Identification of Differentially Expressed Proteins in Sugarcane in Response to Infection by Xanthomonas albilineans Using iTRAQ Quantitative Proteomics

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Growth and Inoculation with X. albilineans
2.2. Quantification of Populations of X. albilineans in Inoculated Sugarcane Leaves
2.3. Total Protein Extraction and Peptide Preparation
2.4. iTRAQ Labeling of Peptides
2.5. High-Performance Liquid Chromatography (HPLC) Fractionation
2.6. Liquid Chromatography Mass Spectrometry (LC-MS/MS) Analysis
2.7. Data Quality Control
2.8. Database Search and Quantitative Proteomic Analysis
2.9. Quantitative Real-Time PCR (qRT-PCR) Analysis
2.10. Statistical Analyses
3. Results
3.1. Pathogen Population Size in the Resistant and the Susceptible Sugarcane Cultivar after Inoculation with X. albilineans
3.2. Overview of Proteomic Profiling of Sugarcane Infected by X. albilineans
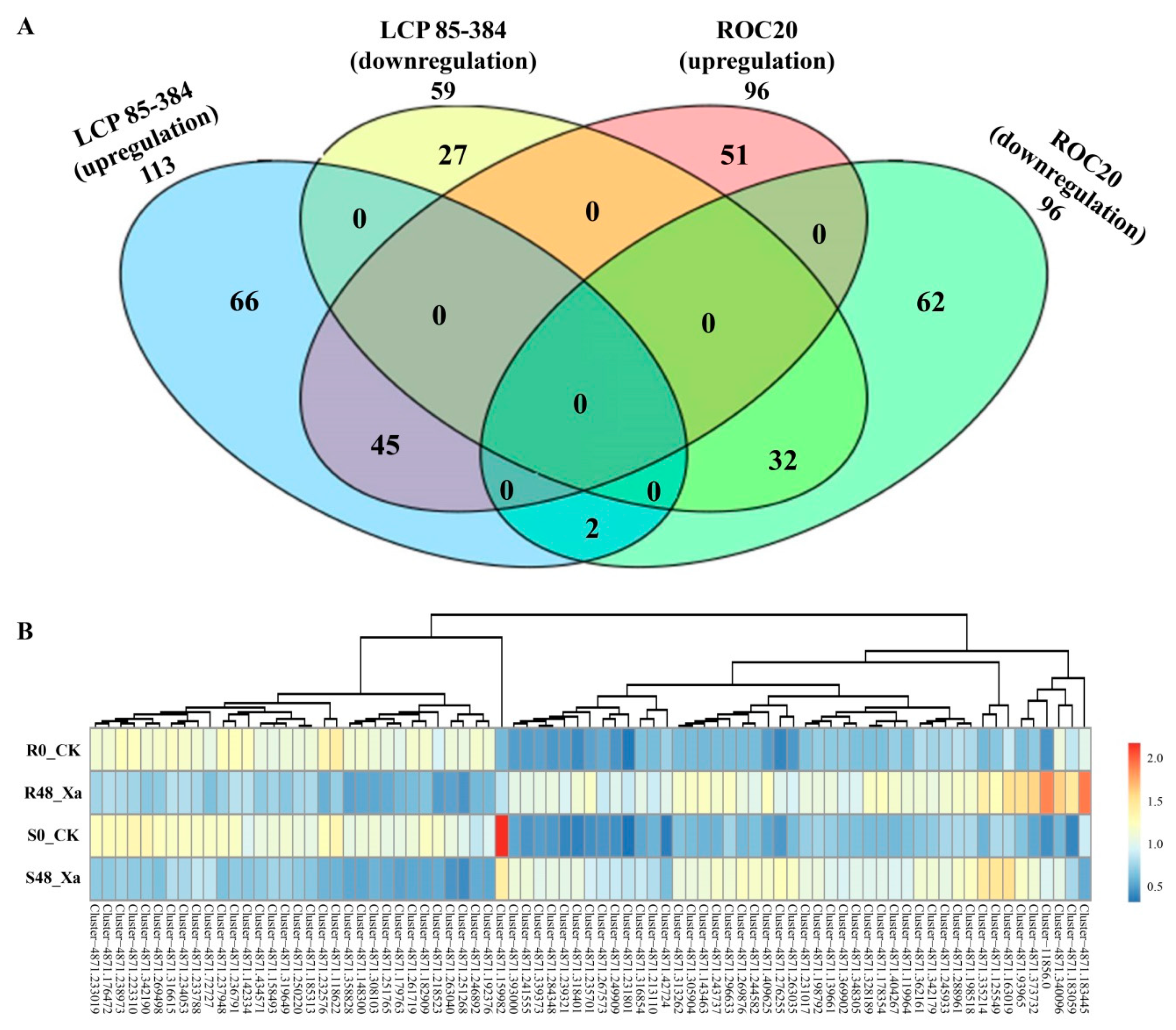
3.3. Identification of Differentially Expressed Proteins (DEPs) in Response to X. albilineans Infection
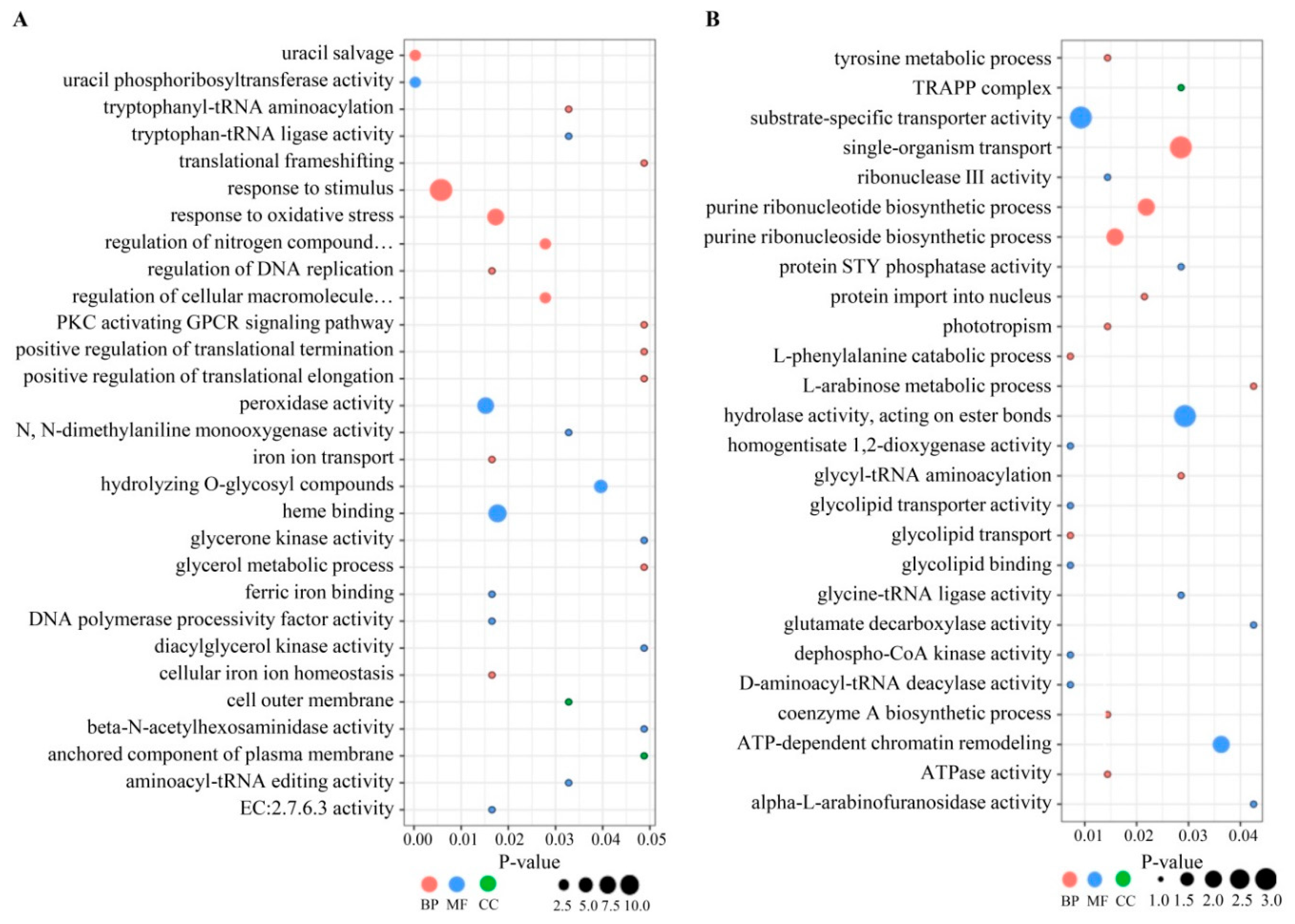
3.4. Gene Annotation of DEPs
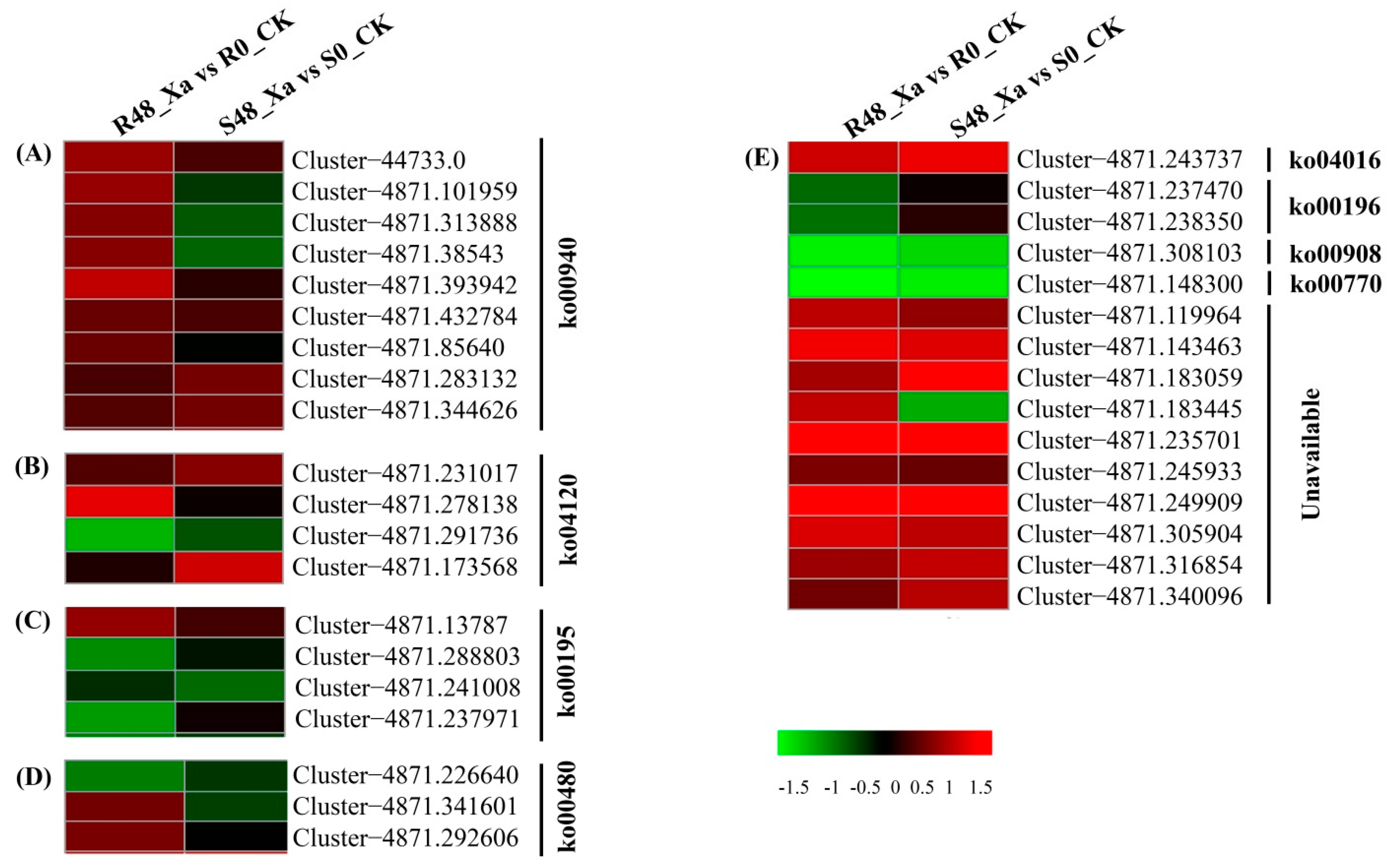
3.5. Functional Classification of DEPs by KEGG Analysis
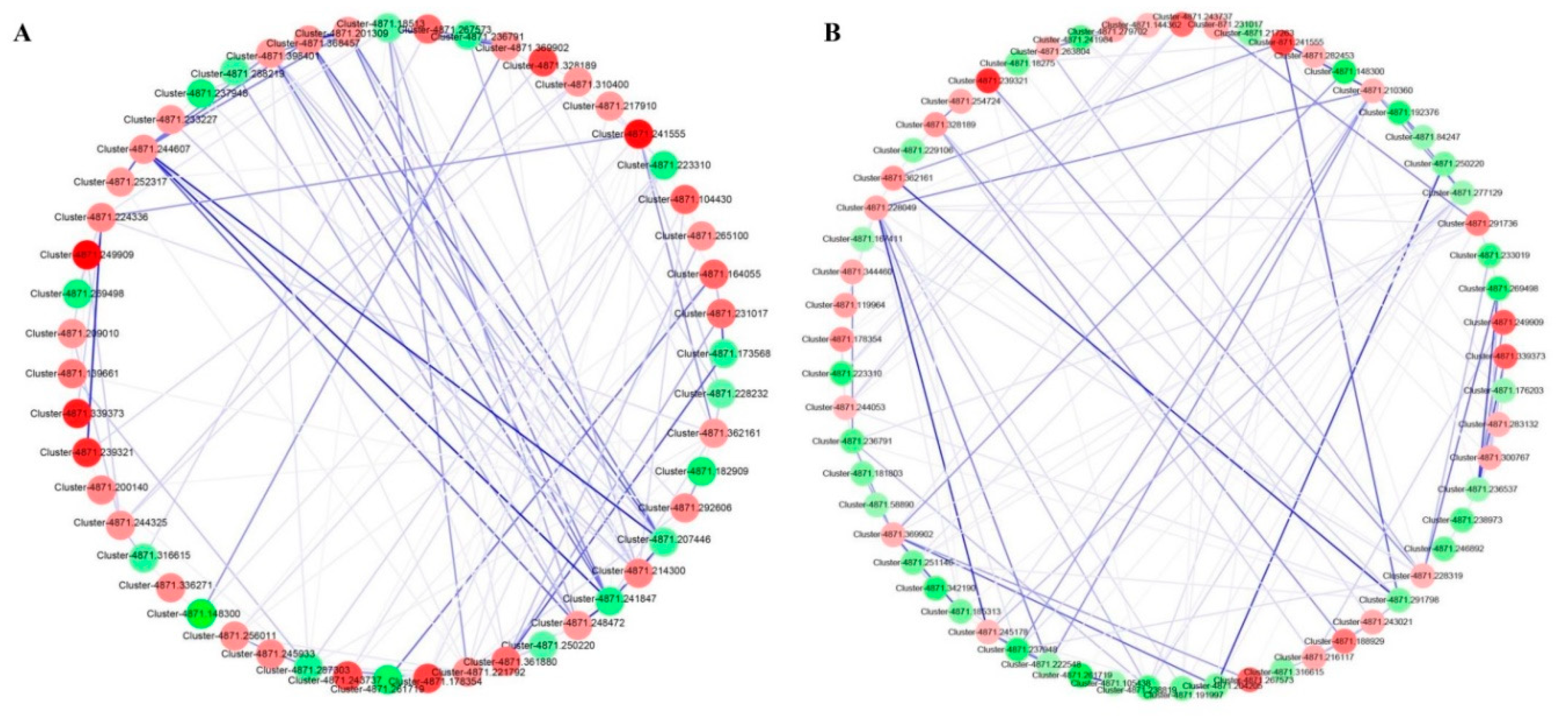
3.6. Protein–Protein Interactions (PPIs) Network Predicted in the STRING Database
3.7. Identification of Plant Defense Genes Triggered by Colonization of Sugarcane by X. albilineans
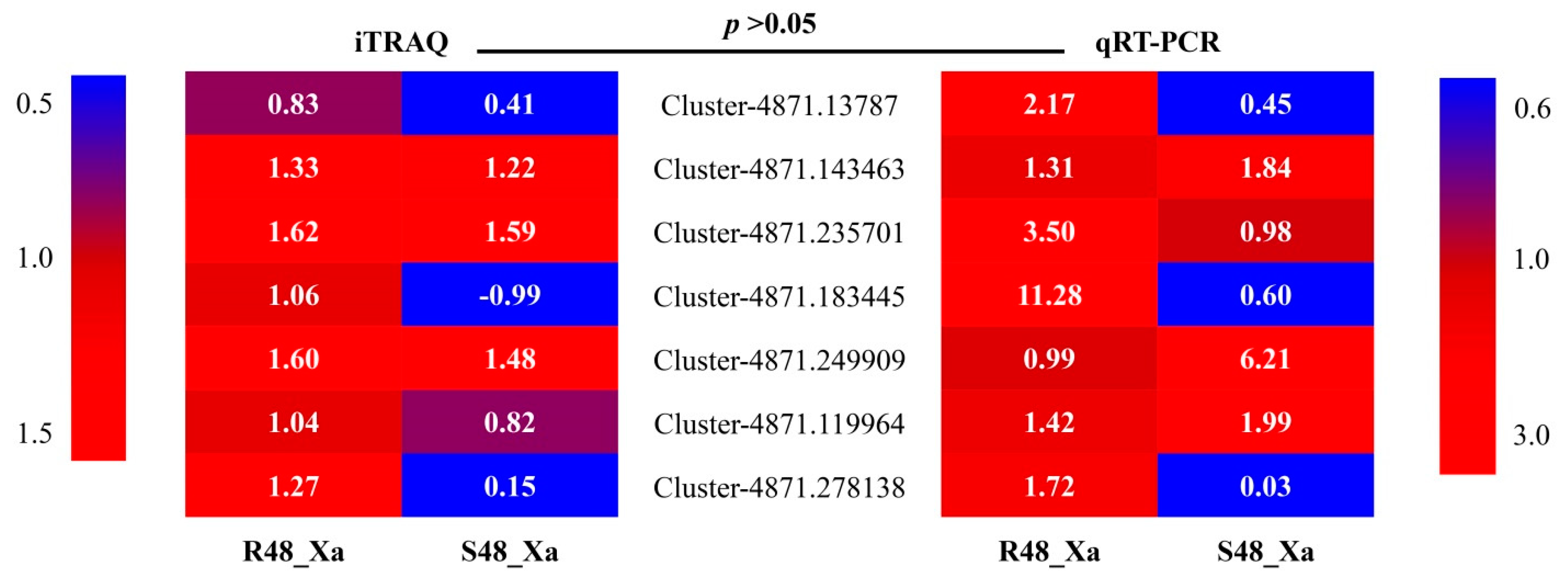
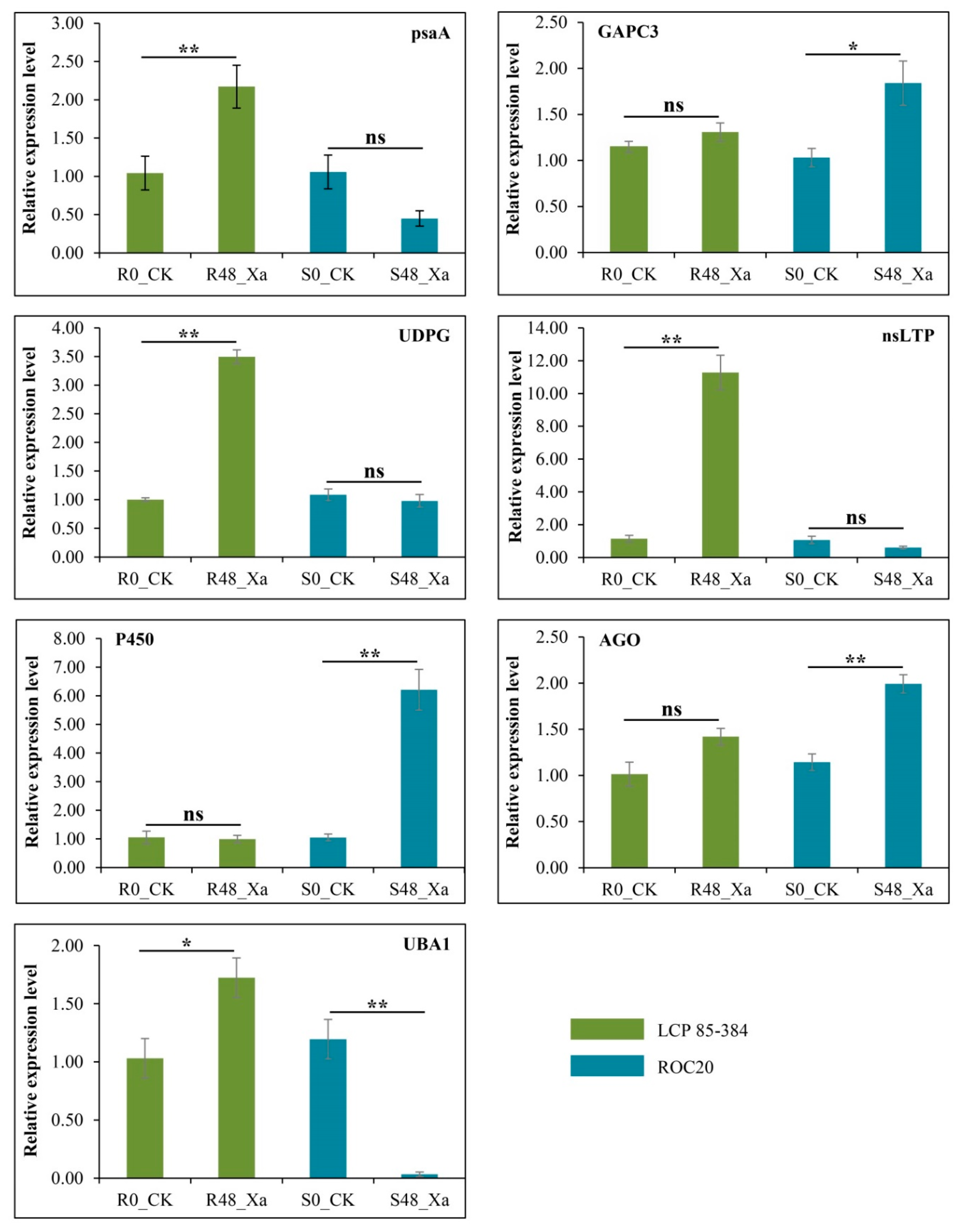
3.8. Transcript Profiling of Seven Selected Genes by qRT-PCR
4. Discussion
4.1. Overall Assessment of DEPs Involved in Response to X. albilineans Infection
4.2. Regulation of Photosynthesis and Glycolytic Pathways of Sugarcane in Response to X. albilineans Infection
4.3. Activation of Plant Innate Immune Systems in Sugarcane after Inoculation with X. albilineans
4.4. High Level Expression of the UDP-Glycosyltransferase in the Sugarcane Cultivar Resistant to Leaf Scald
4.5. Different Expression Patterns of Plant Non-Specific Lipid Transfer Proteins (nsLTPs) in the Leaf Scald Resistant and the Leaf Scald Susceptible Sugarcane Cultivar
4.6. Upregulation of the Plant Cytochrome P450 after Colonization of Sugarcane by X. albilineans
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ali, A.; Khan, M.; Sharif, R.; Mujtaba, M.; Gao, S.J. Sugarcane Omics: An update on the current status of research and crop improvement. Plants 2019, 8, 344. [Google Scholar] [CrossRef] [Green Version]
- D’Hont, A.; Grivet, L.; Feldmann, P.; Rao, S.; Berding, N.; Glaszmann, J.C. Characterisation of the double genome structure of modern sugarcane cultivars (Saccharum spp.) by molecular cytogenetics. Mol. Gen. Genet. 1996, 250, 405–413. [Google Scholar] [CrossRef]
- Zhang, J.S.; Zhang, X.T.; Tang, H.B.; Zhang, Q.; Hua, X.T.; Ma, X.K.; Zhu, F.; Jones, T.; Zhu, X.G.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef] [Green Version]
- Diniz, A.L.; Ferreira, S.S.; Caten, F.T.; Margarido, G.R.A.; Dos Santos, J.M.; Barbosa, G.V.; Carneiro, M.S.; Souza, G.M. Genomic resources for energy cane breeding in the post genomics era. Comput. Struct. Biotechnol. J. 2019, 10, 1404–1414. [Google Scholar] [CrossRef]
- Garsmeur, O.; Droc, G.; Antonise, R.; Grimwood, J.; Potier, B.; Aitken, K.; Jenkins, J.; Martin, G.; Charron, C.; Hervouet, C.; et al. A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat. Commun. 2018, 9, 2638. [Google Scholar] [CrossRef]
- Souza, G.M.; Van Sluys, M.A.; Lembke, C.G.; Lee, H.; Margarido, G.R.A.; Hotta, C.T.; Gaiarsa, J.W.; Diniz, A.L.; Oliveira, M.M.; Ferreira, S.S.; et al. Assembly of the 373k gene space of the polyploid sugarcane genome reveals reservoirs of functional diversity in the world’s leading biomass crop. Gigascience 2019, 8, giz129. [Google Scholar] [CrossRef]
- Lal, M.; Tiwari, A.K.; Gupta, G.N. Commercial scale micropropagation of sugarcane: Constraints and remedies. Sugar Tech. 2015, 17, 339–347. [Google Scholar] [CrossRef]
- Rott, P.C.; Kaye, C.; Naranjo, M.; Shine, J.M.; Sood, S.; Comstock, J.C.; Raid, R.N. Controlling sugarcane diseases in Florida: A challenge in constant evolution. Int. Sugar J. 2018, 2, 274–279. [Google Scholar]
- Rott, P.; Davis, M.J. Leaf scald. In A Guide to Sugarcane Diseases; Rott, P., Bailey, R.A., Comstock, J.C., Croft, B.J., Saumtally, A.S., Eds.; CIRAD/ISSCT, La Librarie du Cirad: Montpellier, France, 2000; pp. 38–44. [Google Scholar]
- Lin, L.H.; Ntambo, M.S.; Rott, P.C.; Wang, Q.N.; Lin, Y.H.; Fu, H.Y.; Gao, S.J. Molecular detection and prevalence of Xanthomonas albilineans, the causal agent of sugarcane leaf scald, in China. Crop. Prot. 2018, 109, 17–23. [Google Scholar] [CrossRef]
- Zhang, R.-Y.; Wang, X.-Y.; Shan, H.-L.; Li, J.; Li, W.-F.; Cang, X.-Y.; Luo, Z.-M.; Yin, J.; Huang, Y.-K. Identification and phylogenetic analysis of Xanthomonas albilineans (Ashby) Downson based on multiple gene sequences in Yunnan province, China. Sugar Tech. 2019, 21, 794–801. [Google Scholar] [CrossRef]
- Ntambo, M.S.; Meng, J.Y.; Rott, P.C.; Royer, M.; Lin, L.H.; Zhang, H.L.; Gao, S.J. Identification and characterization of Xanthomonas albilineans causing sugarcane leaf scald in China using multilocus sequence analysis. Plant Pathol. 2019, 68, 269–277. [Google Scholar] [CrossRef]
- Champoiseau, P.; Daugrois, J.H.; Pieretti, I.; Cociancich, S.; Royer, M.; Rott, P. High variation in pathogenicity of genetically closely related strains of Xanthomonas albilineans, the sugarcane leaf scald pathogen, in Guadeloupe. Phytopathology 2006, 96, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Rott, P.; Marguerettaz, M.; Fleites, L.; Cociancich, S.; Girard, J.C.; Pieretti, I.; Gabriel, D.W.; Royer, M. Unravelling pathogenicity of Xanthomonas albilineans, the causal agent of sugarcane leaf scald. Int. Sugar J. 2011, 113, 490–496. [Google Scholar]
- Hu, J.; Rampitsch, C.; Bykova, N.V. Advances in plant proteomics toward improvement of crop productivity and stress resistancex. Front. Plant Sci. 2015, 6, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosova, K.; Vitamvas, P.; Prasil, I.T.; Renaut, J. Plant proteome changes under abiotic stress-contribution of proteomics studies to understanding plant stress response. J. Proteom. 2011, 74, 1301–1322. [Google Scholar] [CrossRef]
- Barnabas, L.; Ramadass, A.; Amalraj, R.S.; Palaniyandi, M.; Rasappa, V. Sugarcane proteomics: An update on current status, challenges, and future prospects. Proteomics 2015, 15, 1658–1670. [Google Scholar] [CrossRef]
- Ross, P.L.; Huang, Y.L.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [Green Version]
- Zieske, L.R. A perspective on the use of iTRAQ (TM) reagent technology for protein complex and profiling studies. J. Exp. Bot. 2006, 57, 1501–1508. [Google Scholar] [CrossRef]
- Su, Y.C.; Xu, L.P.; Wang, Z.Q.; Peng, Q.; Yang, Y.T.; Chen, Y.; Que, Y.X. Comparative proteomics reveals that central metabolism changes are associated with resistance against Sporisorium scitamineum in sugarcane. BMC Genom. 2016, 17, 800. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.Q.; Singh, P.; Singh, R.K.; Song, X.P.; Li, H.P.; Nong, Y.Y.; Yang, L.T.; Li, Y.R. Proteomic analysis of sugarcane-sporisorium scitamineum interaction based on iTRAQ technique. Acta Agron. Sin. 2019, 45, 55–69. [Google Scholar] [CrossRef]
- Milligan, S.B.; Martin, F.A.; Bischoff, K.P.; Quebedeaux, J.P.; Dufrene, E.O.; Quebedeaux, K.L.; Hoy, J.W.; Reagan, T.E.; Legendre, B.L. Registration of ‘LCP 85-384′ sugarcane. Crop Sci. 1994, 34, 819–820. [Google Scholar] [CrossRef]
- Peng, S.G. Varietal evolution of sugarcane in Taiwan. Southwest China J. Agric. Sci. 1996, 9, 117–124. [Google Scholar]
- Ntambo, M.S.; Meng, J.Y.; Rott, P.C.; Henry, R.J.; Zhang, H.L.; Gao, S.J. Comparative transcriptome profiling of resistant and susceptible sugarcane cultivars in response to infection by Xanthomonas albilineans. Int. J. Mol. Sci. 2019, 20, 6138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garces, F.F.; Gutierrez, A.; Hoy, J.W. Detection and quantification of Xanthomonas albilineans by qPCR and potential characterization of sugarcane resistance to leaf scald. Plant Dis. 2014, 98, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Dixon, R.A. Natural products and plant disease resistance. Nature 2001, 411, 843–847. [Google Scholar] [CrossRef]
- Rojas, C.M.; Senthil-Kumar, M.; Tzin, V.; Mysore, K.S. Regulation of primary plant metabolism during plant-pathogen interactions and its contribution to plant defense. Front. Plant Sci. 2014, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Saddique, M.; Kamran, M.; Shahbaz, M. Differential responses of plants to biotic stress and the role of metabolites. In Plant Metabolites and Regulation under Environmental Stress; Ahmad, P., Ahanger, M.A., Singh, V.P., Tripathi, D.K., Alam, P., Alyemeni, M.N., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 69–87. [Google Scholar] [CrossRef]
- Barón, M.; Flexas, J.; Delucia, E.H. Photosynthetic responses to biotic stress. In Terrestrial Photosynthesis in a Changing Environment: A Molecular, Physiological and Ecological Approach; Flexas, J., Loreto, F., Medrano, H., Eds.; Cambridge University Press: Cambridge, UK, 2012; pp. 331–350. [Google Scholar] [CrossRef]
- Bilgin, D.D.; Zavala, J.A.; Zhu, J.; Clough, S.J.; Ort, D.R.; DeLucia, E.H. Biotic stress globally downregulates photosynthesis genes. Plant Cell Environ. 2010, 33, 1597–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redding, K.; MacMillan, F.; Leibl, W.; Brettel, K.; Hanley, J.; Rutherford, A.W.; Breton, J.; Rochaix, J.D. A systematic survey of conserved histidines in the core subunits of Photosystem I by site-directed mutagenesis reveals the likely axial ligands of P700. EMBO J. 1998, 17, 50–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leister, D.; Schneider, A. From genes to photosynthesis in Arabidopsis thaliana. Int. Rev. Cytol. 2003, 228, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Santa Brigida, A.B.; Rojas, C.A.; Grativol, C.; De Armas, E.M.; Entenza, J.O.; Thiebaut, F.; Lima, M.F.; Farrinelli, L.; Hemerly, A.S.; Lifschitz, S.; et al. Sugarcane transcriptome analysis in response to infection caused by Acidovorax avenae subsp. avenae. PLoS ONE 2016, 11, e0166473. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Z.; Gu, Q.; Li, H.; Han, W.; Shi, Y. Transcriptome analysis of Cucumis sativus infected by Cucurbit chlorotic yellows virus. Virol. J. 2017, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.; Knuesting, J.; Birkholz, O.; Heinisch, J.J.; Scheibe, R. Cytosolic GAPDH as a redox-dependent regulator of energy metabolism. BMC Plant Biol. 2018, 18, 184. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.F.; Deng, R.; Guo, Z.P.; Yang, S.S.; Deng, X.P. Genome-wide identification and characterization of Glyceraldehyde-3-phosphate dehydrogenase genes family in wheat (Triticum aestivum). BMC Genom. 2016, 17, 240. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.Q.; Xie, Y.W.; Liu, G.Y.; Lin, D.Z.; He, C.Z.; Shi, H.T. Molecular identification of GAPDHs in cassava highlights the antagonism of MeGAPCs and MeATG8s in plant disease resistance against cassava bacterial blight. Plant Mol. Biol. 2018, 97, 201–214. [Google Scholar] [CrossRef]
- Goritschnig, S.; Zhang, Y.L.; Li, X. The ubiquitin pathway is required for innate immunity in Arabidopsis. Plant J. 2007, 49, 540–551. [Google Scholar] [CrossRef]
- Marino, D.; Peeters, N.; Rivas, S. Ubiquitination during plant immune signaling. Plant Physiol. 2012, 160, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chai, Q.Y.; Liu, C.H. The ubiquitin system: A critical regulator of innate immunity and pathogen-host interactions. Cell. Mol. Immunol. 2016, 13, 560–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, P.M.; Gosink, M.M.; Carpenter, T.B.; Vierstra, R.D. The ubiquitin-activating enzyme (E1) gene family in Arabidopsis thaliana. Plant J. 1997, 11, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Callis, J. The ubiquitination machinery of the ubiquitin system. Arab. Book 2014, 12, e0174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takizawa, M.; Goto, A.; Watanabe, Y. The tobacco ubiquitin-activating enzymes NtE1A and NtE1B are induced by Tobacco mosaic virus, wounding and stress hormones. Mol. Cells 2005, 19, 228–231. [Google Scholar] [PubMed]
- Zhang, C.Y.; Song, L.; Choudhary, M.K.; Zhou, B.J.; Sun, G.C.; Broderick, K.; Giesler, L.; Zeng, L.R. Genome-wide analysis of genes encoding core components of the ubiquitin system in soybean (Glycine max) reveals a potential role for ubiquitination in host immunity against soybean cyst nematode. BMC Plant Biol. 2018, 18, 149. [Google Scholar] [CrossRef] [PubMed]
- Pumplin, N.; Voinnet, O. RNA silencing suppression by plant pathogens: Defence, counter-defence and counter-counter-defence. Nat. Rev. Microbiol. 2013, 11, 745–760. [Google Scholar] [CrossRef]
- Axtell, M.J. Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, F. Arabidopsis endogenous small RNAs: Highways and byways. Trends Plant Sci. 2006, 11, 460–468. [Google Scholar] [CrossRef]
- Au, P.C.K.; Dennis, E.S.; Wang, M.B. Analysis of argonaute 4-associated long non-coding RNA in Arabidopsis thaliana sheds novel insights into gene regulation through RNA-directed DNA methylation. Genes 2017, 8, 198. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; He, X.; Wang, X.J.; Kohany, O.; Jurka, J.; Hannon, G.J. Distinct catalytic and non-catalytic roles of ARGONAUTE4 in RNA-directed DNA methylation. Nature 2006, 443, 1008–1012. [Google Scholar] [CrossRef]
- Vaucheret, H. Plant ARGONAUTES. Trends Plant Sci. 2008, 13, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Agorio, A.; Vera, P. ARGONAUTE4 is required for resistance to Pseudomonas syringae in Arabidopsis. Plant Cell 2007, 19, 3778–3790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.Y.; Xu, Y.P.; Li, W.; Li, S.S.; Rahman, H.; Cai, X.Z. Genome-wide identification of dicer-like, argonaute, and RNA-dependent RNA polymerase gene families in Brassica species and functional analyses of their Arabidopsis homologs in resistance to Sclerotinia sclerotiorum. Front. Plant Sci. 2016, 7, 1614. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.; Isayenkova, J.; Lim, E.K.; Poppenberger, B. Glycosyltransferases: Managers of small molecules. Curr. Opin. Plant Biol. 2005, 8, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Brazier-Hicks, M.; Gershater, M.; Dixon, D.; Edwards, R. Substrate specificity and safener inducibility of the plant UDP-glucose-dependent family 1 glycosyltransferase super-family. Plant Biotechnol. J. 2018, 16, 337–348. [Google Scholar] [CrossRef]
- Vogt, T.; Jones, P. Glycosyltransferases in plant natural product synthesis: Characterization of a supergene family. Trends Plant Sci. 2000, 5, 380–386. [Google Scholar] [CrossRef]
- Le Roy, J.; Huss, B.; Creach, A.; Hawkins, S.; Neutelings, G. Glycosylation is a major regulator of phenylpropanoid availability and biological activity in plants. Front. Plant Sci. 2016, 7, 735. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.Y.; Hu, F.; Dossa, K.; Wang, Z.K.; Ke, T. Genome-wide analysis of UDP-glycosyltransferase super family in Brassica rapa and Brassica oleracea reveals its evolutionary history and functional characterization. BMC Genom. 2017, 18, 474. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.E.; Tian, L. Phylogenomic analysis of UDP-dependent glycosyltransferases provides insights into the evolutionary landscape of glycosylation in plant metabolism. Plant J. 2019. [Google Scholar] [CrossRef]
- Ma, X.; Du, X.Y.; Liu, G.J.; Yang, Z.D.; Hou, W.Q.; Wang, H.W.; Feng, D.S.; Li, A.F.; Kong, L.R. Cloning and characterization of a novel UDP-glycosyltransferase gene induced by DON from wheat. J. Integr. Agric. 2015, 14, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Ma, X.; Su, P.; Ge, W.; Wu, H.; Guo, X.; Li, A.; Wang, H.; Kong, L. Cloning and characterization of a specific UDP-glycosyltransferase gene induced by DON and Fusarium graminearum. Plant Cell Rep. 2018, 37, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.P.; He, L.Q.; Xiao, J.; Chen, Q.G.; Li, M.H.; Shang, Y.; Zhu, Y.F.; Chen, P.D.; Cao, A.Z.; Wang, X.E. An UDP-glucosyltransferase gene from barley confers disease resistance to Fusarium head blight. Plant Mol. Biol. Rep. 2017, 35, 224–236. [Google Scholar] [CrossRef]
- Zhang, Y.; Huai, D.; Yang, Q.; Cheng, Y.; Ma, M.; Kliebenstein, D.J.; Zhou, Y. Overexpression of three glucosinolate biosynthesis genes in Brassica napus identifies enhanced resistance to Sclerotinia sclerotiorum and Botrytis cinerea. PLoS ONE 2015, 10, e0140491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.P.; Sattely, E.S. Biosynthesis of cabbage phytoalexins from indole glucosinolate. Proc. Natl. Acad. Sci. USA 2017, 114, 1910–1915. [Google Scholar] [CrossRef] [Green Version]
- Fontaniella, B.; Marquez, A.; Rodriguez, C.W.; Pinon, D.; Solas, M.T.; Vicente, C.; Legaz, M.E. A role for sugarcane glycoproteins in the resistance of sugarcane to Ustilago scitaminea. Plant Physiol. Biochem. 2002, 40, 881–889. [Google Scholar] [CrossRef]
- Carvalho, A.D.; Gomes, V.M. Role of plant lipid transfer proteins in plant cell physiology—A concise review. Peptides 2007, 28, 1144–1153. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, X.B.; Lu, C.M.; Zeng, X.H.; Li, Y.J.; Fu, D.H.; Wu, G. Non-specific lipid transfer proteins in plants: Presenting new advances and an integrated functional analysis. J. Exp. Bot. 2015, 66, 5663–5681. [Google Scholar] [CrossRef]
- Palmieri, M.C.; Perazzolli, M.; Matafora, V.; Moretto, M.; Bachi, A.; Pertot, I. Proteomic analysis of grapevine resistance induced by Trichoderma harzianum T39 reveals specific defence pathways activated against downy mildew. J. Exp. Bot. 2012, 63, 6237–6251. [Google Scholar] [CrossRef]
- Dmochowska-Boguta, M.; Alaba, S.; Yanushevska, Y.; Piechota, U.; Lasota, E.; Nadolska-Orczyk, A.; Karlowski, W.M.; Orczyk, W. Pathogen-regulated genes in wheat isogenic lines differing in resistance to brown rust Puccinia triticina. BMC Genom. 2015, 16, 742. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fan, P.; Yun, Z.; Jiang, G.; Zhang, Z.; Jiang, Y. β-aminobutyric acid priming acquisition and defense response of mango fruit to Colletotrichum gloeosporioides infection based on quantitative proteomics. Cells 2019, 8, 1029. [Google Scholar] [CrossRef] [Green Version]
- Rasool, S.; Mohamed, R. Plant cytochrome P450s: Nomenclature and involvement in natural product biosynthesis. Protoplasma 2016, 253, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Ralston, L.; Yu, O. Metabolons involving plant cytochrome P450s. Phytochem. Rev. 2006, 5, 459–472. [Google Scholar] [CrossRef]
- Ghosh, S. Triterpene structural diversification by plant cytochrome P450 enzymes. Front. Plant Sci. 2017, 8, 1886. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.Y.; Tang, W.Q.; He, W.Y.; Ma, X.L.; Vasseur, L.; Baxter, S.W.; Yang, G.; Huang, S.G.; Song, F.Q.; You, M.S. Characterization and expression of the cytochrome P450 gene family in diamondback moth, Plutella xylostella (L.). Sci. Rep. 2015, 5, 8952. [Google Scholar] [CrossRef]
- Pinot, F.; Beisson, F. Cytochrome P450 metabolizing fatty acids in plants: Characterization and physiological roles. FEBS J. 2011, 278, 195–205. [Google Scholar] [CrossRef]
- Kandel, S.; Morant, M.; Benveniste, I.; Blée, E.; Werck-Reichhart, D.; Pinot, F. Cloning, functional expression, and characterization of CYP709C1, the first sub-terminal hydroxylase of long chain fatty acid in plants. J. Biol. Chem. 2005, 280, 35881–35889. [Google Scholar] [CrossRef] [Green Version]
- Stumpe, M.; Feussner, I. Formation of oxylipins by CYP74 enzymes. Phytochem. Rev. 2006, 5, 347–357. [Google Scholar] [CrossRef]
- Koo, A.J.K.; Cooke, T.F.; Howe, G.A. Cytochrome P450 CYP94B3 mediates catabolism and inactivation of the plant hormone jasmonoyl-L-isoleucine. Proc. Natl. Acad. Sci. USA 2011, 108, 9298–9303. [Google Scholar] [CrossRef] [Green Version]
- Kombrink, E. Chemical and genetic exploration of jasmonate biosynthesis and signaling paths. Planta 2012, 236, 1351–1366. [Google Scholar] [CrossRef]
- Heitz, T.; Smirnova, E.; Marquis, V.; Poirier, L. Metabolic control within the jasmonate biochemical pathway. Plant Cell Physiol. 2019. [Google Scholar] [CrossRef]
- Nelson, D.R. Cytochrome P450s in the sugarcane Saccharum spontaneum. Trop. Plant Biol. 2019, 12, 1–8. [Google Scholar] [CrossRef]
- Gentile, A.; Ditt, R.F.; Dias, F.O.; Da Silva, M.J.; Dornelas, M.C.; Menossi, M. Characterization of ScMat1, a putative TFIIH subunit from sugarcane. Plant Cell Rep. 2009, 28, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Huang, L.; Huang, N.; Liu, F.; Su, Y.; Xiao, X.; Ling, H.; Que, Y. RT-PCR amplification and expression analysis of a cytochrome P450 reductase gene from sugarcane. Chin. J. Appl. Environ. Biol. 2016, 22, 173–178. [Google Scholar] [CrossRef]
- Wang, M.; Gu, Z.; Wang, R.; Guo, J.; Ling, N.; Firbank, L.G.; Guo, S. Plant primary metabolism regulated by nitrogen contributes to plant-pathogen interactions. Plant Cell Physiol. 2019, 60, 329–342. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run Name a | Total Spectra | Number of Peptides | Number of Proteins |
|---|---|---|---|
| Run1 | 574,559 | 21,703 | 6126 |
| Run2 | 548,154 | 19,350 | 5463 |
| All | 6891 |
| Pathway ID | Pathway Name | R48_Xa vs. R0_CK | S48_Xa vs. S0_CK | ||
|---|---|---|---|---|---|
| Upregulation | Downregulation | Upregulation | Downregulation | ||
| ko01100 | Metabolic pathways | 27 | 0 | 24 | 11 |
| ko01110 | Biosynthesis of secondary metabolites | 16 | 0 | 13 | 0 |
| ko00940 | Phenylpropanoid biosynthesis | 7 | 0 | 2 | 0 |
| ko03013 | RNA transport | 4 | 0 | 0 | 0 |
| ko00250 | Alanine, aspartate and glutamate metabolism | 3 | 1 | 3 | 0 |
| ko00520 | Amino sugar and nucleotide sugar metabolism | 3 | 1 | 2 | 2 |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 3 | 0 | 0 | 0 |
| ko00480 | Glutathione metabolism | 2 | 1 | 0 | 0 |
| ko00561 | Glycerolipid metabolism | 2 | 0 | 1 | 0 |
| ko03420 | Nucleotide excision repair | 2 | 0 | 1 | 0 |
| ko00511 | Other glycan degradation | 2 | 0 | 2 | 0 |
| ko04146 | Peroxisome | 2 | 0 | 2 | 0 |
| ko00230 | Purine metabolism | 2 | 0 | 3 | 0 |
| ko04120 | Ubiquitin mediated proteolysis | 1 | 1 | 2 | 0 |
| ko00650 | Butanoate metabolism | 1 | 1 | 1 | 0 |
| ko00195 | Photosynthesis | 1 | 2 | 0 | 1 |
| ko04075 | Plant hormone signal transduction | 1 | 0 | 1 | 0 |
| ko00640 | Propanoate metabolism | 1 | 0 | 2 | 0 |
| ko00280 | Valine, leucine and isoleucine degradation | 2 | 0 | 1 | 0 |
| ko00920 | Sulfur metabolism | 0 | 2 | 0 | 3 |
| ko03010 | Ribosome | 0 | 4 | 0 | 2 |
| ko00270 | Cysteine and methionine metabolism | 0 | 2 | 0 | 1 |
| ko01230 | Biosynthesis of amino acids | 0 | 0 | 0 | 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, J.-Y.; Ntambo, M.S.; Rott, P.C.; Fu, H.-Y.; Huang, M.-T.; Zhang, H.-L.; Gao, S.-J. Identification of Differentially Expressed Proteins in Sugarcane in Response to Infection by Xanthomonas albilineans Using iTRAQ Quantitative Proteomics. Microorganisms 2020, 8, 76. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8010076
Meng J-Y, Ntambo MS, Rott PC, Fu H-Y, Huang M-T, Zhang H-L, Gao S-J. Identification of Differentially Expressed Proteins in Sugarcane in Response to Infection by Xanthomonas albilineans Using iTRAQ Quantitative Proteomics. Microorganisms. 2020; 8(1):76. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8010076
Chicago/Turabian StyleMeng, Jian-Yu, Mbuya Sylvain Ntambo, Philippe C. Rott, Hua-Ying Fu, Mei-Ting Huang, Hui-Li Zhang, and San-Ji Gao. 2020. "Identification of Differentially Expressed Proteins in Sugarcane in Response to Infection by Xanthomonas albilineans Using iTRAQ Quantitative Proteomics" Microorganisms 8, no. 1: 76. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8010076