Riboregulation in Nitrogen-Fixing Endosymbiotic Bacteria


1
Intergenomics Group, Departamento de Biología Molecular, Instituto de Biomedicina y Biotecnología de Cantabria, CSIC-Universidad de Cantabria-Sodercan, 39011 Santander, Spain
2
Structure, Dynamics and Function of Rhizobacterial Genomes (Grupo de Ecología Genética de la Rizosfera), Estación Experimental del Zaidín, Consejo Superior de Investigaciones Científicas (CSIC), 18008 Granada, Spain
*
Author to whom correspondence should be addressed.
Microorganisms 2020, 8(3), 384; https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8030384
Submission received: 22 January 2020
/
Revised: 4 March 2020
/
Accepted: 5 March 2020
/
Published: 10 March 2020
(This article belongs to the Special Issue Plant Microbial Interactions)
Abstract
:Small non-coding RNAs (sRNAs) are ubiquitous components of bacterial adaptive regulatory networks underlying stress responses and chronic intracellular infection of eukaryotic hosts. Thus, sRNA-mediated regulation of gene expression is expected to play a major role in the establishment of mutualistic root nodule endosymbiosis between nitrogen-fixing rhizobia and legume plants. However, knowledge about this level of genetic regulation in this group of plant-interacting bacteria is still rather scarce. Here, we review insights into the rhizobial non-coding transcriptome and sRNA-mediated post-transcriptional regulation of symbiotic relevant traits such as nutrient uptake, cell cycle, quorum sensing, or nodule development. We provide details about the transcriptional control and protein-assisted activity mechanisms of the functionally characterized sRNAs involved in these processes. Finally, we discuss the forthcoming research on riboregulation in legume symbionts.
1. Introduction
Some soil-dwelling species of the large classes of α- and β-proteobacteria, collectively referred to as rhizobia, establish mutualistic symbioses with legumes. The outcome of these interactions is the organogenesis of specialized nodule structures on the roots or, less frequently, the stems of their specific host plant. Invading rhizobia colonize nodules intracellularly and differentiate to bacteroids that achieve the nitrogenase-mediated reduction of the atmospheric dinitrogen to ammonia for the benefit of the plant. Symbiotic nitrogen (N) fixation thus renders plant growth independent of exogenously applied combined N, which is commonly provided to crops in the form of pollutant and costly chemical fertilizers. Besides this doubtless agronomical and ecological significance, the rhizobia–legume symbiosis provides a complex biological experimental model to explore the molecular mechanisms underlying bacterial adaptations during chronic intracellular infection of eukaryotic hosts [1,2,3].
The saprophytic and symbiotic competence of free-living rhizobia in soil is largely determined by their capacity to cope efficiently with the abiotic variables shaping this environment, e.g., oligotrophy, drought, salinity or acidity, which are known to influence negatively the physiology of both symbiotic partners and the N fixation process itself [4]. Competitive rhizobial strains actively colonize the rhizosphere of their compatible legume and initiate infection upon synthesis of lipo-chitooligosaccharide signal molecules (i.e., Nod factors) in response to species-specific root exuded flavonoids [3,5]. Nod factor signaling triggers major nodule developmental pathways and root hair invasion through tubular structures made of plant cell-wall material known as infection threads. Besides Nod factors, other signaling molecules of bacterial origin such as surface polysaccharides or effector proteins selectively contribute to the infection of certain legume species by their rhizobial partners [6]. During early symbiotic infection, invading rhizobia elicit a defense response in the host, which is initially featured by the release of reactive oxygen species (ROS) [7]. Compatible rhizobia counteract and survive this oxidative stress by specific mechanisms, being subsequently delivered from the branched infection threads into the cells of the nodule primordia. Inside the plant cells, rhizobia differentiate into N-fixing polyploid bacteroids that end up surrounded by a membrane of plant origin. In some legume lineages, irreversible terminal bacteroid differentiation is directed by plant-secreted nodule-specific cysteine rich (NCR) peptides [8]. Finally, mature bacteroids sense the microoxic environment inside the plant nodule, driving expression of the nitrogenase-coding gene clusters. This complex rhizobial lifestyle demands an adaptive flexibility that is supported by both large genomes and continuous gene expression shifts during host infection, cell cycle progression or in response to nutrient availability [9,10]. To date, symbiotic regulatory networks in rhizobia have been studied almost exclusively from the perspective of the transcriptional control orchestrated by proteins, i.e., transcription factors and alternative RNA polymerase holoenzymes (σ factors) [11]. Indeed, the inventory of genes encoding transcription-related proteins is particularly rich in rhizobial genomes. However, proteins are far from being the only players in the regulation of gene expression in bacteria.
The development of high-throughput sequencing technologies has revolutionized our classic view of the prokaryotic transcriptome. Different, well-established strategies for the generation and deep-sequencing of cDNA libraries (RNA-Seq) allow for genome-wide mapping of transcription start sites (TSS), thus uncovering unexpected operon structures and novel transcribed regions systematically overlooked in the primary genome annotation [12] (Figure 1). Many of these newly discovered transcripts are not translated into proteins, but fulfil important regulatory roles, especially in the adaptation of bacteria to fluctuating environments [13]. Bacterial non-coding transcriptomes typically consist of heterogeneous populations of 50 to 250 nt-long RNA species (sRNAs) with diverse biogenesis pathways and regulatory activity mechanisms. Some untranslated regions at 5’-ends (5’-UTRs) of mRNAs sense shifts in temperature (RNA thermometers) or intracellular levels of certain metabolites (riboswitches) to modulate transcription and/or translation of the downstream coding sequence in a structure-dependent manner. Few other sRNAs mimic sequence or structural DNA or RNA motifs recognized by specific proteins to outcompete them at their cognate cellular targets. Nonetheless, the activity of most sRNAs thus far characterized relies on base pairing interactions to fine-tune translation and/or turnover rates of mRNA targets that can be either antisense-transcribed (asRNAs) or encoded in trans (trans-sRNAs). Complementarity between the asRNA and its mRNA target is perfect and may extend over regions even longer than 200-nt. Conversely, regulation by trans-sRNAs typically involves short and discontinuous series of complementary nucleotides in both molecules, requiring the assistance of proteins (e.g., Hfq) as RNA matchmakers [14]. Regardless of the type of sRNA involved in regulation and the possible effect on translation, formation of RNA duplexes through base pairing usually promote degradation of the mRNA targets by cellular ribonucleases (e.g., RNase E or RNase III). RNA-mediated post-transcriptional regulation of gene expression is ubiquitous in bacteria, influencing virtually any cellular process, e.g., stress responses, biofilm formation, quorum sensing, and other virulence traits [13].
Research on prokaryotic riboregulation has been pioneered in the early post-genomic era by work on the model bacterium Escherichia coli. Soon afterwards, studies in related enterobacteria (i.e., Salmonella spp.) and other clinically relevant microbes revealed the unprecedented prominent roles of sRNAs in the establishment of host-pathogen interactions [13]. In the last decade, functional RNomics is continuously providing new paradigms to the contribution of sRNAs to the ecological specializations of phylogenetically distant bacteria with complex lifestyles, bringing RNA to the forefront of microbial research. In legume symbionts, the knowledge about riboregulation is still scarce and rather limited to Sinorhizobium meliloti, the symbiotic partner of the forage legume alfalfa (Medicago sativa L.) and other related Medicago species [15,16,17]. Here, we summarize and discuss known insights into the structure, function and mechanistic principles of the rhizobial non-coding transcriptome.
2. Deciphering the Rhizobial Non-Coding Transcriptome: From Comparative Genomics to RNA-Seq
Non-protein coding genes largely escape classical genetics screens and the primary annotation of a single bacterial genome sequence, which is essentially limited to the prediction of open reading frames (ORFs), tRNAs, and rRNAs. Before the advent of high-throughput sequencing, computational comparative genomics was thus the tool of choice to identify conserved regions with putative functions in the unannotated portions of the genome. Accordingly, pioneering seminal genome-wide searches for sRNAs in rhizobia relied on the comparison of intergenic sequences (i.e., genomic regions between ORFs; IGRs) from phylogenetically close species to unveil sRNAs of the trans-acting class. Specifically, three studies published almost concurrently used the IGRs from the S. meliloti reference strain 1021 (Rm1021) as queries to interrogate the genomes of α–proteobacterial relatives, i.e., plant symbionts (e.g., Mesorhizobium loti, Rhizobium etli or R. leguminosarum bv. viciae), phytopathogens (Agrobacterium tumefaciens) and animal pathogens (Brucella species) [18,19,20]. All three searches combined genomic comparisons with other known features of trans-sRNAs, namely association with orphan transcription signatures (promoter motifs and/or Rho-independent transcriptional terminators) and conservation of thermodynamically stable secondary structures. Collectively, these approaches predicted more than a hundred IGRs in the three replicons of the Rm1021 genome (chromosome, and pSymA and pSymB megaplasmids) putatively encoding a trans-sRNA. Northern blot hybridization of total RNA, RACE (Rapid Amplification of cDNA Ends) mapping of transcripts boundaries and/or microarray probing experimentally confirmed that a few dozens of these candidate IGRs did express sRNA species from independent transcription units. These studies also anticipated the symbiotic and/or stress-dependent expression of subsets of the identified sRNAs. In the absence of further functional insights, these trans-sRNAs were initially referred to as either Smr [20], Sra [18] or Sm [19] (Table 1). A similar combination of in silico searches and experimental approaches also delivered the first inventories of sRNAs expressed by R. etli CFN42 (termed ReC sRNAs), Bradyrhizobium japonicum USDA110 (BjrC sRNAs) and M. huakuii 7653R (MH_s sRNAs) [21,22,23]. These rhizobia are the symbiotic partners of common bean (Phaseolus vulgaris), soybean (Glycine max), and milkvetch (Astragalus sinicus), respectively.
Inherent limitations to both computational searches and microarray designs necessarily biased these early genome-wide screens for sRNAs in rhizobia to the identification of putative intergenic, trans-acting, conserved riboregulators. Straightforward experimental identification of TSS associated to coding sequences, untranslated mRNA regions, and non-coding RNA genes in the prokaryotic genomes is now feasible with the implementation of oriented differential RNA-Seq (dRNA-Seq) [24] or Cappable-Seq [25] strategies. In particular, dRNA-Seq surveys rediscovered the early-identified sRNAs in S. meliloti and B. japonicum and further uncovered the complex rhizobial transcriptomes by adding hundreds of unknown trans-sRNAs, as well as thousands of mRNA-derived sRNAs and asRNAs [22,26,27,28,29]. Other RNA-Seq studies are conceived to profile specific subpopulations of cellular transcripts supposedly enriched in sRNAs, e.g., RNA species co-immunoprecipitated with the major bacterial RNA chaperone Hfq [30,31]. However, this approach resulted in a minor addition to the sRNA landscape revealed by deep-sequencing of S. meliloti total RNA [32].
Prokaryotic gene prediction pipelines such as EuGen-P have incorporated the novel gene structural features uncovered by dRNA-Seq for the accurate reannotation of the S. meliloti (strains Rm1021 and Rm2011) and B. japonicum USDA110 genomes [27,28,29]. Similar in silico workflows can be used to predict sRNA genes in any bacterial genome [33]. Even though dRNA-Seq mostly serves annotation purposes, comparison of transcripts levels in some datasets identified differentially expressed sRNAs in free-living and nodule endosymbiotic bacteria [26,28,29,32]. In this regard, it is noteworthy the identification of nodule-expressed sRNAs by RNA-Seq based profiling of RNA derived from each developing zone of indeterminate nodules induced on the model legume M. truncatula by Rm2011 [34]. On the other hand, comprehensive mapping of TSS in Rm2011 and USDA110 has facilitated the prediction of motifs putatively recognized by alternative σ factors such as RpoE2 (σE2) or RpoN (σ54) in the promoter regions of some of the identified sRNAs, thus placing these RNA regulators in major stress response and/or symbiotic regulons [27,28,29]. The integration of the updated genome annotation files, primary expression profiles, and promoter predictions (Table 1) provides a solid resource for the forthcoming investigation of the function of sRNAs in plant symbiotic bacteria.
3. Conservation of Rhizobial sRNAs: α-Proteobacterial sRNA Families
Reverse comparative RNomics contributes to unravel sRNA function by identifying either functionally characterized homologs to the query transcripts or conservation patterns potentially related to the particular lifestyle of phylogenetically close bacterial species. Homology predictions typically rely on stochastic covariance models (CMs) capturing both sequence and secondary structure (folding) conservation from a multiple alignment to the query sRNA. CMs can be automatically generated by INFERNAL, which builds RNA families collected by the Rfam database (https://rfam.xfam.org/) [35]. Regardless of their assignment to specific family models, a handful of chromosomally encoded rhizobial sRNAs that occur ubiquitously and exert housekeeping functions in bacteria can be unequivocally identified by the sole comparison of the primary nucleotide sequence. This set includes:
- RNase P (Rfam family model RF00010), which is the ribozyme that cleaves off 5’-extra sequences on tRNA precursors [36].
- tmRNA or SsrA (RF00023), which has a dual function as transfer and mRNA, operating in trans-translation for stalled ribosome recycling [37]. Of note, tmRNA has been the only rhizobial sRNA identified experimentally by a random mutagenesis screen for symbiotic genes in B. japonicum [38]. The S. meliloti tmRNA homolog is expressed in a growth- and stress-dependent manner as an unstable precursor, further processed into two readily detectable 214 and 82 nt-long RNA species likely derived from the mRNA and tRNA domains of the primary transcript [39].
- The 4.5S RNA (RF00169), which associates with the multidomain Ffh protein, in the bacterial SRP (small signal recognition particle) ribonucleoprotein complex, universally required for cotranslational protein targeting [40].
- The 6S RNA or SsrS (RF00013) RNA, which relies on an open promoter-like structure to counteract the activity of the σ70 RNA polymerase holoenzyme by a target mimicry mechanism, thus contributing to the transcriptional reprogramming during transition from exponential to stationary growth [41]. Interestingly, the 6S RNA is also required for the proper multiplication of the pathogen Legionella pneumophila within macrophages [42], which could anticipate novel yet unexplored wider roles of this riboregulator in both virulence and endosymbiosis.
CM-based phylogenetic distribution of other 57 S. meliloti trans-sRNAs without associated functional evidence has been also addressed by several independent studies [43,44,45,46]. In two particular cases where high secondary structure but low sequence conservation hindered construction of the CMs, RNA families were modeled using a complementary thermodynamic matchers (TDMs) based approach [43,44]. TDMs are RNA predictors that ignore sequence conservation in some parts, rather scoring the folding energy of conserved motifs elsewhere in the structure. Altogether, these analyses collected the 57 trans-sRNA input sequences into 43 family models, indicating that several of these sRNA loci occur with different levels of paralogy in the Rm1021 genome. Distribution of the majority of these families was restricted to members of the family Rhizobiaceae within α-proteobacteria, including Agrobacterium species. Only a few chromosomally encoded sRNAs are conserved beyond Rhizobiaceae, occurring in representatives of Brucellaceae or even Bartonellaceae, Xanthobacteriaceae, Beijerinckaceae, or Bradyrhizobiaceae. In contrast, sRNA loci mapping to S. meliloti pSymB or pSymA are almost unique to Sinorhizobium spp. and S. meliloti, respectively, further supporting that these two megaplasmids mostly encode accessory functions, with pSymA being the most recently acquired replicon in genus Sinorhizobium. Overall, the distribution patterns of these α–proteobacterial sRNA families hint at a major contribution of vertical inheritance and frequent ancestral duplications events to the evolution of these class of riboregulators in α-rhizobia. Table 1 compiles known genomic, conservation and expression data of conserved housekeeping and putative regulatory S. meliloti trans-sRNAs.
4. Assigning Functions to Rhizobial Trans-sRNAs
RNA-Seq and comparative genomics have delivered extensive repertoires of sRNAs in S. meliloti and related α–proteobacteria. The challenge now is to decipher the biological functions of this transcriptional output, which can be still regarded as dark matter of rhizobial genomes. In prokaryotes, a large fraction of these transcripts are catalogued as trans-sRNA regulators, which is an intensively investigated class of sRNAs since the early studies of the non-coding transcriptome in classical model bacteria [13]. The biology of bacterial trans-sRNAs is essentially characterised by their differential accumulation in response to environmental signals and target mRNA regulation by protein-assisted base-pairing mechanisms (Figure 2). Therefore, primary studies to unravel trans-sRNA functions must necessarily address expression profiling, transcriptional regulation, gain- and loss-of-function phenotyping, identification of mRNA targets and targeting motifs, and characterization of the associated proteins (e.g., RNA chaperones or RNases). Experimental and in silico approaches to functionally characterize trans-sRNAs in rhizobia are described with detail elsewhere [16,17,47,48]. Expression of trans-sRNAs can be tracked by classical molecular-genetics methods that include the use of promoter-reporter fusions and probing of total RNA on Northern blot membranes [47]. On the other hand, target identification typically relies on comparative genomics-based predictions of most probable conserved sRNA-mRNA base-pairing interactions (e.g., CopraRNA algorithm) followed by experimental validation of the candidates using a two-plasmid genetic reporter assay in vivo [17,47,49,50]. The latter is based on the constitutive co-expression from compatible plasmids in the same cell of the sRNA of interest and a translational fusion of the putative mRNA target to GFP, so that fluorescence of the reporter strains is related to sRNA-mediated translational repression or activation of the mRNA [17,47]. Co-expression of relevant sRNA and mRNA mutant variants in these assays enables the precise mapping of the interacting motifs in both molecules. Alternatively, trans-sRNA targetomes, i.e., the set of mRNA targets, can be characterized on a genome-wide scale by profiling of either transcriptome alterations upon pulse expression of the tested sRNA or the subset of mRNAs picked up by affinity chromatography using an aptamer-tagged (e.g., MS2) version of the sRNA as a bait [51,52]. Remarkably, this latter approach is also suitable to capture in vivo assembled sRNA-protein complexes, which can be further characterized by mass spectrometry analyses [48].
A combination of these strategies has been used to gain insights into the function and activity mechanisms of a handful of trans-sRNAs identified in S. meliloti. In the following paragraphs we describe how these sRNAs contribute to the regulation of cellular processes relevant to both the free-living and endosymbiotic rhizobial states.
4.1. Nutrient Uptake and Metabolism
Rhizobia must cope with a broad range of limiting nutrients in soil and specific nutritional needs in the rhizosphere of their host plants. Therefore, nodulation competitiveness first depends on the adequate regulation and coordination of efficient nutrient uptake and metabolism. Thus, it is not surprising the large number of ABC (ATP-binding cassette) transporters encoded by rhizobial genomes [9,53,54]. Acquisition of amino acids (aa), peptides, metal ions, or sugars through ABC transporters impacts important cellular processes such as differentiation, virulence or conjugation [55]. Remarkably, a selective suite of ABC systems is expressed in Rm1021-induced alfalfa nodules, thus supporting a specialized nutrient exchange between symbiont and host [56].
Nutrient uptake through ABC transporters primarily relies on periplasmic solute binding proteins (SBP) that determine substrate-specificity and are induced by its own ligand, typically two or more molecules [53,55]. This pervasive transcription of transporter genes suggests that rhizobia use post-transcriptional silencing mechanisms to quickly and accurately optimize metabolism with minimal energy costs. As proof of principle, the sRNA rnTrpL, which is produced from transcription attenuation of one of the three tryptophan (trp) biosynthesis operons, trpE(G), directly interacts with the polycistronic trpDC mRNA resulting in negative post-transcriptional regulation in S. meliloti. Considering that trp is the most costly-to-synthesize aa and the conservation of this mechanism in Agrobacterium and Bradyrhizobium but not in E. coli, this precedent provides an example of how soil bacteria have evolved post-transcriptional strategies to coordinate transport and metabolism in complex ecological systems as soils [57].
Riboregulation of nutrient uptake was first described in E. coli and Salmonella by characterization of the GcvB sRNAs, which regulon potentially includes ~1% of all mRNAs in these bacteria, with prevalence of mRNAs from ABC transporters of aa and short peptides, but also containing mRNAs coding for aa biosynthesis-related proteins and transcription factors [58]. Functionally analogous riboregulators were later discovered in the α-proteobacterial species A. tumefaciens, B. abortus and S. meliloti, and were termed AbcR sRNAs (ABC transporter RNA) [59,60,61,62,63]. S. meliloti Rm1021 encodes three AbcR homologs that belong to the αr15 sRNA family, whose members have partial homology to the SuhB sRNA (Rfam model RF00519) [46]. AbcR1 and AbcR2 are Hfq-binding 120 and 114 nt-long transcripts, respectively, that are encoded in tandem in the IGR flanked by the protein-coding genes SMc01226-lsrB (Table 1). Their predicted secondary structures consist of three stem loops (SLs) with two CU-rich anti-Shine Delgarno (aSD) motifs in unpaired regions of the molecules. These motifs are well conserved in AbcR homologs and are putatively responsible for the interaction with the ribosome binding site (RBS) and flanking nucleotides in mRNA targets, which commonly interferes with translation and promotes mRNA decay. The predicted lists of targets for both trans-sRNAs are enriched in ABC transporter mRNAs (35–45% of the top scored). Proteomics and further genetic verification identified prbA, SMa0495 and livK, all coding for the SBP of aa transporters, as common targets of these sRNAs- except livK that is specifically repressed by AbcR1 [32,62].
AbcR1 and AbcR2 expression patterns are divergent, suggesting differential transcriptional regulation from independent promoters. Indeed, both 5’-end RACE mapping and dRNA-Seq consistently identified a TSS for each transcript [20,27]. AbcR1 is accumulated in exponentially growing bacteria and in the nodule invasion zone II, which accommodates undifferentiated actively dividing rhizobia. In contrast, AbcR2 is highly induced in late stationary phase and other stress conditions, but not in nodules [62]. The molecular determinants of the transcriptional regulation of these S. meliloti sRNAs are unknown. Analysis of the abcR2 promoter shows high conservation of motifs recognized by the heat-shock σ factor RpoH, whereas the Brucella abcR1 is directly and positively regulated by the LsrB homolog VltR, encoded by the neighbor gene. Interestingly, while VltR is involved in virulence, LsrB is critical for the successful symbiosis in alfalfa plants by suppression of host defense responses that contribute to nodule premature senescence [64,65,66,67]. Neither AbcR1 nor AbcR2 are required for the competitive and efficient nodulation of S. meliloti on alfalfa roots or free-living growth. However, AbcR homologs in Brucella or Agrobacterium are involved in virulence [61]. Notably, whereas S. meliloti liv ABC system has no contribution to symbiosis, its bra homolog in R. leguminosarum is needed for a Fix+ phenotype in pea [68,69,70]. Therefore, it cannot be ruled out a symbiotic relevance of AbcR sRNAs in other rhizobia–host interactions.
Proper regulation of N and carbon (C) metabolism is also required for successful bacteroid differentiation and N fixation. C storage in nodules occurs in the form of polyhydroxybutyrate (PHB) granules which are produced during the invasion process and degraded during bacteroid differentiation. PHB is likely used as a C source at this stage, being critical for symbiotic performance [71]. MmgR (makes more granules regulator) is an Hfq-dependent S. meliloti 77 nt-long trans-sRNA of the αr8 RNA family (Table 1) linked to PHB metabolism. MmgR homologs have been also identified in S. fredii, R. leguminosarum bv. viciae and R. etli [45]. The predicted MmgR secondary structure shows three SLs and partial homology to SuhB sRNA. An aSD motif within the first loop is predicted to be involved in targeting, but no direct mRNA target of this sRNA has been described to date [45]. MmgR accumulates preferentially upon entry of bacteria into stationary phase when grown in defined minimal medium. In addition, it is induced by nitrate-limiting conditions and repressed by addition of tryptone or aa to the medium. The transcription of mmgR is induced by the global N regulator NtrC, while it is repressed by AniA, a regulator of C flow. As revealed by the loss-of-function phenotype, MmgR preserves synthesis of both PHB-related phasin proteins (PhaP1 and PhaP2) under conditions of C surplus, thereby limiting intracellular accumulation of PHB granules. Together, these data suggest that mmgR expression depends on cellular N and C status [72,73]. However, lack of the PHB synthase coding gene phbC has different effect depending on rhizobial species. Therefore, MmgR may have a different impact in other rhizobia that is worth to investigate [70,74,75,76].
4.2. Quorum Sensing
Bacteria are able to communicate with their pairs and adjust their expression profiles to cell density thanks to the quorum sensing (QS) response. This collective behavior system has been extensively studied in Gram-negative bacteria and it is mainly based on the synthesis of autoinducers, such as acyl-homoserine lactones (AHLs) [77]. These molecules are usually recognized by their cognate LuxR receptors, triggering gene expression changes involved in important biological processes like biofilm formation and virulence. Autoinducers diffuse easily through bacterial membranes and can also stimulate LuxI-type AHLs synthases in neighboring microorganisms, orchestrating transcriptional control. In Vibrio, the model microorganism to study AHLs-based QS regulation, several sRNAs modulating this process have been identified. In the absence of AHLs, V. harveyi expresses five Hfq-dependent homologous QS Regulatory RNAs (Qrr) that regulate multiple mRNAs, including four low cell density master regulators that repress LuxR by four distinct mechanisms [78].
In S. meliloti, AHLs are synthesized by the LuxI-type synthase SinI, which is regulated by other components of the Sin QS system, like SinR and ExpR, under cell density-dependent and stress conditions [79]. Accumulation of sinI mRNA in an hfq knock-out mutant together with co-immunoprecipitation with Hfq anticipated QS riboregulation in S. meliloti [32,80]. On the other hand, the conserved endoribonuclease RNAse E is involved in sinI 5’-UTR degradation, thereby influencing mRNA turnover and AHLs levels [81]. A trans-sRNA that regulates S. meliloti sinI post-transcriptionally has been also unveiled. This sRNA, initially referred to as SmelC587 (Table 1), was identified by inverse bioinformatics target predictions (i.e., using the 5´-UTR of sinI mRNA sequence as query) as a putative sinI base-pair riboregulator [82]. Abundance of SmelC587 transcript does not significantly vary with cell densities but with temperature and salt stress [26] and, accordingly, it was renamed Rhizobial Cold and Salinity stress Riboregulator 1 (RcsR1). Accumulation of this sRNA was very low in the presence of 500 mM NaCl in several Rhizobiaceae, matching the growth defects observed upon RcsR1 overproduction at high salinity conditions [82]. RcsR1 is predicted to fold into three distinct domains: two highly-conserved SLs and a less conserved region that targets the sinI 5’-UTR. This interaction was genetically confirmed in vivo and by in vitro by RNA degradation assays using native RNase E. Interestingly, the results show that RcsR1 competes with the endonuclease for sinI 5’-UTR cleavage site, impairing SinI translation [82]. This work represents an example of negative sRNA-mediated modulation of key bacterial processes under stress signals. Remarkably, RcsR1 is transcribed from an identical position to the trpL-derived attenuator RNA rnTrpL. Ectopic RcsR1 overproduction results in trpDC mRNA level decrease as rnTrpL down-regulates sinI, suggesting a co-regulation of both QS and tryptophan biosynthesis [57].
4.3. Cell Cycle
Cell cycle progression has been widely studied in α-proteobacteria, in which there is a single chromosomal replication event per cycle (Figure 3). Mechanisms to control chromosome replication and cell division in fluctuating environments are of critical relevance for bacterial survival [83]. Accordingly, rhizobia completely rewire cell cycle during the establishment of symbiosis with leguminous plants [34,84]. Control of α-proteobacterial cell cycle relies on several conserved transcription factors regulating the main stages [10]. Among them, DnaA mediates replication initiation of S. meliloti chromosome and GcrA has been reported to control expression of several genes involved in the segregation machinery during S-phase in Caulobacter [85,86]. Interestingly, gcrA-depleted cells resemble bacteroid genome content and morphology in S. meliloti [87], suggesting a similar role of GcrA in cell cycle and also in bacteroid differentiation. Finally, the master cell cycle regulator CtrA controls genes involved in cell division and inhibits replication reinitiation [88]. This essential protein is tightly controlled and negatively regulated, together with GcrA, by the widely-studied plant peptide NCR247, which suggests its involvement in differentiation of N-fixing bacteroids [84]. To date, two S. meliloti trans-sRNAs have been related to the adjustment of cell cycle progression under adverse conditions by different mechanisms (Figure 3); EcpR1 represses dnaA and gcrA expression [87] whereas GspR contributes to ctrA silencing [89].
Target predictions for conserved sRNA families in related α-proteobacteria showed an enrichment of cell cycle-related mRNAs for EcpR1 (formerly SmelC291; Table 1), whereas reverse searches using the 5´-UTR of the ctrA mRNA as a bait ended up with the identification of GspR (formerly SmelC775) as possible CtrA riboregulator. EcpR1 homologs are broadly distributed among Rhizobiales, whilst GspR occurrence is likely restricted to the genus Sinorhizobium [43]. The name of both trans-sRNAs stand for the main phenotypes associated to their gain-of-function. EcpR1 overexpression results into an elongated cell phenotype, along with cell filamentation and genome endoduplication, mimicking the phenotypes of bacteroid and GcrA-depleted cells [87]. Over accumulation of GspR hampers cell growth (growth stop phenotype) in accordance with cell cycle progression defects [89]. Interestingly, these effects are also visible in related species upon overproduction of the respective sRNA homologs. Deletion mutants do not show clear growth phenotypes, however, EcpR1- and GspR- S. meliloti derivatives are outcompeted by their parent strains when co-cultured, suggesting that both sRNAs confer a fitness advantage to bacteria. Expression of both EcpR1 and GspR is induced in growth-limiting stationary phase in rich media and is not detected in mature nodules [87,89]. EcpR1 is preceded by a σ70-dependent promoter and this sRNAs is also activated by other stresses like cold and heat shocks or nutrient deprivation, stimulated by the stringent response signal ppGpp. GspR strongly accumulates in bacteria grown in minimal medium. In agreement with in silico predictions and profiling of their dependent proteomes, EcpR1- and GspR-mediated regulation of cell cycle-related genes was experimentally validated by an eGFP-based genetic reporter system [47]. However, EcpR1 and GspR use different targeting mechanisms for mRNA regulation. The two EcpR1 confirmed mRNA targets (dnaA and gcrA) base pair to the same GC-rich conserved SL in the sRNA, whereas GspR represses expression of ctrA and cspA5 using different domains. Furthermore, considering the targeted mRNA region, dnaA and cspA5 are canonically regulated by sRNA binding to the vicinity of the RBS and the start codon. However, gcrA and ctrA are silenced by a still unknown mechanism that involves base pairing to nucleotides located far upstream of the translation initiation region within the long 5’-UTR of the mRNAs. CspA5 homologs usually act as RNA chaperones facilitating translation, which is probably the observed effect of CspA5 on the long 5’-UTR of the ctrA mRNA. Therefore, GspR and CspA5 are arranged in a coherent feed-forward loop that adds a novel post-transcriptional layer to the intricate CtrA regulation [89]. These results anticipate that RNA-mediated post-transcriptional control of cell cycle genes may be a universal mechanism linking cell growth and stress in bacteria. From a biotechnological perspective, these cell-cycle related sRNAs might be exploited as drug targets in antibacterial therapies.
4.4. Nodule Development and Functioning
Several studies addressing gene expression in legume root nodules have identified highly expressed sRNAs in endosymbiotic rhizobia, thus anticipating riboregulation of bacterial symbiotic traits [21,29,34,90]. Further, differential accumulation of these transcripts in defined zones of indeterminate alfalfa nodules or in developmentally arrested nodules from M. truncatula mutants could be predictive of specific roles of S. meliloti sRNAs in early, intermediate and late symbiotic stages, e.g., infection, bacteroid differentiation or N fixation [34,91]. A small subset of these S. meliloti sRNAs is uniquely expressed in nodule tissues, thus suggesting yet unexplored RNA functions in regulation of genuine symbiotic pathways. However, in most cases expression in nodules concurs with stress-induced accumulation of the sRNA in free-living rhizobia. This is the case of some of the functionally characterized S. meliloti trans-sRNAs described above, e.g., AbcR1, MmgR, EcpR1 or GspR1, whose loss-of-function has none or modest impact in symbiosis [62,72,87,89].
To date, phenotyping of knock-out mutants has revealed putative symbiotic functions for only one chromosomally encoded S. meliloti regulatory trans-sRNA, identified in early genome-wide screens as Smr14C2 or SmelC397, and later renamed NfeR1 (nodule formation efficiency RNA) to reflect this fact [20,26,92]. NfeR1 belongs to the so-called αr14 family of α–proteobacterial sRNAs (Table 1), which is represented in most plant symbionts, phytopathogens and mammal pathogens within Rhizobiaceae, Brucellaceae and Phyllobacteriaceae bacterial families [44,46]. αr14 sRNAs share a predicted secondary structure consisting of three hairpins with hypervariable nucleotide content in the stems and the aSD motif unpaired in all the loops [44,46]. NfeR1 locus occurs in six copies in the S. meliloti Rm1021 and Rm2011 genomes, but in the conditions tested only one of those is transcribed to levels reliably detected by northern blot probing [92]. NfeR1 transcription is driven by a σ70-dependent promoter containing several nucleotide residues that are well-conserved in promoter regions of αr14 sRNAs. This conserved motif is the determinant of both differential expression and high transcription rates, which render NfeR1 as one of the transcripts with highest coverage scores among the identified S. meliloti sRNAs by RNA-Seq surveys [26,27,29,34]. Intracellular NfeR1 levels are particularly high when bacteria are grown in defined glutamate/mannitol medium, upon salt shock and in all steps of the symbiotic interaction between S. meliloti and alfalfa, i.e., rhizoplane colonization, root hair infection, bacteroid differentiation and symbiotic N fixation [34,92]. Linked to this expression profile, NfeR1 loss-of-function compromises S. meliloti survival in hypersaline medium, nodule development and overall symbiotic efficiency [92]. Nodules induced by a S. meliloti nfeR1 deletion mutant on alfalfa roots are apparently colonized by fully differentiated bacteroids, but look round shaped rather than elongated, and smaller than wild-type indeterminate mature nodules. Their histology resembles that of nodules developed by M. truncatula mutants in marker genes for intermediate and late symbiotic stages [91,93] or induced by S. meliloti derivatives engineered to overexpress N assimilation pathways concurrently with symbiotic N fixation [94]. In silico searches for NfeR1 mRNA targets predict that most of the regulatory potential of this sRNA resides in the three conserved aSD motifs, which are indistinctly suited for base-pairing with the translation initiation region of multiple mRNAs, most coding for transport proteins. NfeR1-mediated post-transcriptional silencing of some of these targets and the redundant function of the three identical regulatory motifs have been genetically confirmed [92]. Together, these findings anticipate a pivotal role of NfeR1 in reprogramming important rhizobial metabolic pathways during the free-living to symbiotic transition.
Nodule development is largely controlled by complex organogenesis programs induced by rhizobia in their compatible legume host [95]. In this regard, a very recent study has shown that 18-24 nt-long sRNAs primarily derived from tRNA 3’-ends (tRFs) accumulate in nodules, acting as unprecedented rhizobial signals that modulate soybean nodulation by B. japonicum [96]. These miRNA-like tRFs interfere with the ARGONAUTE1-dependent RNAi machinery to silence plant host target genes involved in root hair curling and nodule formation. This breakthrough report unveils a novel biogenesis pathway for rhizobial sRNAs and provides an example of cross-kingdom communication through RNA trafficking that undoubtedly merits further investigation in this and other symbiotic associations.
5. Antisense Transcription in S. meliloti
Several studies on S. meliloti anticipate a broad impact of cis-acting asRNA regulators on both the free living and host-associated lifestyles of this bacterium [26,27,97]. asRNAs are transcribed opposite to mRNA loci and may overlap either the 5´/3´-UTRs or the coding region of the message (Figure 1). The long and perfect complementarity between asRNAs and their targets favors duplex formation, which can influence transcription, translation and/or turnover rates of the mRNAs. Deep-sequencing of prokaryotic transcriptomes is continuously uncovering pervasive antisense transcription, which likely contributes to the posttranscriptional buffering of gene expression levels [98]. Such RNA-Seq surveys have revealed that S. meliloti expresses more than 3000 asRNAs that are linked to ~35% of the predicted protein-coding genes [26,27]. Nonetheless, functional asRNAs in S. meliloti, like in other microorganisms, were firstly reported associated to extrachromosomal replicons [99].
A systematic screening strategy to identify novel RNA regulators of N fixation revealed that both asRNAs and trans-sRNAs are overrepresented (41% and 24%, respectively) in S. meliloti pSymA symbiotic megaplasmid, with asRNAs particularly biased to nod and nif genes. Further characterization of seven pSymA-borne asRNAs showed that most accumulate in response to different biological conditions, anticipating functional roles [97]. Among them, SMa_asRNA_244, antisense to the of nodD2 3´-UTR, is slightly upregulated at high optical densities and upon luteolin addition (30%). It is worth noting that NodD2 is the only transcriptional regulator of S. meliloti nod genes that is not activated by this plant flavonoid. Several asRNAs opposite to genes encoding the nitrogenase components NifE and NifK show also distinct expression profiles. SmelA031 and SMa_asRNA_277, also accumulate in early and late stationary phase of growth, respectively, while SMa_asRNA_279 responds to other stresses. Interestingly, the three transcripts are repressed in nodules and under low O2 and N2 concentrations, and plants inoculated with S. meliloti overexpressing SmelA031 are moderately affected in N fixation when compared to controls. Nitrogenase activity is extremely sensitive to O2 and needs N2 as substrate to function inside the nodules. Therefore, it is feasible that such post-transcriptional regulation modulates translation of this enzyme to avoid energy loss. These results first evidenced symbiotic-related asRNA regulation in rhizobia, but hundreds of rhizobial asRNAs await functional characterization. There are some works arguing that the majority of cellular asRNAs just arise from noisy transcription and are non-functional [100,101]. However, there is an increasing number of roles assigned to the widespread asRNAs, including both highly specific and global regulation such as DNA-repair and transcription interference [98]. Nevertheless, it is also advisable that, when searching for asRNAs with potential specific regulatory roles, requirements like upstream predicted promoters, high read coverage in reliable RNA-Seq profiling, and informative validated expression patterns are taken into account.
6. Proteins Assisting sRNA Activity
Proteins involved in RNA activity and metabolism are universal players in riboregulation at different levels [102,103,104]. For example, RNA-binding proteins of the CsrA (C storage regulator)/RsmA (regulator of secondary metabolism) family prevent translation by targeting the SD sequence of some mRNAs, and this activity is counteracted by sRNAs of the Csr/Rms class by a target mimicry mechanism [105]. Intriguingly, rhizobial genomes do not encode recognizable homologs of CsrA-like proteins. Other RNA protein partners act as chaperones that promote unwinding of complex secondary structures, e.g., cold-shock proteins (CSPs) [106] or base-pairing between regulatory sRNAs and their targets, e.g., the well-characterized Hfq [107] and the recently discovered ProQ [108]. On the other hand, cellular ribonucleases with diverse substrate preference are the effectors of sRNA-mediated post-transcriptional gene silencing [109]. The role of RNA-binding proteins and ribonucleases in RNA-mediated regulation is best understood in the model E. coli, phylogenetically close enterobacteria and other pathogens [102]. Nonetheless, RNA research has also contributed to gain specific insights into the biology of these proteins in rhizobia [103,110].
6.1. The Rhizobial RNA Chaperone Hfq
The host factor for the replication of the Qβ bacteriophage or Hfq is encoded by 55% of the bacterial genomes sequenced so far [111]. The function of this protein as RNA chaperone extends far beyond its primary role in intracellular RNA phage multiplication, to assist regulation by sRNAs (preferentially trans-sRNAs) and stabilize diverse RNA species including mRNAs and tRNAs [14]. These RNA-binding features place Hfq at a node of bacterial post-transcriptional regulatory networks. All α–rhizobial genomes host an Hfq coding gene that appears to be strongly expressed and autoregulated at the translational level [112,113,114]. The rhizobial Hfq polypeptide has a C-terminal region shorter than that of its enterobacterial counterparts, but retains the core RNA-binding domains of canonical Hfqs [14,115]. This predicts similar quaternary structural arrangements of rhizobial and enterobacterial Hfq proteins into ring-shaped homohexamers that expose two distinct surfaces for RNA binding. Accordingly, E. coli and S. meliloti Hfq homologs have been shown to be functionally interchangeable [112].
Like in enterobacteria, hfq knock-out causes severe pleiotropic phenotypes in S. meliloti that include altered growth, increased sensitivity to abiotic stress, changes in the profile of QS signals, reduced competitiveness for nodulation, compromised survival of bacteroids inside nodule cells and defects in N fixation [80,114,115,116,117]. Late symbiotic deficiencies of these mutants are most likely related to the positive contribution of Hfq to the post-transcriptional control of genes encoding the master regulators of N fixation NifA and FixK in diverse free-living and symbiotic α–proteobacterial diazotrophs by mechanisms that are still poorly understood [113,114,115,117,118]. Nonetheless, this phenotypic pleiotropy can be further explained by the profound Hfq-dependent changes observed in the rhizobial transcriptome and proteome upon free-living growth, as recurrently revealed by several independent studies on S. meliloti [80,115,116,119]. One remarkable observation common to all these reports is the pervasive misregulation of genes encoding ABC transport proteins in bacteria lacking Hfq, with overall upregulation of those presumably involved in uptake of aa and other N sources, such as the broad specificity Aap and Bra systems [70,120]. Although probably involving both common and host-specific regulatory mechanisms, the central role of Hfq in the chronic infection of eukaryotic organisms has been also reported in plant and mammal pathogens within the α–subgroup of proteobacteria such as A. tumefaciens or Brucella species [61,121,122].
Regulation of at least part of the Hfq-dependent mRNAs is expected to involve its cognate partner sRNAs. In S. meliloti, genome-wide profiling of RNA species co-immunoprecipitated with a functional FLAG epitope-tagged Hfq variant (CoIP-RNA) uncovered massive binding of the protein to mRNAs [32,123]. Specifically, nearly 20% of predicted S. meliloti mRNAs were enriched in Hfq CoIP-RNA, most of which functionally related to major symbiotic and stress-response pathways. Many of these mRNAs, such as those encoding NifA, FixK or stress-related alternative RNA polymerase σ factors of extracytoplasmic function (e.g., RpoE2) are positively regulated by Hfq by a mechanism that probably involves protection of the transcripts from degradation by cellular ribonucleases in a trans-acting sRNA-independent manner [103]. Conversely, mRNAs encoding aa transporters that are predicted or experimentally confirmed targets of the functionally characterized trans-sRNAs AbcR1 and AbcR2 are particularly abundant among the Hfq-binding transcripts. In these cases, Hfq most likely acts promoting both trans-sRNA stability and base-pairing to mRNA targets to boost post-transcriptional silencing [103]. Nonetheless, only 14% of trans-sRNAs and 2% of asRNAs currently annotated in the S. meliloti genome were scored as Hfq ligands [26,27,29,32]. These findings indicate a rather more limited impact on riboregulation of the rhizobial than enterobacterial Hfq homolog, anticipating that in the legume symbionts other alternative RNA chaperones may also assist sRNA activity. One possible candidate is the recently identified S. enterica ProQ protein [108,124]. However, the phylogenetic distribution of this protein is more restricted than that of Hfq, being unique to R. leguminosarum bv. viciae among α–rhizobia [125]. Thus, the search for novel RNA chaperones in this group of bacteria remains open.
6.2. Rhizobial Ribonucleases
As major determinants of the steady-state level of all cellular transcripts, ribonucleases (RNases) are critical elements in post-transcriptional regulatory networks [126]. Decay of mRNA upon antisense interaction with regulatory RNAs commonly involves the prevalent prokaryotic endoribonucleases RNase E and/or RNase III, which are specific to single- and double-stranded RNAs (ssRNA and dsRNA), respectively [127]. In some cases, RNase E is recruited by Hfq to the interplay between a trans-sRNA and its target(s) as a component of a multiprotein complex called RNA degradosome that also contains a 3′-exoribonuclease (polynucleotide phosphorylase or PNPase), a RNA helicase (RhlB) and an enolase as the core proteins [126]. On the other hand, RNase III preferentially recognizes RNA duplexes, thus being a major player of gene silencing promoted by asRNAs [128]. RNA decay initiated by RNase E or RNase III results into intermediate products that are further degraded by other cellular endo- and exoribonucleases, e.g., PNPase, RNase II or RNase R [127].
Rhizobial genomes encode a set of more than 20 ribonucleases that include all the aforementioned proteins, but for the vast majority there is a lack of functional information [129]. Nonetheless, several reports have anticipated the involvement of RNase E in trans-sRNA stability and regulation in both Hfq-dependent and independent manner [81,82,87,130,131]. RNase III and RNase J, which is not present in E. coli, are both involved in rRNA maturation but their putative role in riboregulation has not been investigated yet [132,133]. RNase III encoded by the S. meliloti rnc gene has been biochemically and genetically characterized [134]. Its catalytic features resemble those of the E. coli ortholog but with different requirements for optimal activity regarding metal co-factor preference or pH tolerance that could be related to the rhizobial lifestyle. Indeed, although rnc deletion does not compromise viability and morphology of S. meliloti cells, it is detrimental for growth and overall symbiotic performance on alfalfa roots [134].
The gene annotated as SMc01113 in the chromosome of the S. meliloti reference strain Rm1021 is almost ubiquitous in bacteria and has been included in the proposed minimal prokaryotic genome [135]. It encodes a putative metal-dependent hydrolase, later called YbeY, that shares predicted structural similarities with the MID domain of the Argonaute (AGO) proteins involved in eukaryotic RNA silencing, but its function has remained elusive until recently [136]. S. meliloti Hfq and YbeY mutants show strikingly similar free-living and symbiotic phenotypes as well as misregulation of several predicted sRNA-mRNA regulatory pairs [136,137]. These structural and phenotypic features initially hinted at an Hfq-like role of YbeY in riboregulation [136]. However, the E. coli YbeY ortholog was later shown to be a metal-dependent endoribonuclease that specifically acts on ssRNA [138]. Biochemical characterization of S. meliloti YbeY also supports a universal role of this protein as RNase [139,140]. However, unlike its enterobacterial ortholog, S. meliloti YbeY is also competent for cleaving dsRNA [139]. Transcriptome alterations in the S. meliloti YbeY mutant predict a role of this enzyme in trans-sRNA and asRNA mediated silencing of genes involved in nutrient uptake and symbiotic N fixation, respectively [139]. In this regard, both in vivo and in vitro evidences suggest that YbeY is required for AbcR2-dependent down-regulation of the ABC amino acid transporter prbA mRNA. Further supporting a role in dsRNA degradation, S. meliloti YbeY has been also shown to co-purify with several asRNAs and their mRNA partners, including the symbiotically-relevant nifA [139]. It would be interesting to test whether the alteration of asRNA-mediated regulation constitutes one of the main drivers of the symbiotic phenotype associated to YbeY activity.
7. Conclusions and Perspectives
RNA-mediated regulation of gene expression, which is ubiquitous in all living organisms, contributes to fine-tune most, if not all, prokaryotic physiological processes. However, genome annotations of the majority of bacterial species do not include sRNA genes yet, so that the structure and function of the non-coding transcriptomes remains unaccountably unexplored. Knowledge gaining on how the transcriptomes shape the functions of bacteria populating diverse niches or even establish complex relationships with other organisms is now timely and technically feasible. In plant-nodulating rhizobia, most of the knowledge about riboregulation derives from work on the alfalfa symbiont S. meliloti. This bacterium expresses hundreds of trans-sRNAs, but only a handful of those have been characterized and functionally related to free-living and symbiotic rhizobial traits such as metabolism, cell-cycle, quorum sensing or nodule development (Figure 4). Like in other bacterial species, pervasive antisense transcription also occurs in S. meliloti, with a remarkable bias towards symbiotically relevant genes. However, the functional significance of asRNA-mediated regulation has not been investigated further neither at genome-wide scale nor for particular symbiotic genes. Other key players in riboregulation, like RNA-binding proteins and ribonucleases remain also underexplored. Unlike in enterobacteria, the well-characterized RNA-chaperone Hfq seems to have a rather limited role in assisting trans-sRNA activity in rhizobia and the catalytic features of the set of predicted ribonucleases and its impact in RNA silencing are essentially unknown. Therefore, much work remains to be done to characterize the content and function of the sRNAs-associated proteomes.
The knowledge gained in S. meliloti should be necessarily extended to other rhizobia and symbiotic systems. Genomes of other model N-fixing symbionts must be accurately annotated using the most recent RNA-Seq protocols to uncover the primary transcriptome. The resulting catalogs of sRNA genes must be extensively compared and characterized to assess the contribution of riboregulation to the symbiotic diversity. Deciphering the biogenesis, function, and mechanistic principles of all types of sRNAs, i.e., trans-sRNAs, asRNAs, riboswitches, 5’/3’-UTRs, other mRNA leaders, and tRFs will contribute to explore the plasticity of RNA molecules as regulators of gene expression. Of particular interest would be to investigate the RNA trafficking between rhizobia and the plant host cells as a novel layer of dialogue between the symbiotic partners. Finally, the biotechnological implications of post-transcriptional RNA-mediated regulation for the engineering of symbiosis and other environmentally relevant systems should be also explored.
Funding
This research was funded by the Spanish Ministerio de Ciencia, Innovación y Universidades, ERDF co-financed grants BFU2013-48282-C2-2-P and BFU2017-82645-P to J.I.J.-Z.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Poole, P.; Ramachandran, V.; Terpolilli, J. Rhizobia: From saprophytes to endosymbionts. Nat. Rev. Microbiol. 2018, 16, 291. [Google Scholar] [CrossRef] [PubMed]
- Gage, D. Infection and invasion of roots by symbiotic, nitrogen-fixing rhizobia during nodulation of temperate legumes. Microbiol. Mol. Biol. Rev. 2004, 68, 280–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, K.E.; Kobayashi, H.; Walker, G.C. Molecular determinants of a symbiotic chronic infection. Annu. Rev. Genet. 2008, 42, 413–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahran, H.H. Rhizobium-legume symbiosis and nitrogen fixation under severe conditions and in an arid climate. Microbiol. Mol. Biol. Rev. 1999, 63, 968–989. [Google Scholar] [CrossRef] [Green Version]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; Van Der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef]
- Masson-Boivin, C.; Giraud, E.; Perret, X.; Batut, J. Establishing nitrogen-fixing symbiosis with legumes: How many rhizobium recipes? Trends Microbiol. 2009, 17, 458–466. [Google Scholar] [CrossRef]
- Soto, M.J.; Domínguez-Ferreras, A.; Pérez-Mendoza, D.; Sanjuán, J.; Olivares, J. Mutualism versus pathogenesis: The give-and-take in plant-bacteria interactions. Cell. Microbiol. 2009, 11, 381–388. [Google Scholar] [CrossRef]
- Van De Velde, W.; Zehirov, G.; Szatmari, A.; Debreczeny, M.; Ishihara, H.; Kevei, Z.; Farkas, A.; Mikulass, K.; Nagy, A.; Tiricz, H.; et al. Plant Peptides Govern Terminal Differentiation of Bacteria in Symbiosis. Science 2010, 327, 1122–1126. [Google Scholar] [CrossRef]
- Galibert, F.; Finan, T.M.; Long, S.R.; Puhler, A.; Abola, P.; Ampe, F.; Barloy-Hubler, F.; Barnett, M.J.; Becker, A.; Boistard, P.; et al. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 2001, 293, 668–672. [Google Scholar] [CrossRef] [Green Version]
- Brilli, M.; Fondi, M.; Fani, R.; Mengoni, A.; Ferri, L.; Bazzicalupo, M.; Biondi, E.G. The diversity and evolution of cell cycle regulation in alpha-proteobacteria: A comparative genomic analysis. BMC Syst. Biol. 2010, 4, 52. [Google Scholar] [CrossRef] [Green Version]
- Krol, E.; Blom, J.; Winnebald, J.; Berhörster, A.; Barnett, M.J.; Goesmann, A.; Baumbach, J.; Becker, A. RhizoRegNet—A database of rhizobial transcription factors and regulatory networks. J. Biotechnol. 2011, 155, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Cossart, P. Prokaryotic transcriptomics: A new view on regulation, physiology and pathogenicity. Nat. Rev. Genet. 2010, 11, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.G.H.; Romby, P. Small RNAs in bacteria and archaea: Who they are, what they do, and how they do it. In Advances in Genetics; Theodore Friedmann, J.C.D., Stephen, F.G., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 90, pp. 133–208. [Google Scholar]
- Sobrero, P.; Valverde, C. The bacterial protein Hfq: Much more than a mere RNA-binding factor. Crit. Rev. Microbiol. 2012, 38, 276–299. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Zurdo, J.I.; Valverde, C.; Becker, A. Insights into the noncoding RNome of nitrogen-fixing endosymbiotic α-proteobacteria. Mol. Plant-Microbe Interact. 2013, 26, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Zurdo, J.I.; Robledo, M. Unraveling the universe of small RNA regulators in the legume symbiont Sinorhizobium Meliloti. Symbiosis 2015, 67, 43–54. [Google Scholar] [CrossRef]
- Becker, A.; Overlöper, A.; Schlüter, J.-P.; Reinkensmeier, J.; Robledo, M.; Giegerich, R.; Narberhaus, F.; Evguenieva-Hackenberg, E. Riboregulation in plant-associated α-proteobacteria. RNA Biol. 2014, 11, 550–562. [Google Scholar] [CrossRef] [Green Version]
- Ulve, V.M.; Sevin, E.W.; Cheron, A.; Barloy-Hubler, F. Identification of chromosomal alpha-proteobacterial small RNAs by comparative genome analysis and detection in Sinorhizobium meliloti strain 1021. BMC Genom. 2007, 8, 467. [Google Scholar] [CrossRef] [Green Version]
- Valverde, C.; Livny, J.; Schluter, J.P.; Reinkensmeier, J.; Becker, A.; Parisi, G. Prediction of Sinorhizobium meliloti sRNA genes and experimental detection in strain 2011. BMC Genom. 2008, 9, 416. [Google Scholar] [CrossRef] [Green Version]
- del Val, C.; Rivas, E.; Torres-Quesada, O.; Toro, N.; Jiménez-Zurdo, J.I. Identification of differentially expressed small non-coding RNAs in the legume endosymbiont Sinorhizobium meliloti by comparative genomics. Mol. Microbiol. 2007, 66, 1080–1091. [Google Scholar] [CrossRef] [Green Version]
- Vercruysse, M.; Fauvart, M.; Cloots, L.; Engelen, K.; Thijs, I.M.; Marchal, K.; Michiels, J. Genome-wide detection of predicted non-coding RNAs in Rhizobium etli expressed during free-living and host-associated growth using a high-resolution tiling array. BMC Genom. 2010, 11, 53. [Google Scholar] [CrossRef] [Green Version]
- Madhugiri, R.; Pessi, G.; Voss, B.; Hahn, J.; Sharma, C.M.; Reinhardt, R.; Vogel, J.; Hess, W.R.; Fischer, H.-M.; Evguenieva-Hackenberg, E. Small RNAs of the Bradyrhizobium/Rhodopseudomonas lineage and their analysis. RNA Biol. 2012, 9, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuli, X.; Wenlong, Z.; Xiao, W.; Jing, Z.; Baohai, H.; Zhengzheng, Z.; Bin-Guang, M.; Youguo, L. A Genome-Wide Prediction and Identification of Intergenic Small RNAs by Comparative Analysis in Mesorhizobium huakuii 7653R. Front. Microbiol. 2017, 8, 1730. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.M.; Vogel, J. Differential RNA-seq: The approach behind and the biological insight gained. Curr. Opin. Microbiol. 2014, 19, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Ettwiller, L.; Buswell, J.; Yigit, E.; Schildkraut, I. A novel enrichment strategy reveals unprecedented number of novel transcription start sites at single base resolution in a model prokaryote and the gut microbiome. BMC Genom. 2016, 17, 199. [Google Scholar] [CrossRef] [Green Version]
- Schlüter, J.P.; Reinkensmeier, J.; Daschkey, S.; Evguenieva-Hackenberg, E.; Janssen, S.; Janicke, S.; Becker, J.D.; Giegerich, R.; Becker, A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genom. 2010, 11, 245. [Google Scholar] [CrossRef] [Green Version]
- Schlüter, J.P.; Reinkensmeier, J.; Barnett, M.J.; Lang, C.; Krol, E.; Giegerich, R.; Long, S.R.; Becker, A. Global mapping of transcription start sites and promoter motifs in the symbiotic alpha-proteobacterium Sinorhizobium meliloti 1021. BMC Genom. 2013, 14, 156. [Google Scholar] [CrossRef] [Green Version]
- Čuklina, J.; Hahn, J.; Imakaev, M.; Omasits, U.; Förstner, K.U.; Ljubimov, N.; Goebel, M.; Pessi, G.; Fischer, H.-M.; Ahrens, C.H.; et al. Genome-wide transcription start site mapping of Bradyrhizobium japonicum grown free-living or in symbiosis—A rich resource to identify new transcripts, proteins and to study gene regulation. BMC Genom. 2016, 17, 302. [Google Scholar] [CrossRef] [Green Version]
- Sallet, E.; Roux, B.; Sauviac, L.; Jardinaud, M.-F.o.; Carrère, S.; Faraut, T.; de Carvalho-Niebel, F.; Gouzy, J.; Gamas, P.; Capela, D.; et al. Next-generation annotation of prokaryotic genomes with EuGene-P: Application to Sinorhizobium meliloti 2011. DNA Res. 2013, 20, 339–354. [Google Scholar] [CrossRef]
- Sittka, A.; Lucchini, S.; Papenfort, K.; Sharma, C.M.; Rolle, K.; Binnewies, T.T.; Hinton, J.C.D.; Vogel, J. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 2008, 4, e1000163. [Google Scholar] [CrossRef] [Green Version]
- Sittka, A.; Sharma, C.M.; Rolle, K.; Vogel, J. Deep sequencing of Salmonella RNA associated with heterologous Hfq proteins in vivo reveals small RNAs as a major target class and identifies RNA processing phenotypes. RNA Biol. 2009, 6, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Torres-Quesada, O.; Reinkensmeier, J.; Schlüter, J.-P.; Robledo, M.; Peregrina, A.; Giegerich, R.; Toro, N.; Becker, A.; Jimenez-Zurdo, J.I. Genome-wide profiling of Hfq-binding RNAs uncovers extensive post-transcriptional rewiring of major stress response and symbiotic regulons in Sinorhizobium meliloti. RNA Biol. 2014, 11, 563–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, P.R.; Georg, J. Workflow for a Computational Analysis of an sRNA Candidate in Bacteria; Springer: New York, NY, USA, 2018; pp. 3–30. [Google Scholar] [CrossRef]
- Roux, B.; Rodde, N.; Jardinaud, M.-F.; Timmers, T.; Sauviac, L.; Cottret, L.; Carrère, S.; Sallet, E.; Courcelle, E.; Moreau, S.; et al. An integrated analysis of plant and bacterial gene expression in symbiotic root nodules using laser-capture microdissection coupled to RNA sequencing. Plant J. 2014, 77, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2017, 46, D335–D342. [Google Scholar] [CrossRef] [PubMed]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Keiler, K.C. Biology of trans-Translation. Annu. Rev. Microbiol. 2008, 62, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, S.; Kündig, C.; Hennecke, H. Discovery of a rhizobial RNA that is essential for symbiotic root nodule development. J. Bacteriol. 1991, 173, 6373–6382. [Google Scholar] [CrossRef] [Green Version]
- Ulve, V.M.; Cheron, A.; Trautwetter, A.; Fontenelle, C.; Barloy-Hubler, F. Characterization and expression patterns of Sinorhizobium meliloti tmRNA (ssrA). FEMS Microbiol. Lett. 2007, 269, 117–123. [Google Scholar] [CrossRef]
- Keenan, R.J.; Freymann, D.M.; Stroud, R.M.; Walter, P. The signal recognition particle. Annu. Rev. Biochem. 2001, 70, 755–775. [Google Scholar] [CrossRef] [Green Version]
- Wassarman, K.M. 6S RNA, a Global Regulator of Transcription. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Faucher, S.P.; Friedlander, G.; Livny, J.; Margalit, H.; Shuman, H.A. Legionella pneumophila 6S RNA optimizes intracellular multiplication. Proc. Natl. Acad. Sci. USA 2010, 107, 7533–7538. [Google Scholar] [CrossRef] [Green Version]
- Reinkensmeier, J.; Schlüter, J.-P.; Giegerich, R.; Becker, A. Conservation and Occurrence of Trans-Encoded sRNAs in the Rhizobiales. Genes 2011, 2, 925–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinkensmeier, J.; Giegerich, R. Thermodynamic matchers for the construction of the cuckoo RNA family. RNA Biol. 2015, 12, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagares, A., Jr.; Roux, I.; Valverde, C. Phylogenetic distribution and evolutionary pattern of an α-proteobacterial small RNA gene that controls polyhydroxybutyrate accumulation in Sinorhizobium meliloti. Mol. Phylogenetics Evol. 2016, 99, 182–193. [Google Scholar] [CrossRef] [PubMed]
- del Val, C.; Romero-Zaliz, R.; Torres-Quesada, O.; Peregrina, A.; Toro, N.; Jiménez-Zurdo, J.I. A survey of sRNA families in alpha-proteobacteria. RNA Biol. 2012, 9, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Robledo, M.; García-Tomsig, N.I.; Jiménez-Zurdo, J.I. Primary Characterization of Small RNAs in Symbiotic Nitrogen-Fixing Bacteria. In Host-Pathogen Interactions: Methods and Protocols; Medina, C., López-Baena, F.J., Eds.; Springer: New York, NY, USA, 2018; pp. 277–295. [Google Scholar] [CrossRef]
- Robledo, M.; Matia-Gonzalez, A.M.; Garcia-Tomsig, N.I.; Jimenez-Zurdo, J.I. Identification of Small RNA-Protein Partners in Plant Symbiotic Bacteria. Methods Mol. Biol. 2018, 1737, 351–370. [Google Scholar] [CrossRef]
- Wright, P.R.; Georg, J.; Mann, M.; Sorescu, D.A.; Richter, A.S.; Lott, S.; Kleinkauf, R.; Hess, W.R.; Backofen, R. CopraRNA and IntaRNA: Predicting small RNA targets, networks and interaction domains. Nucleic Acids Res. 2014, 42, W119–W123. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.R.; Richter, A.S.; Papenfort, K.; Mann, M.; Vogel, J.; Hess, W.R.; Backofen, R.; Georg, J. Comparative genomics boosts target prediction for bacterial small RNAs. Proc. Natl. Acad. Sci. USA 2013, 110, E3487–E3496. [Google Scholar] [CrossRef] [Green Version]
- Lalaouna, D.; Massé, E. Identification of sRNA interacting with a transcript of interest using MS2-affinity purification coupled with RNA sequencing (MAPS) technology. Genom. Data 2015, 5, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.M.; Vogel, J. Experimental approaches for the discovery and characterization of regulatory small RNA. Curr. Opin. Microbiol. 2009, 12, 536–546. [Google Scholar] [CrossRef]
- Mauchline, T.H.; Fowler, J.E.; East, A.K.; Sartor, A.L.; Zaheer, R.; Hosie, A.H.F.; Poole, P.S.; Finan, T.M. Mapping the Sinorhizobium meliloti 1021 solute-binding protein-dependent transportome. Proc. Natl. Acad. Sci. USA 2006, 103, 17933–17938. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.F. ABC transporters: Physiology, structure and mechanism—An overview. Res. Microbiol. 2001, 152, 205–210. [Google Scholar] [CrossRef]
- Nogales, J.; Munoz, S.; Olivares, J.; Sanjuan, J. Genetic characterization of oligopeptide uptake systems in Sinorhizobium meliloti. FEMS Microbiol. Lett. 2009, 293, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djordjevic, M.A. Sinorhizobium meliloti metabolism in the root nodule: A proteomic perspective. Proteomics 2004, 4, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Melior, H.; Li, S.; Madhugiri, R.; Stotzel, M.; Azarderakhsh, S.; Barth-Weber, S.; Baumgardt, K.; Ziebuhr, J.; Evguenieva-Hackenberg, E. Transcription attenuation-derived small RNA rnTrpL regulates tryptophan biosynthesis gene expression in trans. Nucleic Acids Res. 2019, 47, 6396–6410. [Google Scholar] [CrossRef]
- Miyakoshi, M.; Chao, Y.; Vogel, J. Cross talk between ABC transporter mRNAs via a target mRNA-derived sponge of the GcvB small RNA. EMBO J. 2015, 34, 1478–1492. [Google Scholar] [CrossRef]
- Wilms, I.; Voss, B.; Hess, W.R.; Leichert, L.I.; Narberhaus, F. Small RNA-mediated control of the Agrobacterium tumefaciens GABA binding protein: Small RNA-controlled GABA uptake. Mol. Microbiol. 2011, 80, 492–506. [Google Scholar] [CrossRef]
- Overloper, A.; Kraus, A.; Gurski, R.; Wright, P.R.; Georg, J.; Hess, W.R.; Narberhaus, F. Two separate modules of the conserved regulatory RNA AbcR1 address multiple target mRNAs in and outside of the translation initiation region. RNA Biol. 2014, 11, 624–640. [Google Scholar] [CrossRef] [Green Version]
- Caswell, C.C.; Gaines, J.M.; Roop, R.M., 2nd. The RNA chaperone Hfq independently coordinates expression of the VirB type IV secretion system and the LuxR-type regulator BabR in Brucella abortus 2308. J. Bacteriol. 2012, 194, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Torres-Quesada, O.; Millán, V.; Nisa-Martínez, R.; Bardou, F.; Crespi, M.; Toro, N.; Jiménez-Zurdo, J.I. Independent activity of the homologous small regulatory RNAs AbcR1 and AbcR2 in the legume symbiont Sinorhizobium meliloti. PLoS ONE 2013, 8, e68147. [Google Scholar] [CrossRef]
- Sheehan, L.M.; Caswell, C.C. An account of evolutionary specialization: The AbcR small RNAs in the Rhizobiales. Mol. Microbiol. 2018, 107, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, L.M.; Budnick, J.A.; Blanchard, C.; Dunman, P.M.; Caswell, C.C. A LysR-family transcriptional regulator required for virulence in Brucella abortus is highly conserved among the alpha-proteobacteria. Mol. Microbiol. 2015, 98, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Yao, S.-Y.; Becker, A.; Rüberg, S.; Yu, G.-Q.; Zhu, J.-B.; Cheng, H.-P. Two New Sinorhizobium meliloti LysR-Type Transcriptional Regulators Required for Nodulation. J. Bacteriol. 2005, 187, 4562–4572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Lu, D.; Wang, D.; Luo, L. Sinorhizobium meliloti lsrB is involved in alfalfa root nodule development and nitrogen-fixing bacteroid differentiation. Chin. Sci. Bull. 2013, 58, 4077–4083. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.; Wang, Y.; Luo, L. Transcriptional regulator LsrB of Sinorhizobium meliloti positively regulates the expression of genes involved in lipopolysaccharide biosynthesis. Appl. Environ. Microbiol. 2014, 80, 5265–5273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodwig, E.M.; Hosie, A.H.F.; Bourdès, A.; Findlay, K.; Allaway, D.; Karunakaran, R.; Downie, J.A.; Poole, P.S. Amino-acid cycling drives nitrogen fixation in the legume–Rhizobium symbiosis. Nature 2003, 422, 722–726. [Google Scholar] [CrossRef]
- Prell, J.; White, J.P.; Bourdes, A.; Bunnewell, S.; Bongaerts, R.J.; Poole, P.S. Legumes regulate Rhizobium bacteroid development and persistence by the supply of branched-chain amino acids. Proc. Natl. Acad. Sci. USA 2009, 106, 12477–12482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udvardi, M.; Poole, P.S. Transport and metabolism in legume-rhizobia symbioses. Annu. Rev. Plant Biol. 2013, 64, 781–805. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.M.; Kobayashi, H.; Davies, B.W.; Taga, M.E.; Walker, G.C. How rhizobial symbionts invade plants: The Sinorhizobium–Medicago model. Nat. Rev. Microbiol. 2007, 5, 619–633. [Google Scholar] [CrossRef] [Green Version]
- Lagares, A., Jr.; Borella, G.C.; Linne, U.; Becker, A.; Valverde, C. Regulation of Polyhydroxybutyrate Accumulation in Sinorhizobium meliloti by the Trans-Encoded Small RNA MmgR. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef] [Green Version]
- Ceizel Borella, G.; Lagares, A., Jr.; Valverde, C. Expression of the small regulatory RNA gene mmgR is regulated negatively by AniA and positively by NtrC in Sinorhizobium meliloti 2011. Microbiology 2018, 164, 88–98. [Google Scholar] [CrossRef]
- Wang, C.; Saldanha, M.; Sheng, X.; Shelswell, K.J.; Walsh, K.T.; Sobral, B.W.; Charles, T.C. Roles of poly-3-hydroxybutyrate (PHB) and glycogen in symbiosis of Sinorhizobium meliloti with Medicago sp. Microbiology 2007, 153, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cevallos, M.A.; Encarnación, S.; Leija, A.; Mora, Y.; Mora, J. Genetic and physiological characterization of a Rhizobium etli mutant strain unable to synthesize poly-beta-hydroxybutyrate. J. Bacteriol. 1996, 178, 1646–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodwig, E.M.; Leonard, M.; Marroqui, S.; Wheeler, T.R.; Findlay, K.; Downie, J.A.; Poole, P.S. Role of polyhydroxybutyrate and glycogen as carbon storage compounds in pea and bean bacteroids. Mol. Plant-Microbe Interact. 2005, 18, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Rutherford Steven, T.; Papenfort, K.; Bagert John, D.; van Kessel Julia, C.; Tirrell David, A.; Wingreen Ned, S.; Bassler Bonnie, L. A Qrr Noncoding RNA Deploys Four Different Regulatory Mechanisms to Optimize Quorum-Sensing Dynamics. Cell 2015, 160, 228–240. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, M.; Meyer, S.; Becker, A. Novel Sinorhizobium meliloti quorum sensing positive and negative regulatory feedback mechanisms respond to phosphate availability. Mol. Microbiol. 2009, 74, 1238–1256. [Google Scholar] [CrossRef]
- Gao, M.; Barnett, M.J.; Long, S.R.; Teplitski, M. Role of the Sinorhizobium meliloti global regulator Hfq in gene regulation and symbiosis. Mol. Plant-Microbe Interact. 2010, 23, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Baumgardt, K.; Charoenpanich, P.; McIntosh, M.; Schikora, A.; Stein, E.; Thalmann, S.; Kogel, K.-H.; Klug, G.; Becker, A.; Evguenieva-Hackenberg, E. RNase E Affects the Expression of the Acyl-Homoserine Lactone Synthase Gene sinI in Sinorhizobium meliloti. J. Bacteriol. 2014, 196, 1435–1447. [Google Scholar] [CrossRef] [Green Version]
- Baumgardt, K.; Šmídová, K.; Rahn, H.; Lochnit, G.; Robledo, M.; Evguenieva-Hackenberg, E. The stress-related, rhizobial small RNA RcsR1 destabilizes the autoinducer synthase encoding mRNA sinI in Sinorhizobium meliloti. RNA Biol. 2016, 13, 486–499. [Google Scholar] [CrossRef] [Green Version]
- Jonas, K. To divide or not to divide: Control of the bacterial cell cycle by environmental cues. Curr. Opin. Microbiol. 2014, 18, 54–60. [Google Scholar] [CrossRef]
- Penterman, J.; Abo, R.P.; De Nisco, N.J.; Arnold, M.F.F.; Longhi, R.; Zanda, M.; Walker, G.C. Host plant peptides elicit a transcriptional response to control the Sinorhizobium meliloti cell cycle during symbiosis. Proc. Natl. Acad. Sci. USA 2014, 111, 3561–3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frage, B.; Döhlemann, J.; Robledo, M.; Lucena, D.; Sobetzko, P.; Graumann, P.L.; Becker, A. Spatiotemporal choreography of chromosome and megaplasmids in the Sinorhizobium meliloti cell cycle. Mol. Microbiol. 2016, 100, 808–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtzendorff, J.; Hung, D.; Brende, P.; Reisenauer, A.; Viollier, P.H.; McAdams, H.H.; Shapiro, L. Oscillating Global Regulators Control the Genetic Circuit Driving a Bacterial Cell Cycle. Science 2004, 304, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Robledo, M.; Frage, B.; Wright, P.R.; Becker, A. A stress-induced small RNA modulates alpha-rhizobial cell cycle progression. PLoS Genet. 2015, 11, e1005153. [Google Scholar] [CrossRef] [Green Version]
- Pini, F.; De Nisco, N.J.; Ferri, L.; Penterman, J.; Fioravanti, A.; Brilli, M.; Mengoni, A.; Bazzicalupo, M.; Viollier, P.H.; Walker, G.C.; et al. Cell Cycle Control by the Master Regulator CtrA in Sinorhizobium meliloti. PLoS Genet. 2015, 11, e1005232. [Google Scholar] [CrossRef] [Green Version]
- Robledo, M.; Schlüter, J.-P.; Loehr, L.O.; Linne, U.; Albaum, S.P.; Jiménez-Zurdo, J.I.; Becker, A. An sRNA and Cold Shock Protein Homolog-Based Feedforward Loop Post-transcriptionally Controls Cell Cycle Master Regulator CtrA. Front. Microbiol. 2018, 9, 763. [Google Scholar] [CrossRef]
- Barnett, M.J.; Toman, C.J.; Fisher, R.F.; Long, S.R. A dual-genome Symbiosis Chip for coordinate study of signal exchange and development in a prokaryote-host interaction. Proc. Natl. Acad. Sci. USA 2004, 101, 16636–16641. [Google Scholar] [CrossRef] [Green Version]
- Lang, C.; Long, S.R. Transcriptomic analysis of Sinorhizobium meliloti and Medicago truncatula symbiosis using nitrogen fixation–deficient nodules. Mol. Plant-Microbe Interact. 2015, 28, 856–868. [Google Scholar] [CrossRef] [Green Version]
- Robledo, M.; Peregrina, A.; Millán, V.; García-Tomsig, N.I.; Torres-Quesada, O.; Mateos, P.F.; Becker, A.; Jiménez-Zurdo, J.I. A conserved α-proteobacterial small RNA contributes to osmoadaptation and symbiotic efficiency of rhizobia on legume roots. Environ. Microbiol. 2017, 19, 2661–2680. [Google Scholar] [CrossRef]
- Starker, C.G.; Parra-Colmenares, A.L.; Smith, L.; Mitra, R.M.; Long, S.R. Nitrogen fixation mutants of Medicago truncatula fail to support plant and bacterial symbiotic gene expression. Plant Physiol. 2006, 140, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Patriarca, E.J.; Tatè, R.; Iaccarino, M. Key Role of Bacterial NH(4)(+) Metabolism in Rhizobium-Plant Symbiosis. Microbiol. Mol. Biol. Rev. 2002, 66, 203–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, B.J.; Mens, C.; Hastwell, A.H.; Zhang, M.; Su, H.; Jones, C.H.; Chu, X.; Gresshoff, P.M. Legume nodulation: The host controls the party. Plant Cell Environ. 2019, 42, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, B.; Wang, X.; Duan, J.; Ma, J. Rhizobial tRNA-derived small RNAs are signal molecules regulating plant nodulation. Science 2019, 365, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Robledo, M.; Jiménez-Zurdo, J.I.; Becker, A. Antisense transcription of symbiotic genes in Sinorhizobium meliloti. Symbiosis 2015, 67, 55–67. [Google Scholar] [CrossRef]
- Georg, J.; Hess, W.R. Widespread Antisense Transcription in Prokaryotes. Microbiol. Spectr. 2018, 6, 191–210. [Google Scholar] [CrossRef]
- MacLellan, S.R.; Smallbone, L.A.; Sibley, C.D.; Finan, T.M. The expression of a novel antisense gene mediates incompatibility within the large repABC family of α-proteobacterial plasmids. Mol. Microbiol. 2005, 55, 611–623. [Google Scholar] [CrossRef]
- Raghavan, R.; Sloan, D.B.; Ochman, H. Antisense Transcription Is Pervasive but Rarely Conserved in Enteric Bacteria. mBio 2012, 3, e00156-12. [Google Scholar] [CrossRef] [Green Version]
- Lybecker, M.; Bilusic, I.; Raghavan, R. Pervasive transcription: Detecting functional RNAs in bacteria. Transcription 2014, 5, e944039. [Google Scholar] [CrossRef] [Green Version]
- Holmqvist, E.; Vogel, J. RNA-binding proteins in bacteria. Nat. Rev. Microbiol. 2018. [Google Scholar] [CrossRef]
- Jiménez-Zurdo, J.I.; Robledo, M. RNA silencing in plant symbiotic bacteria: Insights from a protein-centric view. RNA Biol. 2017, 14, 1672–1677. [Google Scholar] [CrossRef]
- De Lay, N.; Schu, D.J.; Gottesman, S. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J. Biol. Chem. 2013, 288, 7996–8003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakulskas, C.A.; Potts, A.H.; Babitzke, P.; Ahmer, B.M.M.; Romeo, T. Regulation of Bacterial Virulence by Csr (Rsm) Systems. Microbiol. Mol. Biol. Rev. 2015, 79, 193–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, C.J.; Menendez-Gil, P.; Catalan-Moreno, A.; Vergara-Irigaray, M.; García, B.; Segura, V.; Irurzun, N.; Villanueva, M.; Ruiz de los Mozos, I.; Solano, C.; et al. The regulon of the RNA chaperone CspA and its auto-regulation in Staphylococcus aureus. Nucleic Acids Res. 2018, 46, 1345–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, E.G. Cycling of RNAs on Hfq. RNA Biol. 2013, 10, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnov, A.; Förstner, K.U.; Holmqvist, E.; Otto, A.; Günster, R.; Becher, D.; Reinhardt, R.; Vogel, J. Grad-seq guides the discovery of ProQ as a major small RNA-binding protein. Proc. Natl. Acad. Sci. USA 2016, 113, 11591–11596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, I.J.; Saramago, M.; Dressaire, C.; Domingues, S.; Viegas, S.C.; Arraiano, C.M. Importance and key events of prokaryotic RNA decay: The ultimate fate of an RNA molecule. Wiley Interdiscip. Rev. RNA 2011, 2, 818–836. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Zurdo, J.I.; Torres-Quesada, O.; Valverde, C.; Sobrero, P. Contribution of the RNA Chaperone Hfq to Environmental Fitness and Symbiosis in Sinorhizobium meliloti; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2015; pp. 737–745. [Google Scholar]
- Vogel, J.; Luisi, B.F. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 2011, 9, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Sobrero, P.; Valverde, C. Evidences of autoregulation of hfq expression in Sinorhizobium meliloti strain 2011. Arch. Microbiol. 2011, 193, 629–639. [Google Scholar] [CrossRef]
- Kaminski, P.A.; Desnoues, N.; Elmerich, C. The expression of nifA in Azorhizobium caulinodans requires a gene product homologous to Escherichia coli HF-I, an RNA-binding protein involved in the replication of phage Q beta RNA. Proc. Natl. Acad. Sci. USA 1994, 91, 4663–4667. [Google Scholar] [CrossRef] [Green Version]
- Barra-Bily, L.; Pandey, S.P.; Trautwetter, A.; Blanco, C.; Walker, G.C. The Sinorhizobium meliloti RNA chaperone Hfq mediates symbiosis of Sinorhizobium meliloti and alfalfa. J. Bacteriol. 2010, 192, 1710–1718. [Google Scholar] [CrossRef] [Green Version]
- Torres-Quesada, O.; Oruezabal, R.I.; Peregrina, A.; Jofre, E.; Lloret, J.; Rivilla, R.; Toro, N.; Jimenez-Zurdo, J.I. The Sinorhizobium meliloti RNA chaperone Hfq influences central carbon metabolism and the symbiotic interaction with alfalfa. BMC Microbiol. 2010, 10, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobrero, P.; Schluter, J.P.; Lanner, U.; Schlosser, A.; Becker, A.; Valverde, C. Quantitative proteomic analysis of the Hfq-regulon in Sinorhizobium meliloti 2011. PLoS ONE 2012, 7, e48494. [Google Scholar] [CrossRef]
- Gao, M.; Nguyen, H.; Salas González, I.; Teplitski, M. Regulation of fixLJ by Hfq controls symbiotically important genes in Sinorhizobium meliloti. Mol Plant-Microbe Interact. 2016, 29, 844–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, P.A.; Elmerich, C. The control of Azorhizobium caulinodans nifA expression by oxygen, ammonia and by the HF-I-like protein, NrfA. Mol. Microbiol. 1998, 28, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Barra-Bily, L.; Fontenelle, C.; Jan, G.; Flechard, M.; Trautwetter, A.; Pandey, S.P.; Walker, G.C.; Blanco, C. Proteomic alterations explain phenotypic changes in Sinorhizobium meliloti lacking the RNA chaperone Hfq. J. Bacteriol. 2010, 192, 1719–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulley, G.; White, J.P.; Karunakaran, R.; Prell, J.; Bourdes, A.; Bunnewell, S.; Hill, L.; Poole, P.S. Mutation of GOGAT prevents pea bacteroid formation and N2 fixation by globally downregulating transport of organic nitrogen sources. Mol. Microbiol. 2011, 80, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Roop, R.M.N.; Robertson, G.T.; Ferguson, G.P.; Milford, L.E.; Winkler, M.E.; Walker, G.C. Seeking a niche: Putative contributions of the hfq and bacA gene products to the successful adaptation of the brucellae to their intracellular home. Vet. Microbiol. 2002, 90, 349–363. [Google Scholar] [CrossRef]
- Wilms, I.; Moller, P.; Stock, A.M.; Gurski, R.; Lai, E.M.; Narberhaus, F. Hfq influences multiple transport systems and virulence in the plant pathogen Agrobacterium tumefaciens. J. Bacteriol. 2012, 194, 5209–5217. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Benge, A.; Mesa, J.M.; Javier, R.; Liu, F.-X. Use of RNA Immunoprecipitation Method for Determining Sinorhizobium meliloti RNA-Hfq Protein Associations In Vivo. Biol. Proced. Online 2018, 20, 8. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, A.; Wang, C.; Drewry, L.L.; Vogel, J. Molecular mechanism of mRNA repression in trans by a ProQ-dependent small RNA. EMBO J. 2017, 36, 1029–1045. [Google Scholar] [CrossRef]
- Olejniczak, M.; Storz, G. ProQ/FinO-domain proteins: Another ubiquitous family of RNA matchmakers? Mol. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arraiano, C.M.; Andrade, J.M.; Domingues, S.; Guinote, I.B.; Malecki, M.; Matos, R.G.; Moreira, R.N.; Pobre, V.; Reis, F.P.; Saramago, M.; et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol. Rev. 2010, 34, 883–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalaouna, D.; Simoneau-Roy, M.; Lafontaine, D.; Massé, E. Regulatory RNAs and target mRNA decay in prokaryotes. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2013, 1829, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Lasa, I.; Toledo-Arana, A.; Dobin, A.; Villanueva, M.; de los Mozos, I.R.; Vergara-Irigaray, M.; Segura, V.; Fagegaltier, D.; Penadés, J.R.; Valle, J.; et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 20172–20177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.; Barnett, M.J.; Capela, D.; Dondrup, M.; Kamp, P.B.; Krol, E.; Linke, B.; Ruberg, S.; Runte, K.; Schroeder, B.K.; et al. A portal for rhizobial genomes: RhizoGATE integrates a Sinorhizobium meliloti genome annotation update with postgenome data. J. Biotechnol. 2009, 140, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, B.; Holscher, M.; Baumgarth, B.; Kalbfleisch, A.; Kaya, C.; Hess, W.R.; Becker, A.; Evguenieva-Hackenberg, E. Expression of small RNAs in Rhizobiales and protection of a small RNA and its degradation products by Hfq in Sinorhizobium meliloti. Biochem. Biophys. Res. Commun. 2009, 390, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hong, G. Post-transcriptional regulation of NifA expression by Hfq and RNase E complex in Rhizobium leguminosarum bv. viciae. Acta Biochim. Biophys. Sin. 2009, 41, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Baumgardt, K.; Melior, H.; Madhugiri, R.; Thalmann, S.; Schikora, A.; McIntosh, M.; Becker, A.; Evguenieva-Hackenberg, E. RNase E and RNase J are needed for S-adenosylmethionine homeostasis in Sinorhizobium meliloti. Microbiology 2017, 163, 570–583. [Google Scholar] [CrossRef]
- Madhugiri, R.; Evguenieva-Hackenberg, E. RNase J is involved in the 5′-end maturation of 16S rRNA and 23S rRNA in Sinorhizobium meliloti. FEBS Lett. 2009, 583, 2339–2342. [Google Scholar] [CrossRef] [Green Version]
- Saramago, M.; Robledo, M.; Matos, R.G.; Jiménez-Zurdo, J.I.; Arraiano, C.M. Sinorhizobium meliloti RNase III: Catalytic Features and Impact on Symbiosis. Front. Genet. 2018, 9. [Google Scholar] [CrossRef]
- Gil, R.; Silva, F.J.; Peretó, J.; Moya, A. Determination of the core of a minimal bacterial gene set. Microbiol. Mol. Biol. Rev. 2004, 68, 518–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, S.P.; Minesinger, B.K.; Kumar, J.; Walker, G.C. A highly conserved protein of unknown function in Sinorhizobium meliloti affects sRNA regulation similar to Hfq. Nucleic Acids Res. 2011, 39, 4691–4708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, B.W.; Walker, G.C. A highly conserved protein of unknown function is required by Sinorhizobium meliloti for symbiosis and environmental stress protection. J. Bacteriol. 2008, 190, 1118–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob Asha, I.; Köhrer, C.; Davies Bryan, W.; RajBhandary Uttam, L.; Walker Graham, C. Conserved bacterial RNase YbeY plays key roles in 70S ribosome quality control and 16S rRNA maturation. Mol. Cell 2013, 49, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saramago, M.; Peregrina, A.; Robledo, M.; Matos, R.G.; Hilker, R.; Serrania, J.; Becker, A.; Arraiano, C.M.; Jiménez-Zurdo, J.I. Sinorhizobium meliloti YbeY is an endoribonuclease with unprecedented catalytic features, acting as silencing enzyme in riboregulation. Nucleic Acids Res. 2017, 45, 1371–1391. [Google Scholar] [CrossRef] [PubMed]
- Babu, V.M.P.; Sankari, S.; Budnick, J.A.; Caswell, C.C.; Walker, G.C. Sinorhizobium meliloti YbeY is a zinc-dependent single-strand specific endoribonuclease that plays an important role in 16S ribosomal RNA processing. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The prokaryotic non-coding transcriptome as revealed by dRNA-Seq. Identified Transcription Start Sites (TSSs; shown by arrows) can be assigned to mRNA and the different sRNA types (trans-sRNAs, asRNAs and mRNA-derived sense-sRNAs). See text for further details.
Figure 1.
The prokaryotic non-coding transcriptome as revealed by dRNA-Seq. Identified Transcription Start Sites (TSSs; shown by arrows) can be assigned to mRNA and the different sRNA types (trans-sRNAs, asRNAs and mRNA-derived sense-sRNAs). See text for further details.

Figure 2.
Key features of bacterial trans-sRNA biology. Differential regulation, target recognition and association with RNA chaperones and ribonucleases.
Figure 2.
Key features of bacterial trans-sRNA biology. Differential regulation, target recognition and association with RNA chaperones and ribonucleases.

Figure 3.
Activity mechanisms of S. meliloti cell-cycle trans-sRNAs. EcpR1 (left) is transcriptionally induced by the alarmone ppGpp and represses the early cell cycle transcriptional regulators DnaA and GcrA, restraining transition to G2 phase. GspR (right) regulates the expression of the gene encoding the essential cell division protein CtrA, preventing cytokinesis, and the cold-shock protein homolog CspA5, which is a CtrA activator.
Figure 3.
Activity mechanisms of S. meliloti cell-cycle trans-sRNAs. EcpR1 (left) is transcriptionally induced by the alarmone ppGpp and represses the early cell cycle transcriptional regulators DnaA and GcrA, restraining transition to G2 phase. GspR (right) regulates the expression of the gene encoding the essential cell division protein CtrA, preventing cytokinesis, and the cold-shock protein homolog CspA5, which is a CtrA activator.

Figure 4.
Cell processes that are riboregulated in rhizobia and the sRNAs involved.


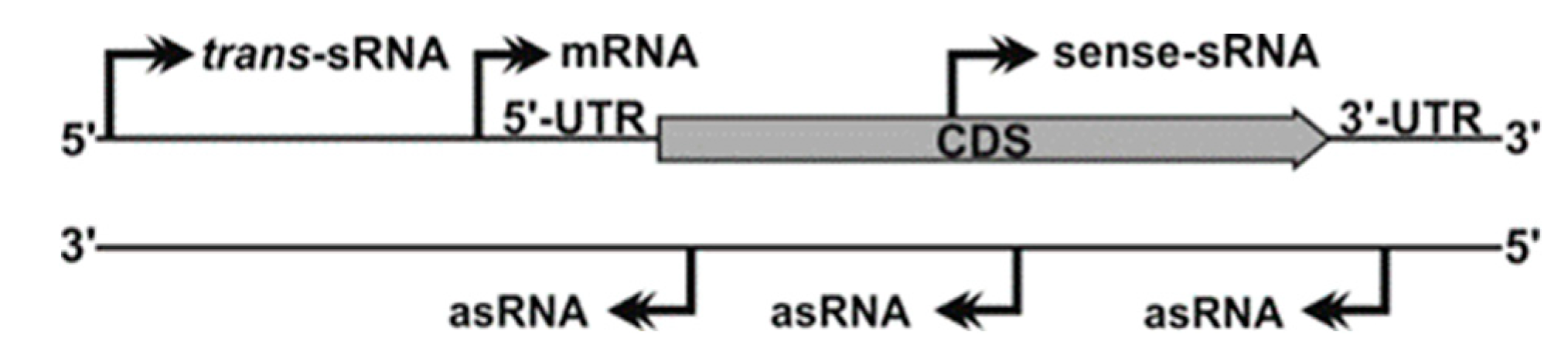{kind=link}
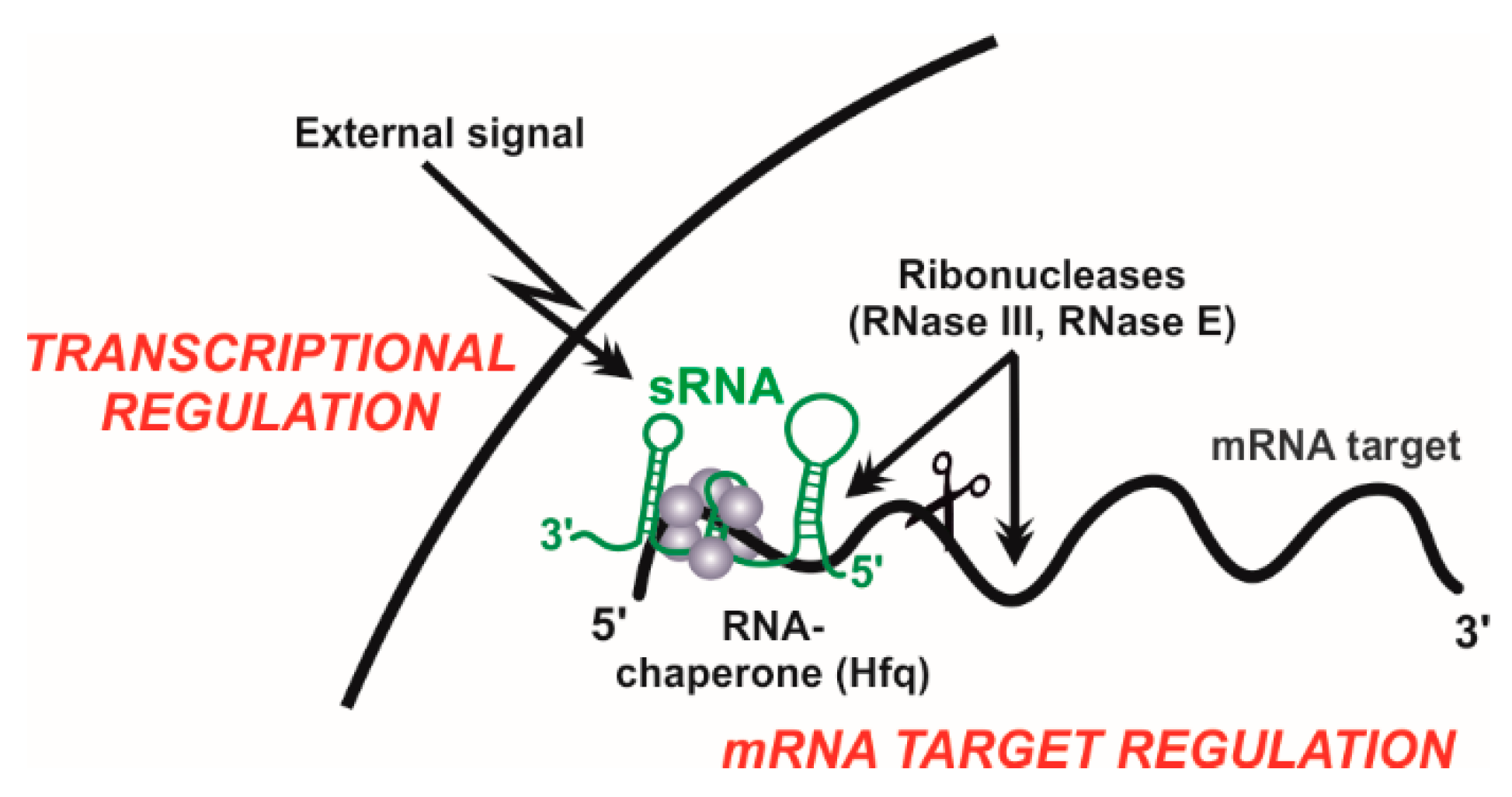{kind=link}
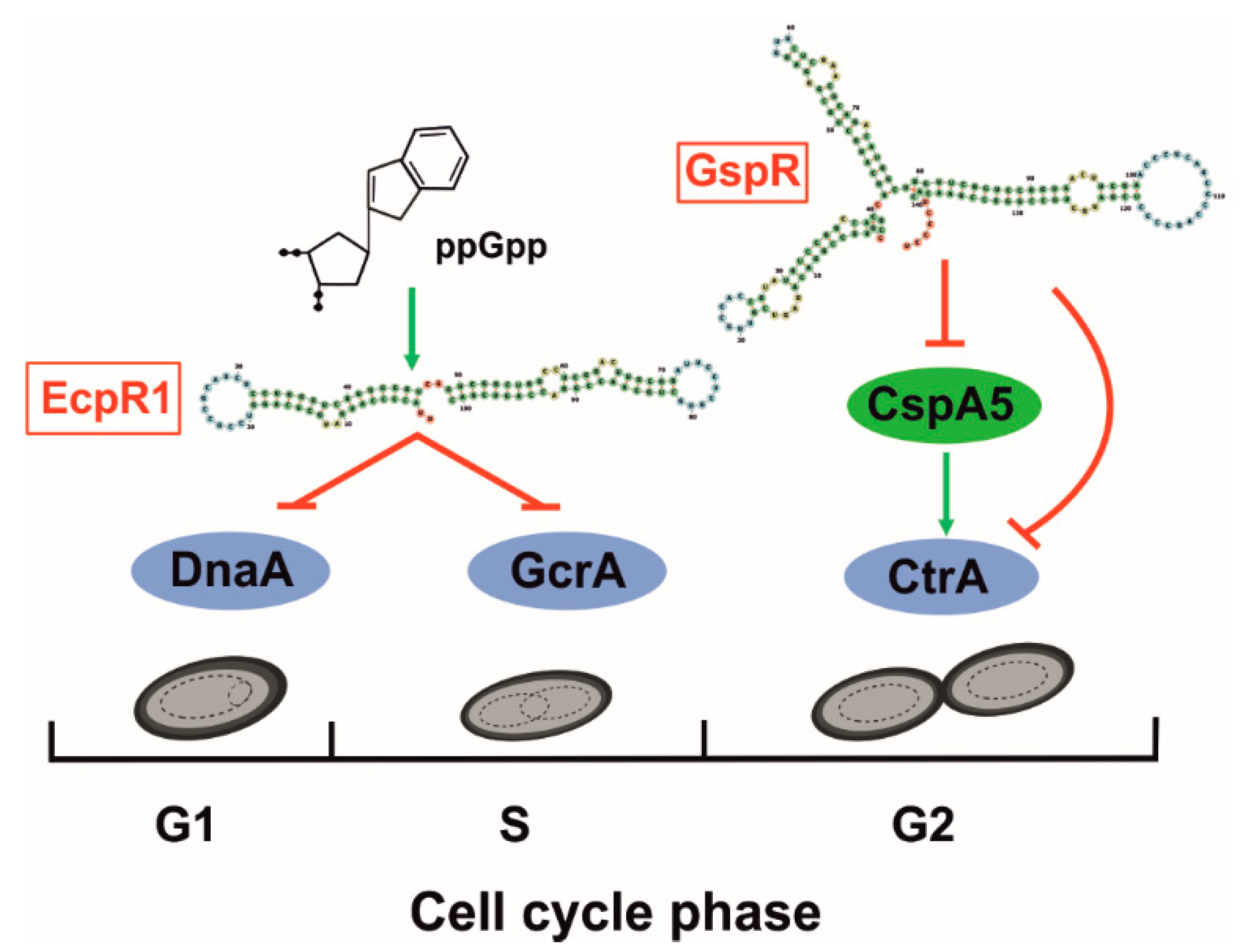{kind=link}
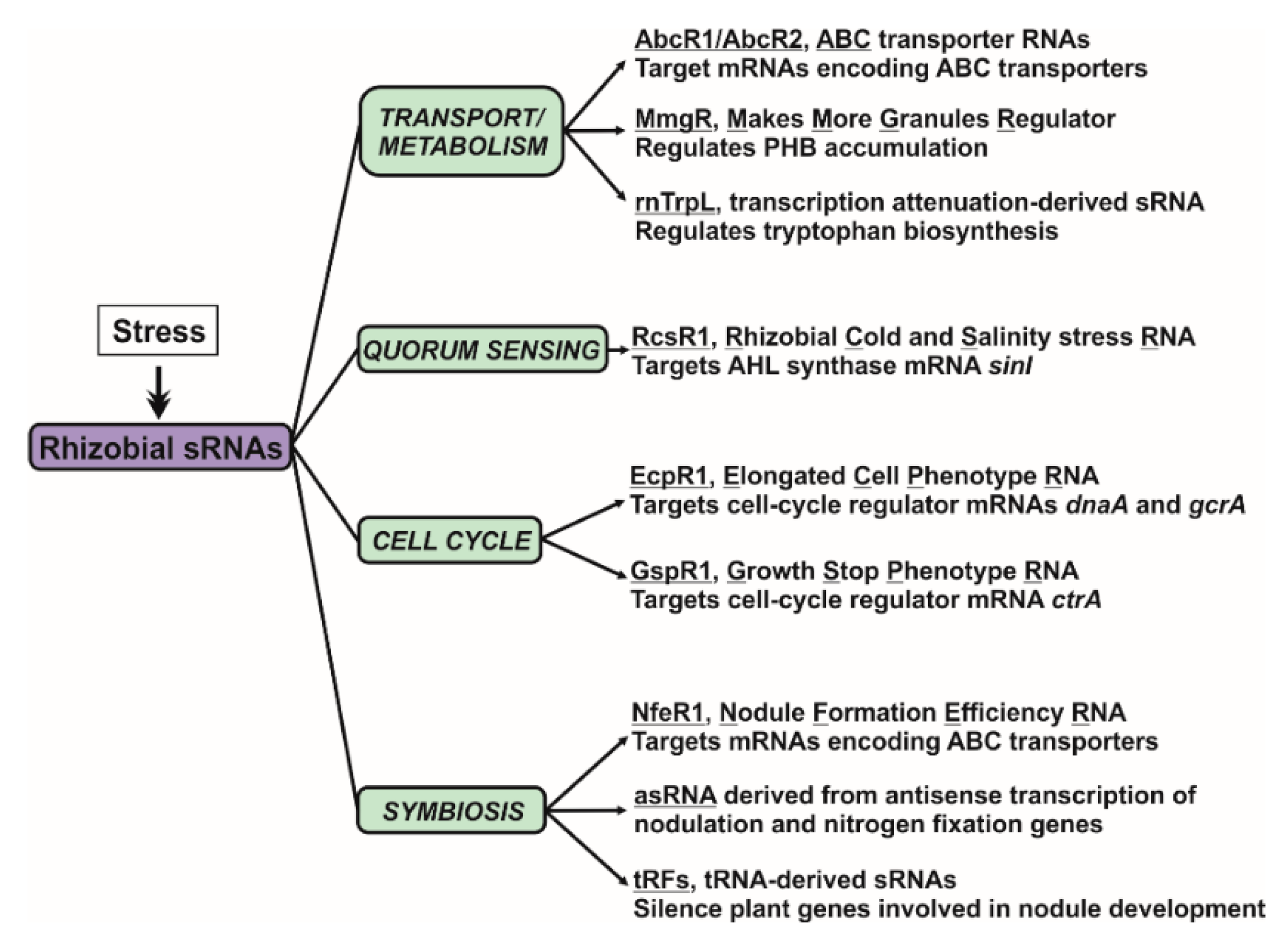{kind=link}
Table 1.
Identification, conservation and functional data of S. meliloti sRNAs.
| Symbionts | Pathogens | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name/s in S. meliloti 1021 and (S. meliloti 2011) a | Genome Copies | Coordinates in S. meliloti 1021 Genome | Strand | Predicted Promoter Motifs [27] | Hfq Co-IP RNA [32] | Expression in Nodules b [34] | S. meliloti | S. fredii | S. medicae | R. leguminosarum | R. etli | M. loti | B. japonicum | Agrobacterium sp. | Brucella sp. | Others | |
| Housekeeping | SMc04478/tmRNA/ssrA | 1 | 2291143–2291463 | + | RpoD | ||||||||||||
| SmelC035/sra05/4.5S/SRP–RNA/ffs (SMc06075) | 1 | 259927–260032 | + | RpoD RpoN | ZII/IZ/ZIII | ||||||||||||
| SmelC667/sra56/Smr22C/6S RNA/ssrs (SMc06867) | 1 | 2972090–2972252 | - | RpoD | IZ/ZIII | ||||||||||||
| SmelC809/sra50/SmrC19/RNase P/rnpB (SMc06867) | 1 | 2356793–2357134 | - | RpoD | IZ/ZIII | ||||||||||||
| trans-sRNAs | SmelC411/sm3/sra41/smrC15/AbcR2 (SMc06510) | 3 | 1698731–1698617 | - | RpoD RpoH1/2 | IZ/ZIII | |||||||||||
| SmelC412/sm3’/sra41/smrC16/AbcR1 (SMc06511) | 3 | 1698937–1698817 | - | RpoD | ZII/IZ/ZIII | ||||||||||||
| SmelC397/smrC14/NfeR1 (SMc06496) | 6 | 1667613–1667492 | - | RpoD | IZ/ZIII | ||||||||||||
| SmelC689/MmgR (SMc06891) | 3 | 3046712–3046789 | + | RpoD RpoN | IZ/ZIII | ||||||||||||
| SmelC706/smrC45/SpeF (SMc06909) | 1 | 3105298–3105445 | - | RpoH1/2 | ZIII | ||||||||||||
| SmelA075/smA8 (SMa6506) | 5 | 1220693–1220808 | + | RpoD | ZII/IZ | ||||||||||||
| SmelC023/sra03/smrC7 (SMc06056) | 1 | 201679–201825 | + | RpoD RpoH1/2 | ZII/ZIII | ||||||||||||
| SmelC289/sra32/smrC9 (SMc06412) | 1 | 1398278–1398426 | - | RpoD | ZII | ||||||||||||
| SmelC291/sra33/smrC10/EcpR1 (SMc06416+SMc06418) | 1 | 1411678–1411848 | + | ZII/IZ | |||||||||||||
| SmelC671/sm84 (SMc06875) | 1 | 2986421–2986520 | + | ZII | |||||||||||||
| SmelA099/smrA6 (SMa6570) | 1 | 1328175–1328334 | - | ZII/IZ/ZIII | |||||||||||||
| SmelB053/smrB35 (SMb23147) | 3 | 577730–577873 | + | RpoD RpoH1/2 | ZII | ||||||||||||
| SmelC151 (SMc06261) | 1 | 843451–843524 | + | IZ/ZIII | |||||||||||||
| SmelC587/sm104/RcsR1 (rnTrpL?) | 1 | 2575832–2575947 | - | ||||||||||||||
| SmelC165 | 1 | 910182–910297 | + | RpoN | |||||||||||||
| SmelC416/sm138 (SMc06519+) | 1 | 1718814–1718919 | - | ||||||||||||||
| SmelC500 (SMc06663) | 1 | 2180099–2180274 | - | ZII | |||||||||||||
| SmelC507 (SMc06677) | 1 | 2206348–2206579 | + | RpoD RpoH1/2 | ZII | ||||||||||||
| SmelC549/sm4 (SMc06721) | 1 | 2371597–2371855 | + | RpoD RpoH1/2 | ZII/IZ(ZIII | ||||||||||||
| SmelC601 (SMc06778) | 1 | 2625313–2625439 | - | RpoE2 | ZII/IZ/ZIII | ||||||||||||
| SmelC775/GspR1 (SMc07200) | 1 | 3509658–3509803 | + | ZII | |||||||||||||
| SmelA003 | 4 | 15179–15268 | + | ||||||||||||||
| SmelA033/smA3a/smrA2 | 1 | 512140–512221 | - | RpoD | |||||||||||||
| SmelB003 (SMb23003) | 1 | 30240–30345 | + | RpoH1 | IZ/ZIII | ||||||||||||
| SmelB008 | 1 | 65071–65147 | + | RpoE2 | |||||||||||||
| SmelB009 | 1 | 65186–65266 | - | RpoH1/2 | |||||||||||||
| SmelB033 (SMb23075) | 1 | 379319–379391 | - | RpoN | IZ | ||||||||||||
| SmelB044/smrB3b (SMb23116) | 2 | 541771–541909 | - | ZII/IZ | |||||||||||||
| SmelB075 (SMb23231) | 1 | 800730–800793 | - | RpoH1/2 | ZII | ||||||||||||
| SmelB126/smB9 (SMb23362) | 4 | 1325476–1325586 | + | RpoE2 | ZII/ZIII | ||||||||||||
| SmelC434/sm118 (SMc06565) | 1 | 1821211–1821366 | + | ZII | |||||||||||||
| SmelA001 (SMa6000) | 1 | 1002–1054 | + | RpoE2 | IZ/ZIII | ||||||||||||
| SmelA014 (SMa6078) | 1 | 206669–206772 | + | RpoH1/2 | ZII | ||||||||||||
| SmelA018 (SMa6091) | 1 | 234850–234930 | - | RpoE2 | ZIII | ||||||||||||
| SmelA019 | 1 | 235259–235355 | + | RpoH1/2 | ZII/IZ/ZIII | ||||||||||||
| SmelA020 (SMa6094) | 1 | 235393–235497 | - | RpoH1/2 | ZII/IZ/ZIII | ||||||||||||
| SmelA054 (SMa6367) | 1 | 911299–911374 | + | ZII | |||||||||||||
| SmelA056 (SMa6389) | 1 | 954480–954620 | - | ZII | |||||||||||||
| SmelC032 | 1 | 241172–241247 | + | ||||||||||||||
| SmelC749 (SMc07132) | 1 | 3383055–3383121 | - | ZIII | |||||||||||||
a sRNAs identified in the chromosome (SmelC) and megaplasmids pSymA (SmelA) and pSymB (SmelB). In bold, sRNAs with assigned functions as described in the text. b Nodule zones: ZII, invasion zone; IZ, bacteroid differentiation zone; ZIII, N fixation zone. Shadowed boxes stand for the presence/conservation of a given sRNA, +, positive/-, negative DNA strand.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Robledo, M.; García-Tomsig, N.I.; Jiménez-Zurdo, J.I. Riboregulation in Nitrogen-Fixing Endosymbiotic Bacteria. Microorganisms 2020, 8, 384. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8030384
AMA Style
Robledo M, García-Tomsig NI, Jiménez-Zurdo JI. Riboregulation in Nitrogen-Fixing Endosymbiotic Bacteria. Microorganisms. 2020; 8(3):384. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8030384
Chicago/Turabian StyleRobledo, Marta, Natalia I. García-Tomsig, and José I. Jiménez-Zurdo. 2020. "Riboregulation in Nitrogen-Fixing Endosymbiotic Bacteria" Microorganisms 8, no. 3: 384. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8030384
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.