al. A Novel Chaphamaparvovirus Is the Etiological Agent of Hepatitis Outbreaks in Pheasants (Phasianus colchicus) Characterized by High Mortality. Transbound. Emerg. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ahn, E.H.; Kang, S.S.; Liu, X.; Alam, A.; Ye, K. Gut Dysbiosis Contributes to Amyloid Pathology, Associated with C/EBPβ/AEP Signaling Activation in Alzheimer’s Disease Mouse Model. Sci. Adv. 2020, 6, eaba0466. [Google Scholar] [CrossRef]
- Giovannini, M.G.; Lana, D.; Traini, C.; Vannucchi, M.G. The Microbiota–Gut–Brain Axis and Alzheimer Disease. From Dysbiosis to Neurodegeneration: Focus on the Central Nervous System Glial Cells. J. Clin. Med. 2021, 10, 2358. [Google Scholar] [CrossRef]
- Radaic, A.; Kapila, Y.L. The Oralome and Its Dysbiosis: New Insights into Oral Microbiome-Host Interactions. Comput. Struct. Biotechnol. J. 2021, 19, 1335–1360. [Google Scholar] [CrossRef]
- Gradisteanu Pircalabioru, G.; Corcionivoschi, N.; Gundogdu, O.; Chifiriuc, M.-C.; Marutescu, L.G.; Ispas, B.; Savu, O. Dysbiosis in the Development of Type I Diabetes and Associated Complications: From Mechanisms to Targeted Gut Microbes Manipulation Therapies. Int. J. Mol. Sci. 2021, 22, 2763. [Google Scholar] [CrossRef]
- Chen, J.; Domingue, J.C.; Sears, C.L. Microbiota Dysbiosis in Select Human Cancers: Evidence of Association and Causality. Semin. Immunol. 2017, 32, 25–34. [Google Scholar] [CrossRef]
- Mascitti, M.; Togni, L.; Troiano, G.; Caponio, V.C.A.; Gissi, D.B.; Montebugnoli, L.; Procaccini, M.; Lo Muzio, L.; Santarelli, A. Beyond Head and Neck Cancer: The Relationship Between Oral Microbiota and Tumour Development in Distant Organs. Front. Cell. Infect. Microbiol. 2019, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Pride, D.T.; Salzman, J.; Haynes, M.; Rohwer, F.; Davis-Long, C.; White, R.A.; Loomer, P.; Armitage, G.C.; Relman, D.A. Evidence of a Robust Resident Bacteriophage Population Revealed through Analysis of the Human Salivary Virome. ISME J. 2012, 6, 915–926. [Google Scholar] [CrossRef]
- Soffritti, I.; D’Accolti, M.; Fabbri, C.; Passaro, A.; Manfredini, R.; Zuliani, G.; Libanore, M.; Franchi, M.; Contini, C.; Caselli, E. Oral Microbiome Dysbiosis Is Associated With Symptoms Severity and Local Immune/Inflammatory Response in COVID-19 Patients: A Cross-Sectional Study. Front. Microbiol. 2021, 12, 687513. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz Peña, M.J.; Gonzalez-Granado, L.I.; Garcia-Heredia, I.; Carballa, L.M.; Martinez-Garcia, M. Minimal-Moderate Variation of Human Oral Virome and Microbiome in IgA Deficiency. Sci. Rep. 2021, 11, 14913. [Google Scholar] [CrossRef] [PubMed]
- Parras, M.; López-Bueno, A. Methods for Enrichment and Sequencing of Oral Viral Assemblages: Saliva, Oral Mucosa, and Dental Plaque Viromes: Methods and Protocols. In The Human Virome; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1838, pp. 143–161. ISBN 978-1-4939-8681-1. [Google Scholar]
- Martínez, A.; Kuraji, R.; Kapila, Y.L. The Human Oral Virome: Shedding Light on the Dark Matter. Periodontol. 2000 2021, 87, 282–298. [Google Scholar] [CrossRef]
- Kapila, Y.L. Oral Health’s Inextricable Connection to Systemic Health: Special Populations Bring to Bear Multimodal Relationships and Factors Connecting Periodontal Disease to Systemic Diseases and Conditions. Periodontol. 2000 2021, 87, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.X.; Min, N.; Wong, E.P.Y.; Chong, C.Y.; Chu, J.J.H. Characterization of Oral Virome and Microbiome Revealed Distinctive Microbiome Disruptions in Paediatric Patients with Hand, Foot and Mouth Disease. Npj Biofilms Microbiomes 2021, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Ly, M.; Abeles, S.R.; Boehm, T.K.; Robles-Sikisaka, R.; Naidu, M.; Santiago-Rodriguez, T.; Pride, D.T. Altered Oral Viral Ecology in Association with Periodontal Disease. mBio 2014, 5, e01133-14. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz Peña, M.J.; Martinez-Hernandez, F.; Garcia-Heredia, I.; Lluesma Gomez, M.; Fornas, Ò.; Martinez-Garcia, M. Deciphering the Human Virome with Single-Virus Genomics and Metagenomics. Viruses 2018, 10, 113. [Google Scholar] [CrossRef]
- Edlund, A.; Santiago-Rodriguez, T.M.; Boehm, T.K.; Pride, D.T. Bacteriophage and Their Potential Roles in the Human Oral Cavity. J. Oral Microbiol. 2015, 7, 27423. [Google Scholar] [CrossRef]
- Pérez-Brocal, V.; Moya, A. The Analysis of the Oral DNA Virome Reveals Which Viruses Are Widespread and Rare among Healthy Young Adults in Valencia (Spain). PLoS ONE 2018, 13, e0191867. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe 2019, 26, 527–541.e5. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic Analyses of an Uncultured Viral Community from Human Feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef] [PubMed]
- Aggarwala, V.; Liang, G.; Bushman, F.D. Viral Communities of the Human Gut: Metagenomic Analysis of Composition and Dynamics. Mob. DNA 2017, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Manrique, P.; Bolduc, B.; Walk, S.T.; van der Oost, J.; de Vos, W.M.; Young, M.J. Healthy Human Gut Phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 2020, 28, 724–740.e8. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Yang, K.; Niu, J.; Zuo, T.; Sun, Y.; Xu, Z.; Tang, W.; Liu, Q.; Zhang, J.; Ng, E.K.W.; Wong, S.K.H.; et al. Alterations in the Gut Virome in Obesity and Type 2 Diabetes Mellitus. Gastroenterology 2021, 161, 1257–1269.e13. [Google Scholar] [CrossRef]
- Hasan, M.R.; Rahman, M.; Khan, T.; Saeed, A.; Sundararaju, S.; Flores, A.; Hawken, P.; Rawat, A.; Elkum, N.; Hussain, K.; et al. Virome-Wide Serological Profiling Reveals Association of Herpesviruses with Obesity. Sci. Rep. 2021, 11, 2562. [Google Scholar] [CrossRef]
- Liang, G.; Conrad, M.A.; Kelsen, J.R.; Kessler, L.R.; Breton, J.; Albenberg, L.G.; Marakos, S.; Galgano, A.; Devas, N.; Erlichman, J.; et al. Dynamics of the Stool Virome in Very Early-Onset Inflammatory Bowel Disease. J. Crohns Colitis 2020, 14, 1600–1610. [Google Scholar] [CrossRef]
- Fulci, V.; Stronati, L.; Cucchiara, S.; Laudadio, I.; Carissimi, C. Emerging Roles of Gut Virome in Pediatric Diseases. Int. J. Mol. Sci. 2021, 22, 4127. [Google Scholar] [CrossRef]
- Lindfors, K.; Lin, J.; Lee, H.-S.; Hyöty, H.; Nykter, M.; Kurppa, K.; Liu, E.; Koletzko, S.; Rewers, M.; Hagopian, W.; et al. Metagenomics of the Faecal Virome Indicate a Cumulative Effect of Enterovirus and Gluten Amount on the Risk of Coeliac Disease Autoimmunity in Genetically at Risk Children: The TEDDY Study. Gut 2020, 69, 1416–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Mouzan, M.; Assiri, A.; Al Sarkhy, A.; Alasmi, M.; Saeed, A.; Al-Hussaini, A.; AlSaleem, B.; Al Mofarreh, M. Viral Dysbiosis in Children with New-Onset Celiac Disease. PLoS ONE 2022, 17, e0262108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Härkönen, T.; Hämäläinen, A.-M.; et al. Intestinal Virome Changes Precede Autoimmunity in Type I Diabetes-Susceptible Children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef] [PubMed]
- Kramná, L.; Kolářová, K.; Oikarinen, S.; Pursiheimo, J.-P.; Ilonen, J.; Simell, O.; Knip, M.; Veijola, R.; Hyöty, H.; Cinek, O. Gut Virome Sequencing in Children with Early Islet Autoimmunity. Diabetes Care 2015, 38, 930–933. [Google Scholar] [CrossRef]
- Cinek, O.; Kramna, L.; Lin, J.; Oikarinen, S.; Kolarova, K.; Ilonen, J.; Simell, O.; Veijola, R.; Autio, R.; Hyöty, H. Imbalance of Bacteriome Profiles within the Finnish Diabetes Prediction and Prevention Study: Parallel Use of 16S Profiling and Virome Sequencing in Stool Samples from Children with Islet Autoimmunity and Matched Controls. Pediatr. Diabetes 2017, 18, 588–598. [Google Scholar] [CrossRef]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early Life Dynamics of the Human Gut Virome and Bacterial Microbiome in Infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef]
- Moreno-Gallego, J.L.; Chou, S.-P.; Di Rienzi, S.C.; Goodrich, J.K.; Spector, T.D.; Bell, J.T.; Youngblut, N.D.; Hewson, I.; Reyes, A.; Ley, R.E. Virome Diversity Correlates with Intestinal Microbiome Diversity in Adult Monozygotic Twins. Cell Host Microbe 2019, 25, 261–272.e5. [Google Scholar] [CrossRef]
- Monaco, C.L.; Gootenberg, D.B.; Zhao, G.; Handley, S.A.; Ghebremichael, M.S.; Lim, E.S.; Lankowski, A.; Baldridge, M.T.; Wilen, C.B.; Flagg, M.; et al. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 2016, 19, 311–322. [Google Scholar] [CrossRef]
- Cao, J.; Wang, C.; Zhang, Y.; Lei, G.; Xu, K.; Zhao, N.; Lu, J.; Meng, F.; Yu, L.; Yan, J.; et al. Integrated Gut Virome and Bacteriome Dynamics in COVID-19 Patients. Null 2021, 13, 1887722. [Google Scholar] [CrossRef]
- Morse, S.S.; Mazet, J.A.; Woolhouse, M.; Parrish, C.R.; Carroll, D.; Karesh, W.B.; Zambrana-Torrelio, C.; Lipkin, W.I.; Daszak, P. Prediction and Prevention of the next Pandemic Zoonosis. Lancet 2012, 380, 1956–1965. [Google Scholar] [CrossRef]
- Carroll, D.; Morzaria, S.; Briand, S.; Johnson, C.K.; Morens, D.; Sumption, K.; Tomori, O.; Wacharphaueasadee, S. Preventing the next Pandemic: The Power of a Global Viral Surveillance Network. BMJ 2021, 372, n485. [Google Scholar] [CrossRef] [PubMed]
- Honigsbaum, M. Revisiting the 1957 and 1968 Influenza Pandemics. Lancet 2020, 395, 1824–1826. [Google Scholar] [CrossRef]
- Ladner, J.T. Genomic Signatures for Predicting the Zoonotic Potential of Novel Viruses. PLoS Biol. 2021, 19, e3001403. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Yi, S.V. On the Origin and Evolution of SARS-CoV-2. Exp. Mol. Med. 2021, 53, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Rohr, J.R.; Barrett, C.B.; Civitello, D.J.; Craft, M.E.; Delius, B.; DeLeo, G.A.; Hudson, P.J.; Jouanard, N.; Nguyen, K.H.; Ostfeld, R.S.; et al. Emerging Human Infectious Diseases and the Links to Global Food Production. Nat. Sustain. 2019, 2, 445–456. [Google Scholar] [CrossRef]
- Naguib, M.M.; Li, R.; Ling, J.; Grace, D.; Nguyen-Viet, H.; Lindahl, J.F. Live and Wet Markets: Food Access versus the Risk of Disease Emergence. Trends Microbiol. 2021, 29, 573–581. [Google Scholar] [CrossRef]
- Aguirre, A.A.; Catherina, R.; Frye, H.; Shelley, L. Illicit Wildlife Trade, Wet Markets, and COVID-19: Preventing Future Pandemics. World Med. Health Policy 2020, 12, 256–265. [Google Scholar] [CrossRef]
- Tollefson, J. Why Deforestation and Extinctions Make Pandemics More Likely. Nature 2020, 584, 175–177. [Google Scholar] [CrossRef]
- Brancalion, P.H.S.; Broadbent, E.N.; de-Miguel, S.; Cardil, A.; Rosa, M.R.; Almeida, C.T.; Almeida, D.R.A.; Chakravarty, S.; Zhou, M.; Gamarra, J.G.P.; et al. Emerging Threats Linking Tropical Deforestation and the COVID-19 Pandemic. Perspect. Ecol. Conserv. 2020, 18, 243–246. [Google Scholar] [CrossRef]
- White, R.J.; Razgour, O. Emerging Zoonotic Diseases Originating in Mammals: A Systematic Review of Effects of Anthropogenic Land-Use Change. Mammal. Rev. 2020, 50, 336–352. [Google Scholar] [CrossRef]
- Liyanaarachchi, D.R.; Rajakaruna, R.S.; Dikkumbura, A.W.; Rajapakse, R.P.V.J. Ticks Infesting Wild and Domestic Animals and Humans of Sri Lanka with New Host Records. Acta Trop. 2015, 142, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Chaves, L.S.M.; Fry, J.; Malik, A.; Geschke, A.; Sallum, M.A.M.; Lenzen, M. Global Consumption and International Trade in Deforestation-Associated Commodities Could Influence Malaria Risk. Nat. Commun. 2020, 11, 1258. [Google Scholar] [CrossRef] [PubMed]
- Ilacqua, R.C.; Medeiros-Sousa, A.R.; Ramos, D.G.; Obara, M.T.; Ceretti-Junior, W.; Mucci, L.F.; Marrelli, M.T.; Laporta, G.Z. Reemergence of Yellow Fever in Brazil: The Role of Distinct Landscape Fragmentation Thresholds. J. Environ. Public Health 2021, 2021, e8230789. [Google Scholar] [CrossRef] [PubMed]
- Jonas, O.; Seifman, R. Do We Need a Global Virome Project? Lancet Glob. Health 2019, 7, e1314–e1316. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, A.M.; Esposito, M.M.; Ptashnik, A. Phylogenetic Diversity of Animal Oral and Gastrointestinal Viromes Useful in Surveillance of Zoonoses. Microorganisms 2022, 10, 1815. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10091815
Esposito AM, Esposito MM, Ptashnik A. Phylogenetic Diversity of Animal Oral and Gastrointestinal Viromes Useful in Surveillance of Zoonoses. Microorganisms. 2022; 10(9):1815. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10091815
Chicago/Turabian StyleEsposito, Anthony Michael, Michelle Marie Esposito, and Albert Ptashnik. 2022. "Phylogenetic Diversity of Animal Oral and Gastrointestinal Viromes Useful in Surveillance of Zoonoses" Microorganisms 10, no. 9: 1815. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms10091815