Epidemiological Surveillance of Norovirus and Rotavirus in Sewage (2016–2017) in Valencia (Spain)

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Treatment
2.2. RNA Extraction and Molecular Detection of Noroviruses and Rotaviruses
2.3. Sequencing and Phylogenetic Analysis
3. Results
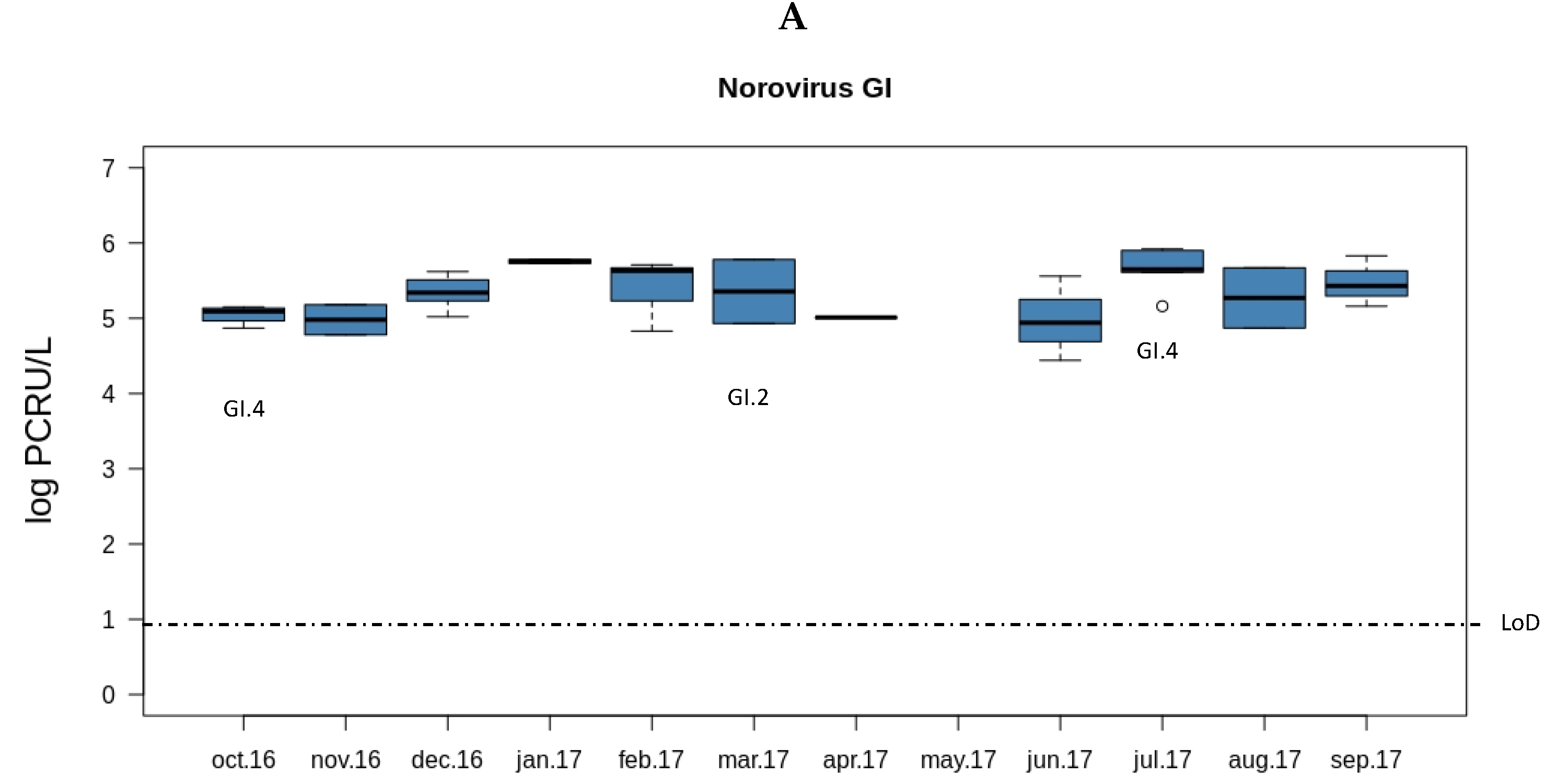
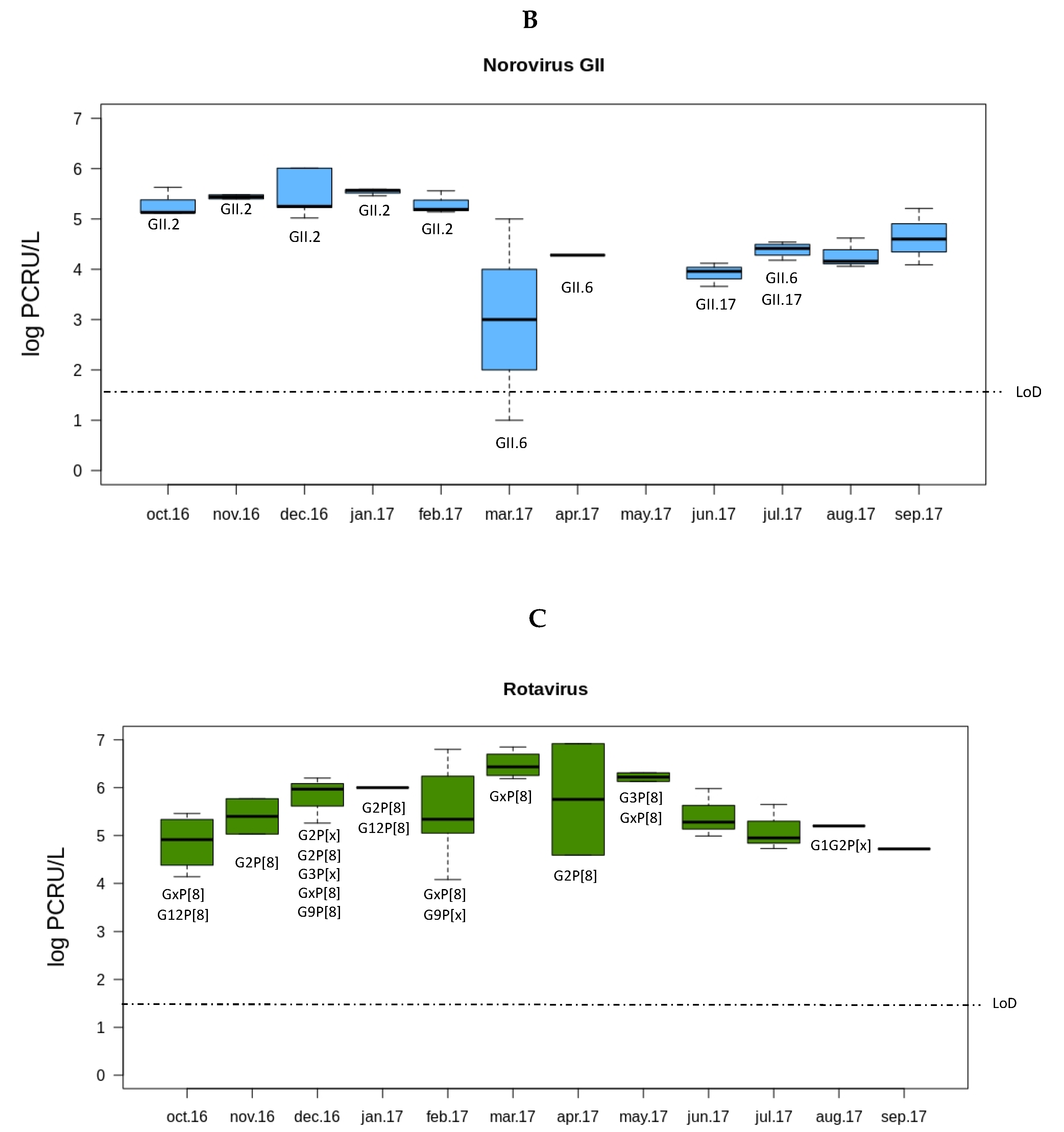
3.1. Prevalence of Rotaviruses and Noroviruses in Sewage Samples from Valencia
3.2. Rotavirus and Norovirus Genotypes from Sewage Samples
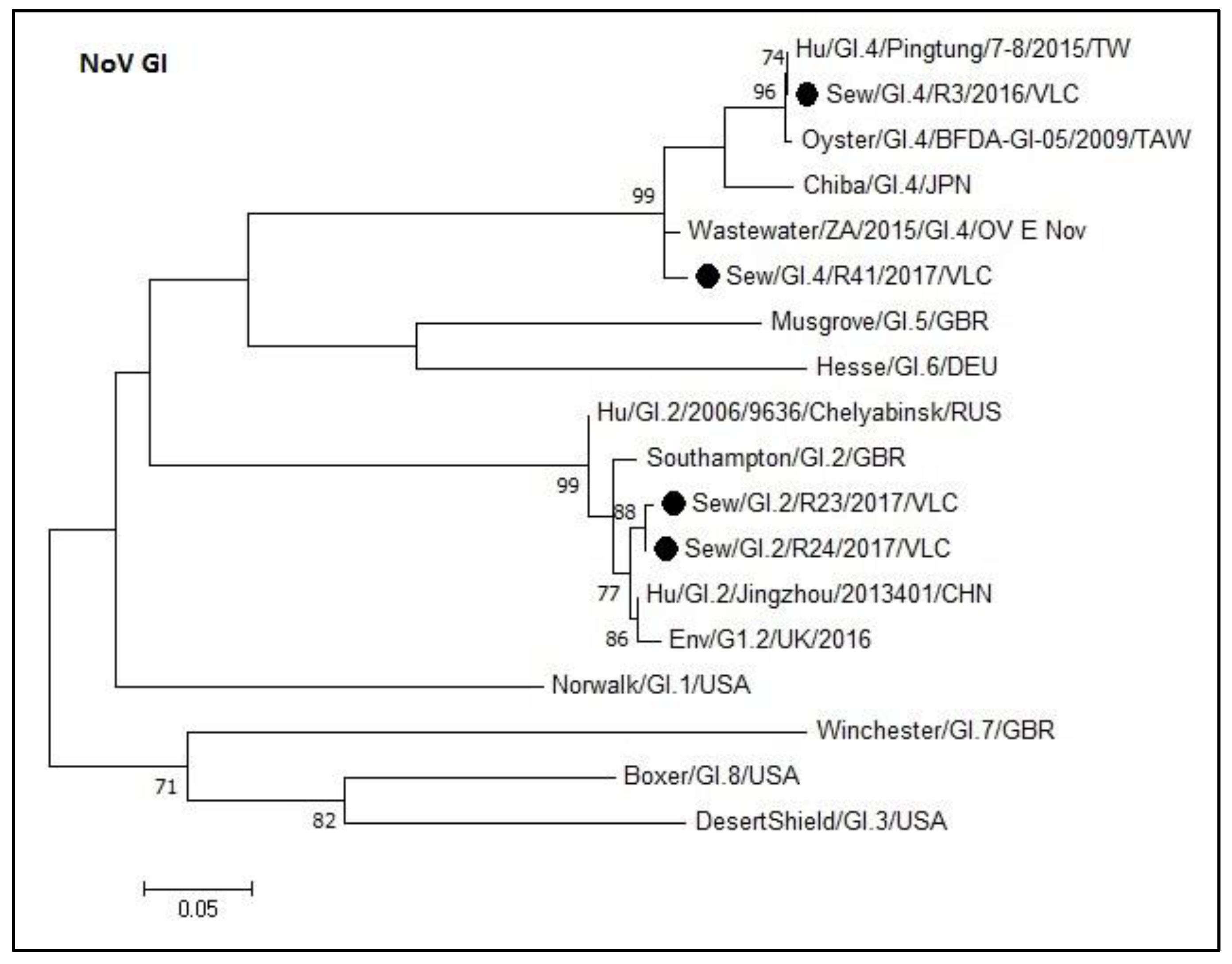
3.3. Phylogenetic Analyses of Sewage Detected Norovirus VP1 Coding Gene
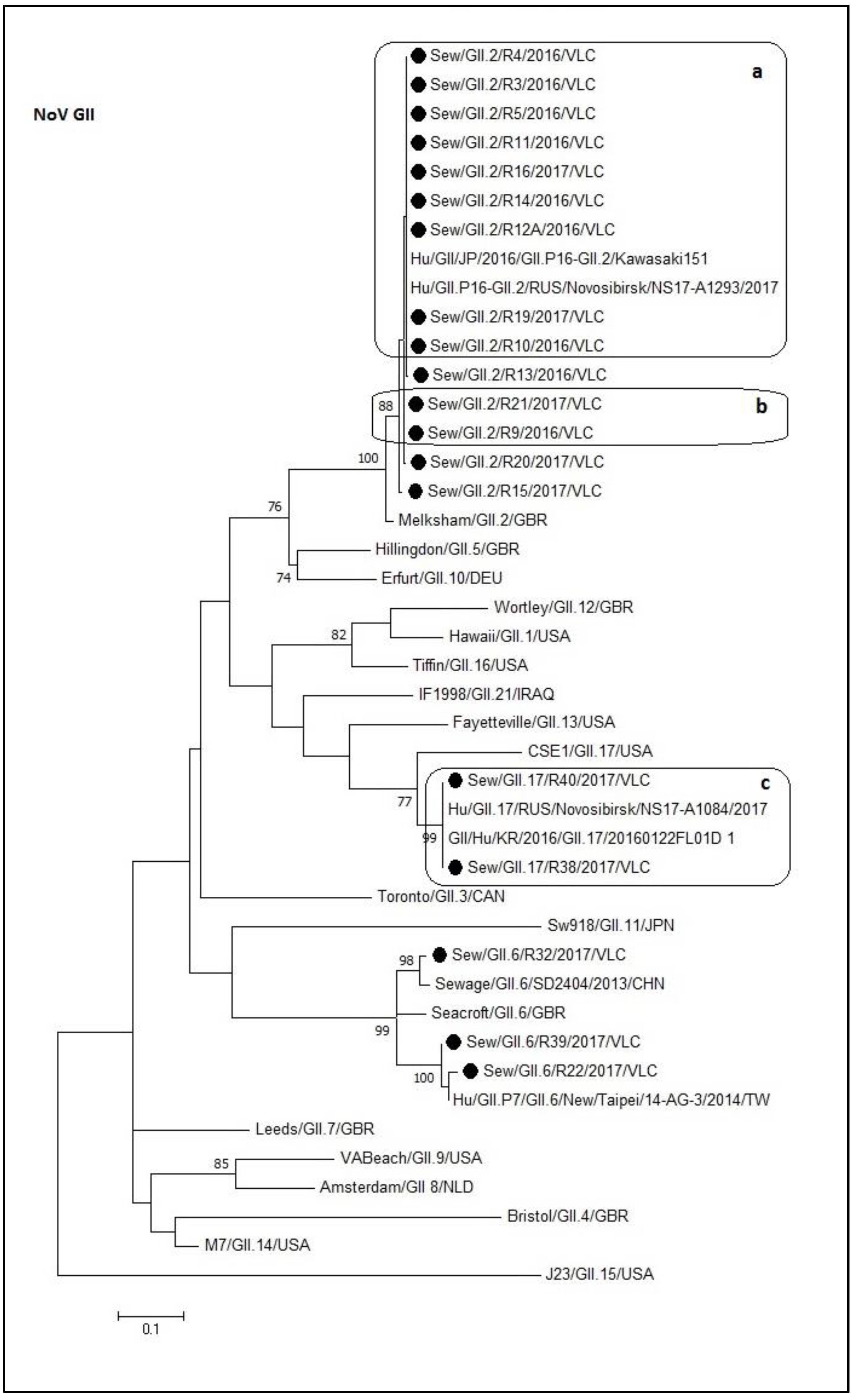
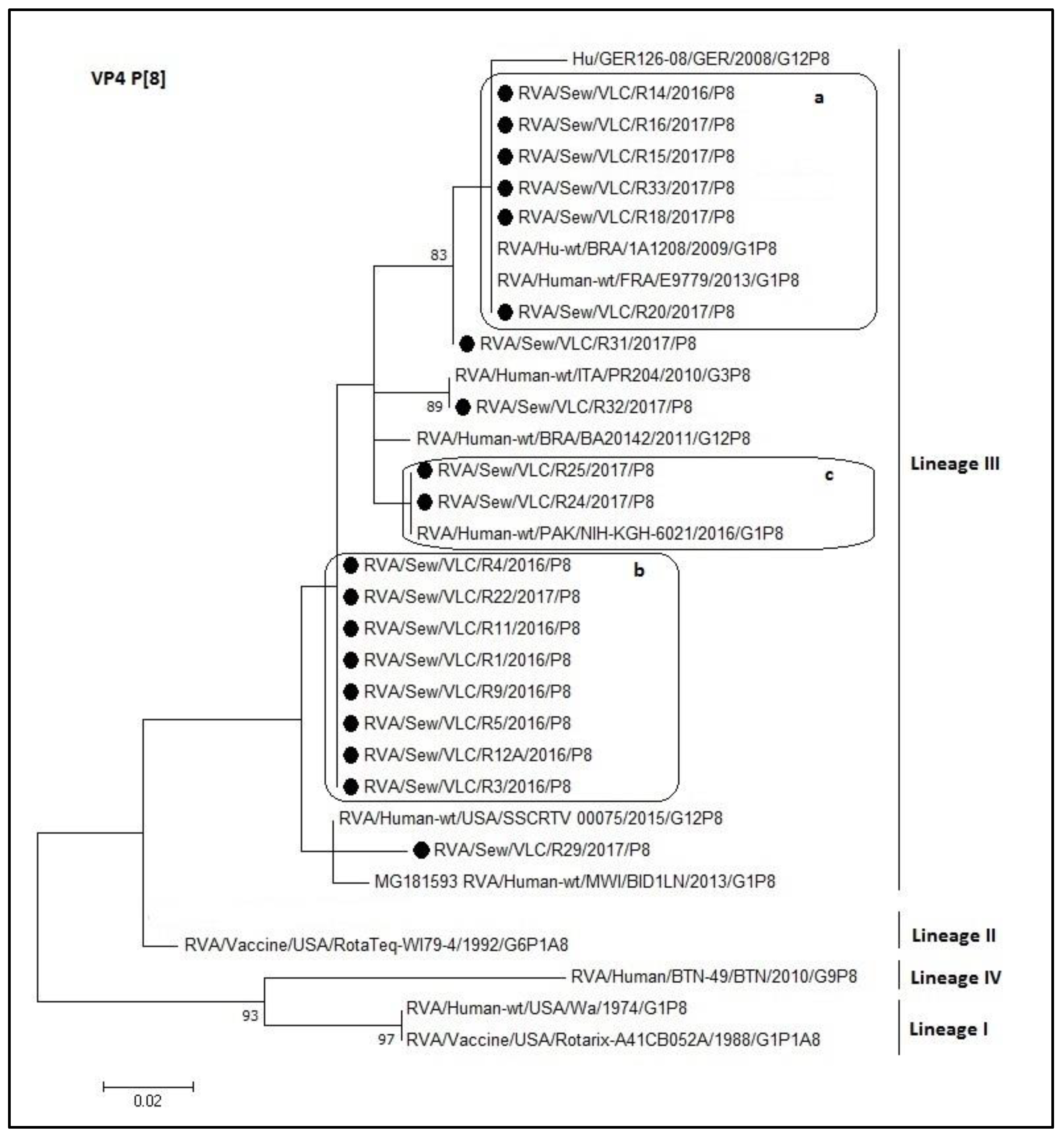
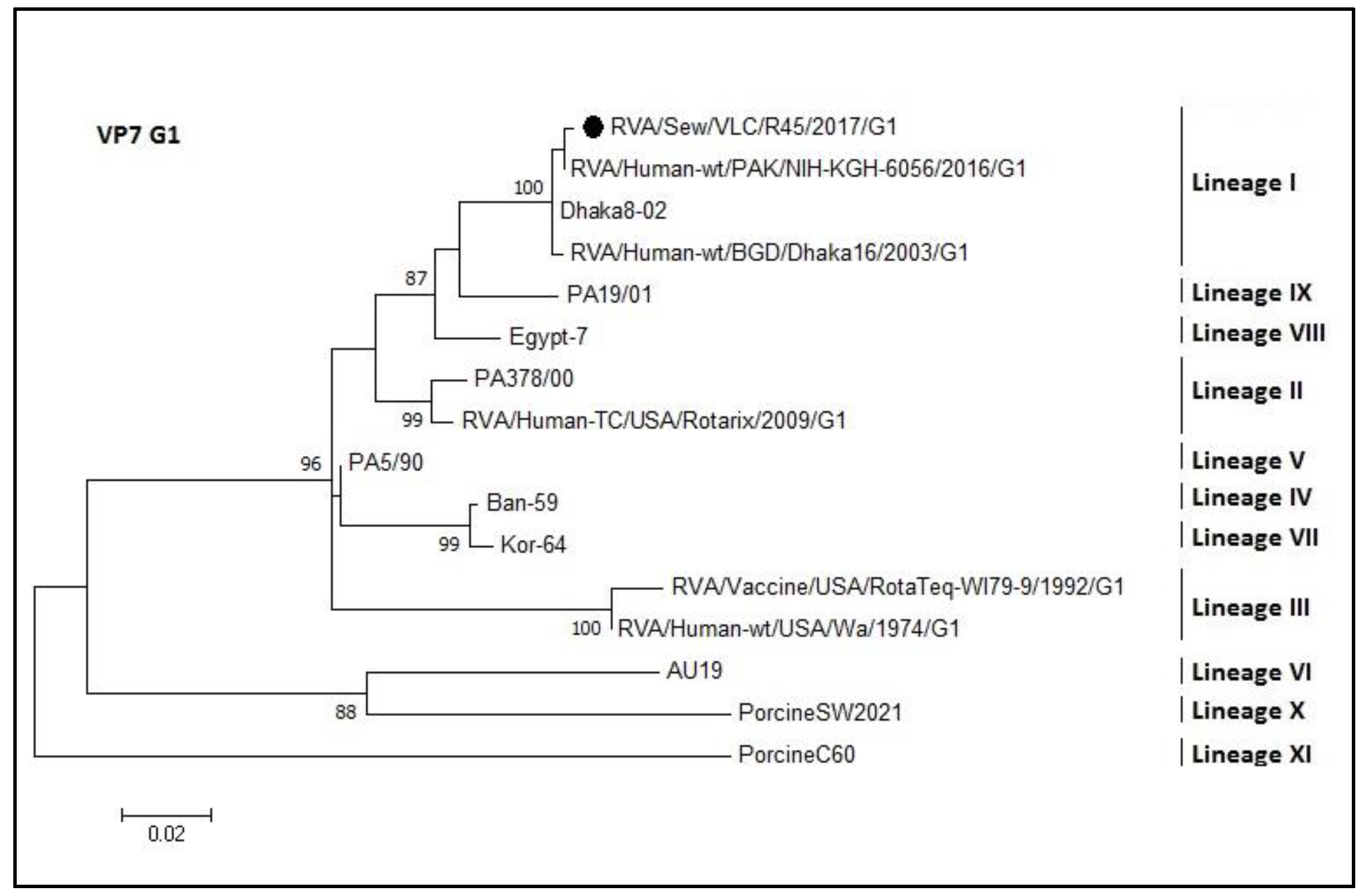
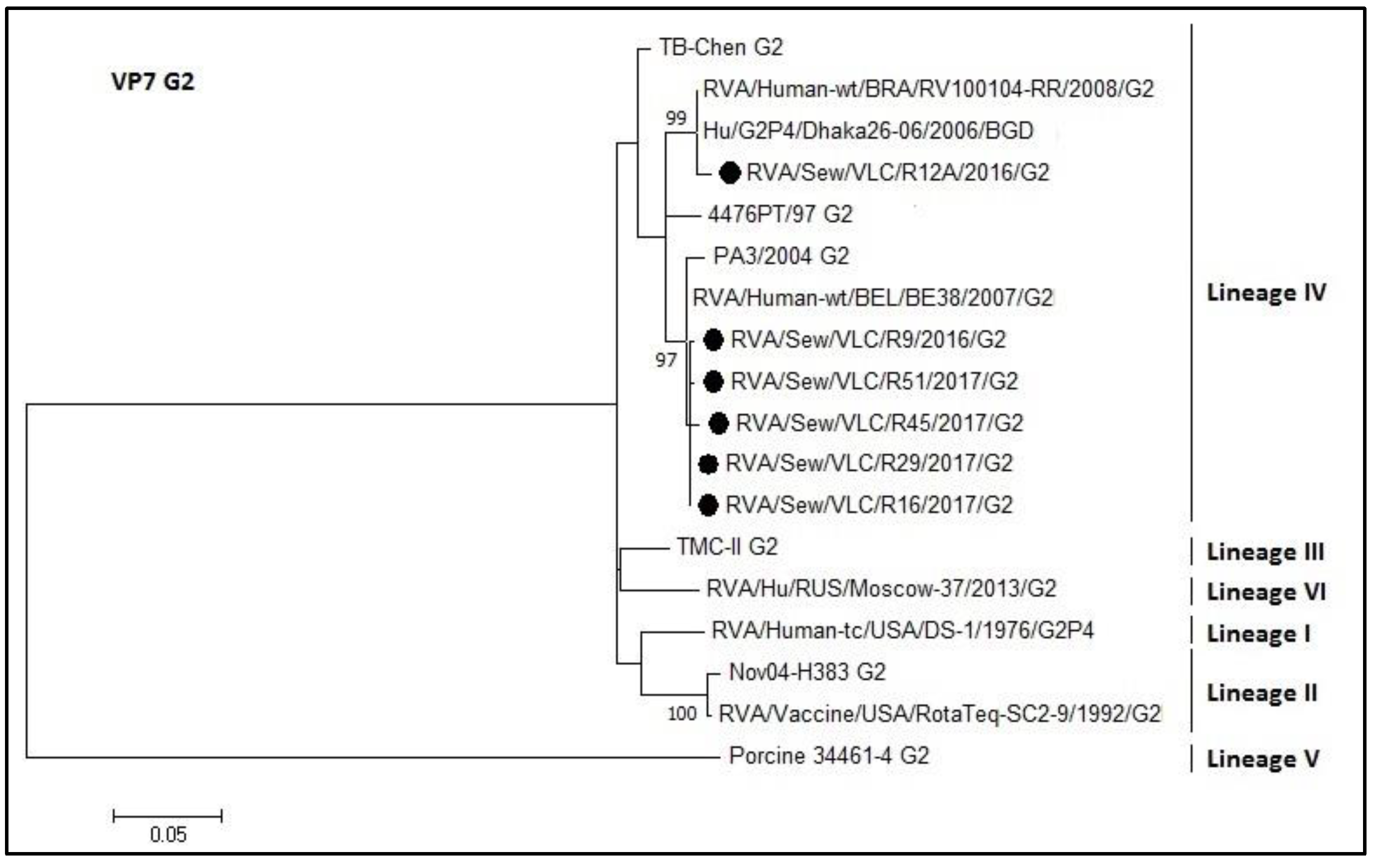
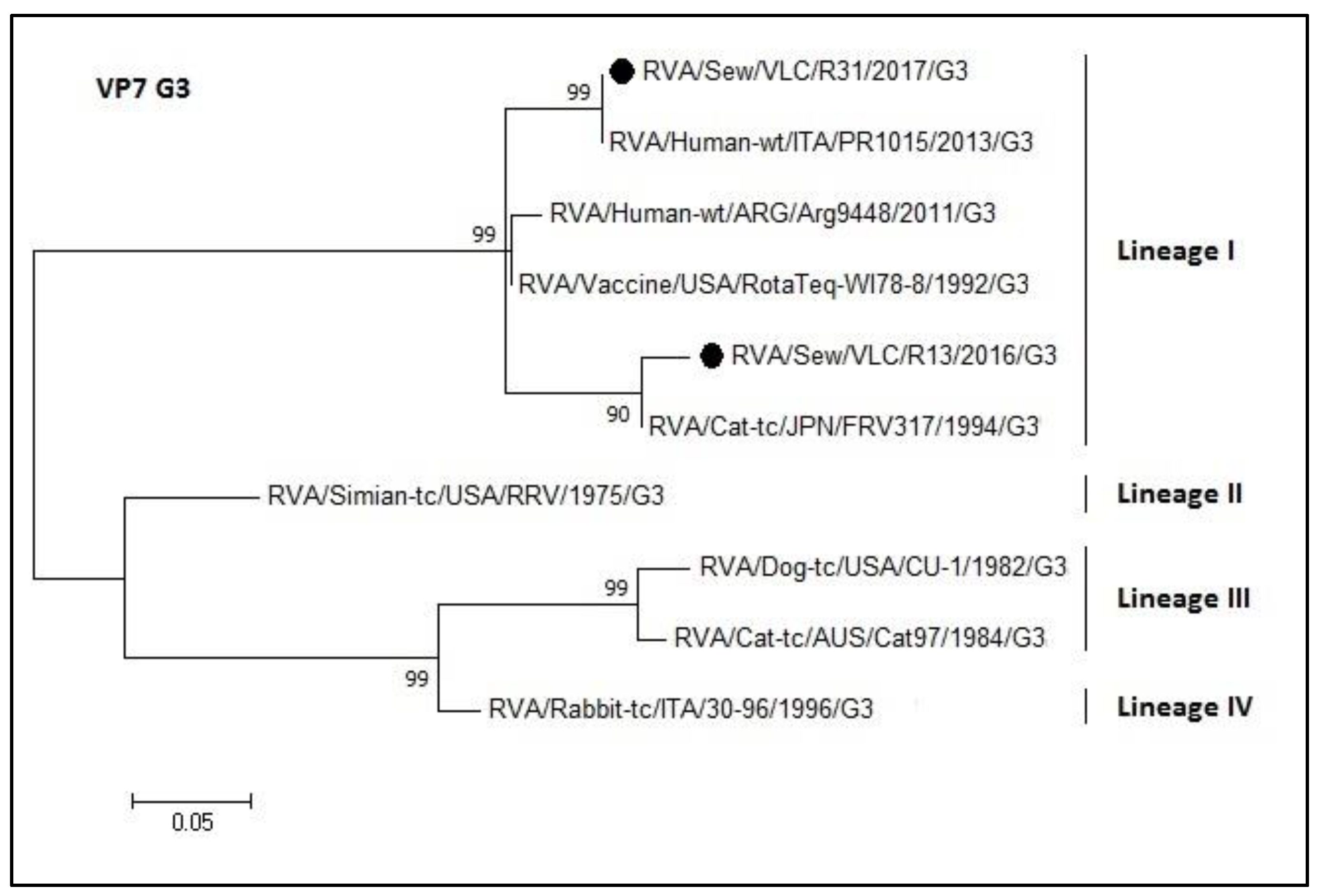
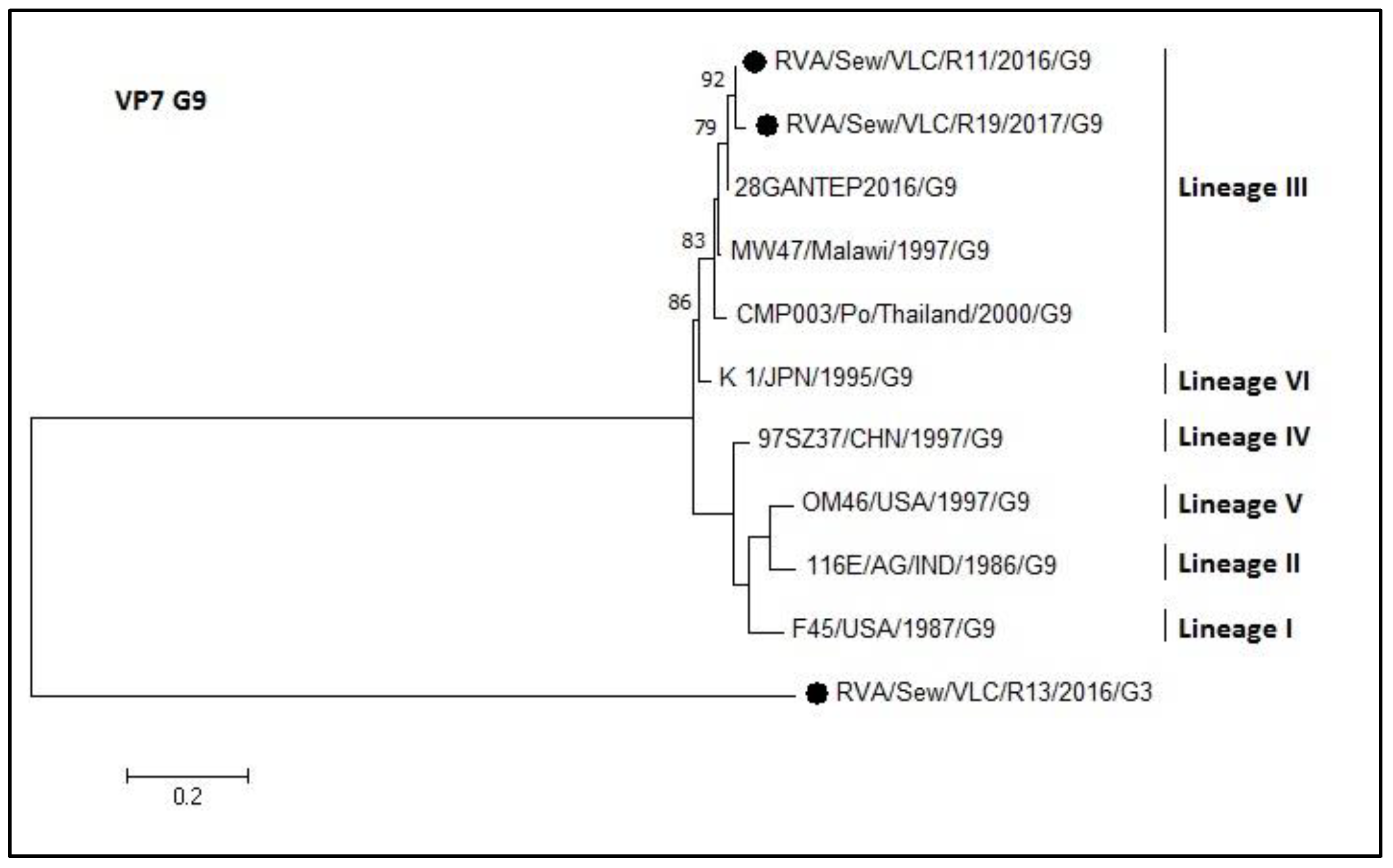
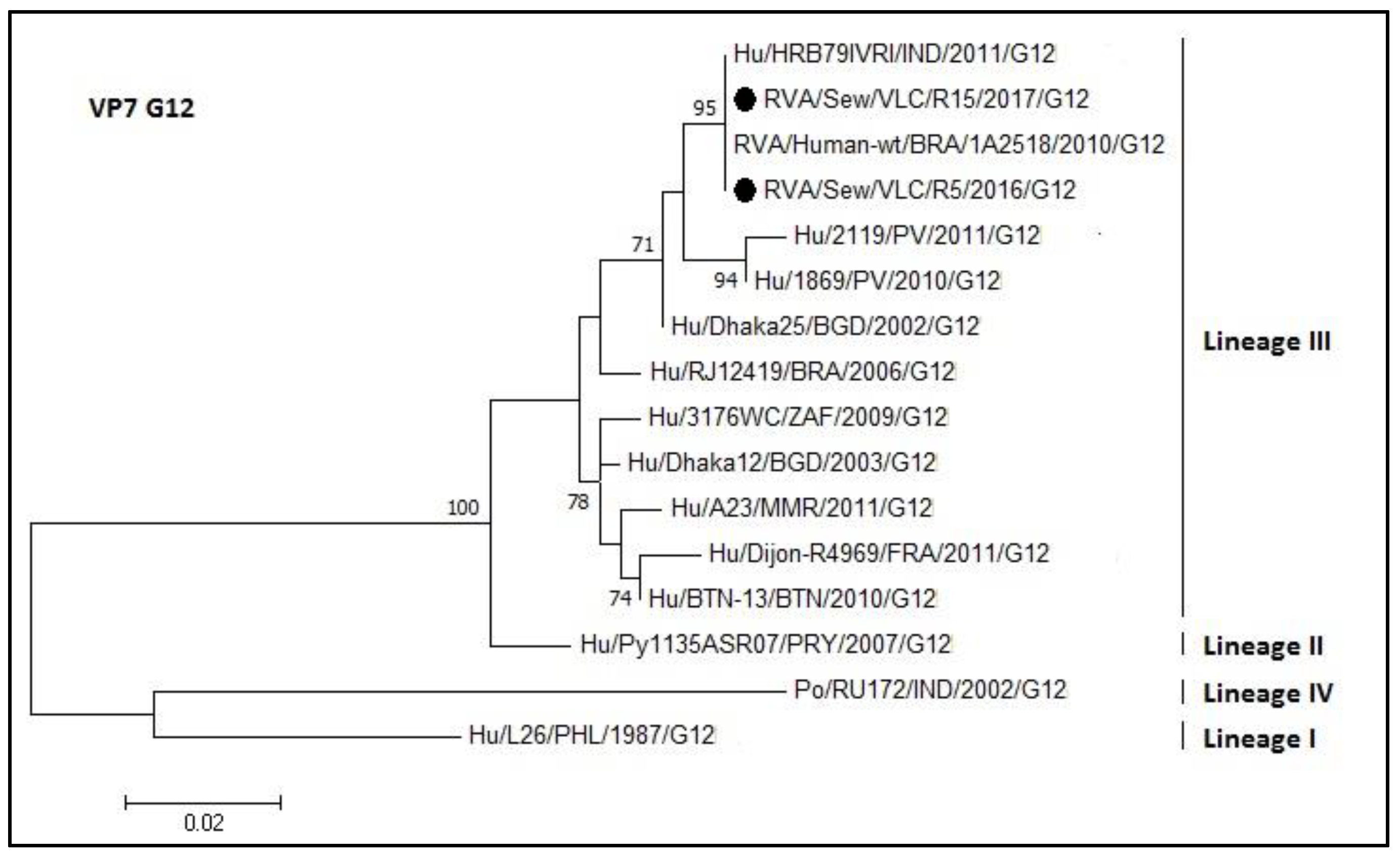
3.4. Phylogenetic Analyses of Sewage Detected Rotavirus VP4 and VP7 Coding Genes
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- GBD 2015 Mortality; Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef] [Green Version]
- Bartnicki, E.; Cunha, J.B.; Kolawole, A.O.; Wobus, C.E. Recent advances in understanding noroviruses. F1000Research 2017, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Widdowson, M.A.; Glass, R.I.; Akazawa, K.; Vinjé, J.; Parashar, U.D. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 2008, 14, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Miettinen, I. Persistence of Norovirus GII Genome in Drinking Water and Wastewater at Different Temperatures. Pathogens 2017, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Seitz, S.R.; Leon, J.S.; Schwab, K.J.; Lyon, G.M.; Dowd, M.; McDaniels, M.; Abdulhafid, G.; Fernandez, M.L.; Lindesmith, L.C.; Baric, R.S.; et al. Norovirus Infectivity in Humans and Persistence in Water. Appl. Environ. Microbiol. 2011, 77, 6884–6888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teunis, P.F.M.; Moe, C.L.; Liu, P.; Miller, S.E.; Lindesmith, L.; Baric, R.S.; Le Pendu, J.; Calderon, R.L. Norwalk virus: How infectious is it? J. Med. Virol. 2008, 80, 1468–1476. [Google Scholar] [CrossRef]
- Havelaar, A.H.; Kirk, M.D.; Torgerson, P.R.; Gibb, H.J.; Hald, T.; Lake, R.J.; Praet, N.; Bellinger, D.C.; de Silva, N.R.; Gargouri, N.; et al. World Health Organization Global Estimates and Regional Comparisons of the Burden of Foodborne Disease in 2010. PLoS Med. 2015, 12, e1001923. [Google Scholar] [CrossRef] [Green Version]
- Lopman, B.A.; Steele, D.; Kirkwood, C.D.; Parashar, U.D. The Vast and Varied Global Burden of Norovirus: Prospects for Prevention and Control. PLoS Med. 2016, 13, e1001999. [Google Scholar] [CrossRef]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.-W.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100. [Google Scholar] [CrossRef]
- Fauquet, C.M.; Mayo, M.a.; Maniloff, J.; Desselberger, U.; Ball, L.a. Virus Taxonomy: VIIIth Report of the International Committee on Taxonomy of Viruses. Virus Res. 2005. [Google Scholar]
- Xi, J.; Graham, D.; Wang, K.; Estes, M. Norwalk virus genome cloning and characterization. Science 1990, 250, 1580–1583. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.V.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray Crystallographic Structure of the Norwalk Virus Capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Bányai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RCWG Rotavirus Classification Working Group. Available online: https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification/rcwg (accessed on 7 November 2019).
- Buesa, J.; Rodriguez-Díaz, J. The Molecular Virology of Enteric Viruses. In Viruses in Foods; Springer International Publishing: Cham, Switzerland, 2016; pp. 59–130. [Google Scholar]
- Lodder, W.J.; de Roda Husman, A.M. Presence of Noroviruses and Other Enteric Viruses in Sewage and Surface Waters in The Netherlands. Appl. Environ. Microbiol. 2005, 71, 1453–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, H.; Haramoto, E.; Oguma, K.; Yamashita, H.; Tajima, A.; Nakajima, H.; Ohgaki, S. One-year monthly quantitative survey of noroviruses, enteroviruses, and adenoviruses in wastewater collected from six plants in Japan. Water Res. 2008, 42, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Diaz, J.; Querales, L.; Caraballo, L.; Vizzi, E.; Liprandi, F.; Takiff, H.; Betancourt, W.Q. Detection and Characterization of Waterborne Gastroenteritis Viruses in Urban Sewage and Sewage-Polluted River Waters in Caracas, Venezuela. Appl. Environ. Microbiol. 2009. [Google Scholar] [CrossRef] [Green Version]
- Bosch, A.; Guix, S.; Sano, D.; Pintó, R.M. New tools for the study and direct surveillance of viral pathogens in water. Curr. Opin. Biotechnol. 2008, 19, 295–301. [Google Scholar] [CrossRef]
- Pérez-Sautu, U.; Sano, D.; Guix, S.; Kasimir, G.; Pintó, R.M.; Bosch, A. Human norovirus occurrence and diversity in the Llobregat river catchment, Spain. Environ. Microbiol. 2012, 14, 494–502. [Google Scholar] [CrossRef]
- Miagostovich, M.P.; Ferreira, F.F.M.; Guimaraes, F.R.; Fumian, T.M.; Diniz-Mendes, L.; Luz, S.L.B.; Silva, L.A.; Leite, J.P.G. Molecular Detection and Characterization of Gastroenteritis Viruses Occurring Naturally in the Stream Waters of Manaus, Central Amazonia, Brazil. Appl. Environ. Microbiol. 2008, 74, 375–382. [Google Scholar] [CrossRef] [Green Version]
- Betancourt, W.Q.; Querales, L.; Sulbaran, Y.F.; Rodriguez-Diaz, J.; Caraballo, L.; Pujol, F.H. Molecular Characterization of Sewage-Borne Pathogens and Detection of Sewage Markers in an Urban Stream in Caracas, Venezuela. Appl. Environ. Microbiol. 2010. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Ferrando, E.; Randazzo, W.; Pérez-Cataluña, A.; Sánchez, G. HEV Occurrence in Waste and Drinking Water Treatment Plants. Front. Microbiol. 2020, 10, 2937. [Google Scholar] [CrossRef] [PubMed]
- Alcala, A.; Vizzi, E.; Rodriguez-Diaz, J.; Zambrano, J.L.; Betancourt, W.; Liprandi, F. Molecular Detection and Characterization of Aichi Viruses in Sewage-Polluted Waters of Venezuela. Appl. Environ. Microbiol. 2010, 76, 4113–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, G.; Elizaquível, P.; Aznar, R. A single method for recovery and concentration of enteric viruses and bacteria from fresh-cut vegetables. Int. J. Food Microbiol. 2012, 152, 9–13. [Google Scholar] [CrossRef]
- Randazzo, W.; Piqueras, J.; Evtoski, Z.; Sastre, G.; Sancho, R.; Gonzalez, C.; Sánchez, G. Interlaboratory Comparative Study to Detect Potentially Infectious Human Enteric Viruses in Influent and Effluent Waters. Food Environ. Virol. 2019, 11, 350–363. [Google Scholar] [CrossRef] [PubMed]
- ISO/TS. Microbiology of Food and Animal Feed. Horizontal Method for Determination of Hepatitis A Virus and Norovirus in Food Using Real-Time RT-PCR Title; ISO/TS: Geneva, Switzerland, 2013. [Google Scholar]
- ISO/TS. Microbiology of Food and Animal Feed. Horizontal Method for Determination of Hepatitis A Virus and Norovirus in Food Using Real-time RT-PCR. Part 1: Method for Quantification; ISO/TS: Geneva, Switzerland, 2017. [Google Scholar]
- Jothikumar, N.; Kang, G.; Hill, V.R. Broadly reactive TaqMan®assay for real-time RT-PCR detection of rotavirus in clinical and environmental samples. J. Virol. Methods 2009, 155, 126–131. [Google Scholar] [CrossRef]
- Pinto, R.M.; Costafreda, M.I.; Bosch, A. Risk Assessment in Shellfish-Borne Outbreaks of Hepatitis A. Appl. Environ. Microbiol. 2009, 75, 7350–7355. [Google Scholar] [CrossRef] [Green Version]
- Randazzo, W.; Piqueras, J.; Rodríguez-Díaz, J.; Aznar, R.; Sánchez, G. Improving efficiency of viability-qPCR for selective detection of infectious HAV in food and water samples. J. Appl. Microbiol. 2017, 124, 958–964. [Google Scholar] [CrossRef]
- Randazzo, W.; López-Gálvez, F.; Allende, A.; Aznar, R.; Sánchez, G. Evaluation of viability PCR performance for assessing norovirus infectivity in fresh-cut vegetables and irrigation water. Int. J. Food Microbiol. 2016, 229, 1–6. [Google Scholar] [CrossRef]
- Eurorotanet EuroRotaNet. Available online: https://www.eurorotanet.com/project-information/documents-and-methods/ (accessed on 8 October 2019).
- Iturriza-Gómara, M.; Kang, G.; Gray, J. Rotavirus genotyping: Keeping up with an evolving population of human rotaviruses. J. Clin. Virol. 2004, 31, 259–265. [Google Scholar] [CrossRef]
- Kageyama, T.; Shinohara, M.; Uchida, K.; Fukushi, S.; Hoshino, F.B.; Kojima, S.; Takai, R.; Oka, T.; Takeda, N.; Katayama, K. Coexistence of multiple genotypes, including newly identified genotypes, in outbreaks of gastroenteritis due to Norovirus in Japan. J. Clin. Microbiol. 2004, 42, 2988–2995. [Google Scholar] [CrossRef] [Green Version]
- Kojima, S.; Kageyama, T.; Fukushi, S.; Hoshino, F.B.; Shinohara, M.; Uchida, K.; Natori, K.; Takeda, N.; Katayama, K. Genogroup-specific PCR primers for detection of Norwalk-like viruses. J. Virol. Methods 2002, 100, 107–114. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholas, K.B.; Nicholas, H.B., Jr. GeneDoc: A tool for editing and annotating multiple sequence alignments. IEEE Softw. 1997. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar]
- Hoque, S.A.; Thongprachum, A.; Takanashi, S.; Mostafa, S.M.; Saito, H.; Anwar, K.S.; Nomura, A.; Hoque, S.A.; Begum, R.; Sultana, U.N.; et al. Alarming Situation of Spreading Enteric Viruses Through Sewage Water in Dhaka City: Molecular Epidemiological Evidences. Food Environ. Virol. 2019, 11, 65–75. [Google Scholar] [CrossRef]
- Randazzo, W.; Khezri, M.; Ollivier, J.; Le Guyader, F.S.; Rodríguez-Díaz, J.; Aznar, R.; Sánchez, G. Optimization of PMAxx pretreatment to distinguish between human norovirus with intact and altered capsids in shellfish and sewage samples. Int. J. Food Microbiol. 2018, 266, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Strubbia, S.; Phan, M.V.T.; Schaeffer, J.; Koopmans, M.; Cotten, M.; Le Guyader, F.S. Characterization of Norovirus and Other Human Enteric Viruses in Sewage and Stool Samples Through Next-Generation Sequencing. Food Environ. Virol. 2019, 11, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Cassi, X.; Timoneda, N.; Martínez-Puchol, S.; Rusiñol, M.; Rodriguez-Manzano, J.; Figuerola, N.; Bofill-Mas, S.; Abril, J.F.; Girones, R. Metagenomics for the study of viruses in urban sewage as a tool for public health surveillance. Sci. Total Environ. 2018, 618, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Puchol, S.; Rusiñol, M.; Fernández-Cassi, X.; Timoneda, N.; Itarte, M.; Andrés, C.; Antón, A.; Abril, J.F.; Girones, R.; Bofill-Mas, S. Characterisation of the sewage virome: Comparison of NGS tools and occurrence of significant pathogens. Sci. Total Environ. 2020, 713, 136604. [Google Scholar] [CrossRef] [PubMed]
- Oshiki, M.; Miura, T.; Kazama, S.; Segawa, T.; Ishii, S.; Hatamoto, M.; Yamaguchi, T.; Kubota, K.; Iguchi, A.; Tagawa, T.; et al. Microfluidic PCR Amplification and MiSeq Amplicon Sequencing Techniques for High-Throughput Detection and Genotyping of Human Pathogenic RNA Viruses in Human Feces, Sewage, and Oysters. Front. Microbiol. 2018, 9, 830. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Parra, G.I.; Levenson, E.A.; Green, K.Y. A large outbreak of acute gastroenteritis in Shippensburg, Pennsylvania, 1972 revisited: Evidence for common source exposure to a recombinant GII.Pg/GII.3 norovirus. Epidemiol. Infect. 2017, 145, 1591–1596. [Google Scholar] [CrossRef] [Green Version]
- Chan, M.C.W.; Hu, Y.; Chen, H.; Podkolzin, A.T.; Zaytseva, E.V.; Komano, J.; Sakon, N.; Poovorawan, Y.; Vongpunsawad, S.; Thanusuwannasak, T.; et al. Global spread of norovirus gii.17 kawasaki 308, 2014–2016. Emerg. Infect. Dis. 2017, 23, 1354–1359. [Google Scholar] [CrossRef]
- Green, K.Y.; Belliot, G.; Taylor, J.L.; Valdesuso, J.; Lew, J.F.; Kapikian, A.Z.; Lin, F.C. A Predominant Role for Norwalk-like Viruses as Agents of Epidemic Gastroenteritis in Maryland Nursing Homes for the Elderly. J. Infect. Dis. 2002, 185, 133–146. [Google Scholar] [CrossRef]
- Boon, D.; Mahar, J.E.; Abente, E.J.; Kirkwood, C.D.; Purcell, R.H.; Kapikian, A.Z.; Green, K.Y.; Bok, K. Comparative Evolution of GII.3 and GII.4 Norovirus over a 31-Year Period. J. Virol. 2011, 85, 8656–8666. [Google Scholar] [CrossRef] [Green Version]
- Hardy, M.E.; Kramer, S.F.; Treanor, J.J.; Estes, M.K. Human calicivirus genogroup II capsid sequence diversity revealed by analyses of the prototype Snow Mountain agent. Arch. Virol. 1997, 142, 1469–1479. [Google Scholar] [CrossRef]
- Santiso-Bellón, C.; Monzó, C.; Fuentes-Trillo, A.; Vila-Vicent, S.; da Silva Ribeiro de Andrade, J.; Gozalbo-Rovira, R.; Buesa, J.; Chaves, F.J.; Rodríguez-Díaz, J. Nearly Complete Genome Sequences of Human Norovirus Belonging to Several Genotypes from Valencia, Spain. Microbiol. Resour. Announc. 2019, 8, e00641-19. [Google Scholar] [CrossRef] [Green Version]
- Ao, Y.; Cong, X.; Jin, M.; Sun, X.; Wei, X.; Wang, J.; Zhang, Q.; Song, J.; Yu, J.; Cui, J.; et al. Genetic Analysis of Re-Emerging GII.P16–GII.2 Noroviruses in 2016–2017 in China. J. Infect. Dis. 2018, 218, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medici, M.C.; Tummolo, F.; Martella, V.; De Conto, F.; Arcangeletti, M.C.; Pinardi, F.; Ferraglia, F.; Chezzi, C.; Calderaro, A. Emergence of novel recombinant GII.P16-GII.2 and GII. P16-GII.4 Sydney 2012 norovirus strains in Italy, winter 2016/2017. New Microbiol. 2018, 41, 71–72. [Google Scholar]
- Niendorf, S.; Jacobsen, S.; Faber, M.; Eis-Hübinger, A.M.; Hofmann, J.; Zimmermann, O.; Höhne, M.; Bock, C.T. Steep rise in norovirus cases and emergence of a new recombinant strain GII.P16-GII.2, Germany, winter 2016. Eurosurveillance 2017. [Google Scholar]
- Thongprachum, A.; Okitsu, S.; Khamrin, P.; Maneekarn, N.; Hayakawa, S.; Ushijima, H. Emergence of norovirus GII.2 and its novel recombination during the gastroenteritis outbreak in Japanese children in mid-2016. Infect. Genet. Evol. 2017, 51, 86–88. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Rotavirus vaccines WHO position paper - January 2013. Wkly. Epidemiol. Rec. 2013, 88, 49–64. [Google Scholar]
- Kirkwood, C.D. Genetic and Antigenic Diversity of Human Rotaviruses: Potential Impact on Vaccination Programs. J. Infect. Dis. 2010, 202 (Suppl.: S43-8). [Google Scholar] [CrossRef]
- Iturriza-Gómara, M.; Dallman, T.; Bányai, K.; Böttiger, B.; Buesa, J.; Dietrich, S.; Fiore, L.; Johansen, K.; Koopmans, M.; KORSUN, N.; et al. Rotavirus genotypes co-circulating in Europe between 2006 and 2009 as determined by EuroRotaNet, a pan-European collaborative strain surveillance network. Epidemiol. Infect. 2011, 139, 895–909. [Google Scholar] [CrossRef]
- Estes, M.K.; Kapikian, A.Z. Rotaviruses. In Fields’ Virology; Knipe, D.M., Lippincott, H.P., Eds.; Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1918–1974. ISBN 0781760607. [Google Scholar]
- Bányai, K.; László, B.; Duque, J.; Steele, A.D.; Nelson, E.A.S.; Gentsch, J.R.; Parashar, U.D. Systematic review of regional and temporal trends in global rotavirus strain diversity in the pre rotavirus vaccine era: Insights for understanding the impact of rotavirus vaccination programs. Vaccine 2012, 30, 122–130. [Google Scholar] [CrossRef]
- Delogu, R.; Ianiro, G.; Camilloni, B.; Fiore, L.; Ruggeri, F.M. Unexpected spreading of G12P[8] rotavirus strains among young children in a small area of central Italy. J. Med. Virol. 2015, 87, 1292–1302. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Heylen, E.; Zeller, M.; Rahman, M.; Lemey, P.; Van Ranst, M. Phylodynamic analyses of rotavirus genotypes G9 and G12 underscore their potential for swift global spread. Mol. Biol. Evol. 2010, 27, 2431–2436. [Google Scholar] [CrossRef] [Green Version]
- Mijatovic-Rustempasic, S.; Teel, E.N.; Kerin, T.K.; Hull, J.J.; Roy, S.; Weinberg, G.A.; Payne, D.C.; Parashar, U.D.; Gentsch, J.R.; Bowen, M.D. Genetic analysis of G12P[8] rotaviruses detected in the largest U.S. G12 genotype outbreak on record. Infect. Genet. Evol. 2014, 21, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Ortín, R.; Vila-Vicent, S.; Carmona-Vicente, N.; Santiso-Bellón, C.; Rodríguez-Díaz, J.; Buesa, J. Histo-Blood Group Antigens in Children with Symptomatic Rotavirus Infection. Viruses 2019, 11, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Purpose | Primer or Probe Name | Sequence | Nucleotide Positions |
|---|---|---|---|---|
| Norovirus GI | qPCR | QNIF4 | 5′-CGC TGG ATG CGN TTC CAT-3′ | 5291–5308 |
| Norovirus GI | qPCR | NV1LCR | 5′-CCT TAG ACG CCA TCA TCA TTT AC-3′ | 5354–5376 |
| Norovirus GI | qPCR | NVGG1p | VIC-5′-TGG ACA GGA GAY CGC RAT CT-3′-Quencher | 5321–5340 |
| Norovirus GII | qPCR | QNIF2 | 5′-ATG TTC AGR TGG ATG AGR TTC TCW GA-3′ | 5012–5037 |
| Norovirus GII | qPCR | COG2R | 5′-TCG ACG CCA TCT TCA TTC ACA-3′ | 5080–5100 |
| Norovirus GII | qPCR | QNIFs | FAM-5′-AGC ACG TGG GAG GGC GAT CG-3′-Quencher | 5042–5061 |
| Mengovirus | qPCR | Mengo 110 | 5′-GCG GGT CCT GCC GAA AGT-3′ | 110–127 |
| Mengovirus | qPCR | Mengo 209 | 5′-GAA GTA ACA TAT AGA CAG ACG CAC AC-3′ | 245–270 |
| Mengovirus | qPCR | Mengo 147 | FAM-5′-ATC ACA TTA CTG GCC GAA GC-3′-Quencher | 209–227 |
| Norovirus GI | 1st PCR | COG1F | 5′-CGY TGG ATG CGN TTY CAT GA-3′ | 5291–5311 |
| Norovirus GI | 1st PCR/2nd PCR | * G1SKR | 5′-CCA ACC CAR CCA TTR TAC A-3′ | 5653–5671 |
| Norovirus GI | 2nd PCR | * G1SKF | 5′-CTG CCC GAA TTY GTA AAT GA-3′ | 5342–5361 |
| Norovirus GII | 1st PCR | COG2F | 5′-CAR GAR BCN ATG TTY AGR TGG ATG AG-3′ | 5003–5029 |
| Norovirus GII | 1st PCR/2nd PCR | * G2SKR | 5′-CCR CCN GCA TRH CCR TTR TAC AT-3′ | 5379–5401 |
| Norovirus GII | 2nd PCR | * G2SKF | 5′-CNT GGG AGG GCG ATC GCA A-3′ | 5058–5076 |
| Rotavirus NSP3 | qPCR | JVKF | 5′-CAG TGG TTG ATG CTC AAG ATG GA-3′ | 17–39 |
| Rotavirus NSP3 | qPCR | JVKR | 5′-TCA TTG TAA TCA TAT TGA ATA CCC A-3′ | 123–147 |
| Rotavirus NSP3 | qPCR | JVKP | FAM-5′-ACA ACT GCA GCT TCA AAA GAA GWG T-3-Quencher | 72–96 |
| Rotavirus VP7 | 1st PCR | VP7-F | 5′-ATG TAT GGT ATT GAA TAT ACC AC-3′ | 51–71 |
| Rotavirus VP7 | 1st PCR/2nd PCR | * VP7-R | 5′-AAC TTG CCA CCA TTT TTT CC-3′ | 914–932 |
| Rotavirus VP7 | 2nd PCR | * G1 | 5′-CAA GTA CTC AAA TCA ATG ATG G-3′ | 314–335 |
| Rotavirus VP7 | 2nd PCR | * G2 | 5′-CAA TGA TAT TAA CAC ATT TTC TGT G-3′ | 411–435 |
| Rotavirus VP7 | 2nd PCR | * G3 | 5′-ACG AAC TCA ACA CGA GAG G-3′ | 250–269 |
| Rotavirus VP7 | 2nd PCR | G4 | 5′-CGT TTC TGG TGA GGA GTT G-3′ | 480–499 |
| Rotavirus VP7 | 2nd PCR | G8 | 5′-GTC ACA CCA TTT GTA AAT TCG-3′ | 178–198 |
| Rotavirus VP7 | 2nd PCR | * G9 | 5′-CTT GAT GTG ACT AYA AAT AC-3′ | 757–776 |
| Rotavirus VP7 | 2nd PCR | G10 | 5′-ATG TCA GAC TAC ARA TAC TGG-3′ | 666–687 |
| Rotavirus VP7 | 2nd PCR | * G12 | 5′-GGT TAT GTA ATC CGA TGG ACG-3′ | 548–567 |
| Rotavirus VP4 | 1st PCR/2nd PCR | * VP4-F | 5′-TAT GCT CCA GTN AAT TGG-3′ | 132–149 |
| Rotavirus VP4 | 1st PCR | VP4-R | 5′-ATT GCA TTT CTT TCC ATA ATG-3′ | 775–795 |
| Rotavirus VP4 | 2nd PCR | P[4]-R | 5′-CTA TTG TTA GAG GTT AGA GTC-3′ | 474–494 |
| Rotavirus VP4 | 2nd PCR | P[6]-R | 5′-TGT TGA TTA GTT GGA TTC AA-3′ | 259–278 |
| Rotavirus VP4 | 2nd PCR | * P[8]-R | 5′-TCT ACT GGR TTR CAN TGC-3′ | 339–356 |
| Rotavirus VP4 | 2nd PCR | P[9]-R | 5′-TGA GAC ATG CAA TTG GAC-3′ | 385–402 |
| Rotavirus VP4 | 2nd PCR | P[10]-R | 5′-ATC ATA GTT AGT AGT CGG-3′ | 575–594 |
| Rotavirus VP4 | 2nd PCR | P[11]-R | 5′-GTA AAC ATC CAG AAT GTG-3′ | 305–323 |
| Sample | Sample | Sampling Date | Norovirus | Rotavirus | Recovery Efficiency | ||||
|---|---|---|---|---|---|---|---|---|---|
| GI Norovirus | GII Norovirus | ||||||||
| Genotype | log PCRU/L ± SD | Genotype | log PCRU/L ± SD | Genotype | log PCRU/L ± SD | % | |||
| R1 | #1 | 29.9.16 | ND | 4.98 ± 0.43 | ND | 6.13 ± 0.05 | GxP[8] | 5.21 ± 0.08 | 1.47 |
| R3 | #2 | 18.10.16 | GI.4 | 4.74 ± 0.32 | GII.2 | 6.12 ± 0.35 | GxP[8] | 5.46 ± 0.14 | 5.11 |
| R4 | #3 | 20.10.16 | ND | 5.01 ± 0.25 | GII.2 | 6.60 ± 0.01 | GxP[8] | 4.62 ± 0.18 | 2.22 |
| R5 | #4 | 27.10.16 | ND | 4.92 ± 0.06 | GII.2 | 6.13 ± 0.12 | G12P[8] | 4.14 ± 0.10 | 4.37 |
| R9 | #5 | 17.11.2016 | ND | 4.66 ± 0.64 | GII.2 | 6.39 ± 0.00 | G2P[8] | 5.03 ± 0.06 | 7.73 |
| R10 | #6 | 24.11.2016 | ND | 5.04 ± 0.28 | GII.2 | 6.46 ± 0.11 | ND | 5.77 ± 0.22 | 6.39 |
| R11 | #7 | 2.12.2016 | ND | 4.89 ± 0.06 | GII.2 | 6.24 ± 0.15 | G9P[8] | 5.26 | 1.37 |
| R12A | #8 | 6.12.2016 | ND | 5.47 ± 0.43 | GII.2 | 6.95 ± 0.10 | G2P[8] | <LOD | 7.62 |
| R13 | #9 | 21.12.2016 | ND | 5.09 ± 0.13 | GII.2 | 6.02 ± 0.00 | G3P[x] | 5.97 | 6.73 |
| R14 | #10 | 30.12.2016 | ND | 5.36 ± 0.19 | GII.2 | 6.95 ± 0.05 | GxP[8] | 6.20 ± 0.12 | 7.38 |
| R51 | #11 | 15.12.2016 | ND | 5.19 ± 0.32 | ND | 6.22 ± 0.13 | G2P[x] | <LOD | 8.54 |
| R15 | #12 | 4.01.2017 | ND | 5.62 ± 0.19 | GII.2 | 6.44 ± 0.10 | G12P[8] | <LOQ | 7.73 |
| R16 | #13 | 11.01.2017 | ND | 5.57 ± 0.08 | GII.2 | 6.56 ± 0.06 | G2P[8] | 6.00 ± 0.10 | 7.44 |
| R17 | #14 | 18.01.2017 | ND | <LOD | ND | 6.54 ± 0.18 | ND | <LOD | 8.38 |
| R18 | #15 | 15.02.2017 | ND | <LOD | ND | <LOD | GxP[8] | 6.24 ± 0.05 | 3.68 |
| R19 | #16 | 16.02.2017 | ND | 4.70 ± 0.21 | GII.2 | 6.14 ± 0.09 | G9P[x] | 5.34 ± 0.04 | 4.79 |
| R20 | #17 | 2.02.2017 | ND | 5.55 ± 0.04 | GII.2 | 6.53 ± 0.34 | GxP[8] | 5.05 ± 0.09 | 10.82 |
| R21 | #18 | 22.02.2017 | ND | 5.47 ± 0.08 | GII.2 | 6.19 ± 0.76 | ND | 4.08 ± 0.23 | 4.39 |
| R22 | #19 | 02.03.2017 | ND | <LOD | GII.6 | 5.38 ± 0.21 | GxP[8] | 6.32 ± 0.81 | 18.39 |
| R23 | #20 | 08.03.2017 | GI.2 | <LOD | ND | 6.25 | ND | <LOD | 20.50 |
| R24 | #21 | 15.03.2017 | GI.2 | 5.62 ± 0.05 | ND | 5.01 ± 1.71 | GxP[8] | 6.55 ± 0.25 | 12.50 |
| R25 | #22 | 23.03.2017 | ND | 4.80 ± 0.02 | ND | 5.82 ± 0.05 | GxP[8] | 6.85 ± 0.22 | 11.39 |
| R26 | #23 | 30.03.2017 | ND | <LOD | ND | <LOD | ND | 6.19 ± 0.08 | 1.18 |
| R27 | #24 | 06.04.2017 | ND | <LOD | ND | <LOD | ND | <LOD | 1.97 |
| R28 | #25 | 12.04.2017 | ND | 4.87 ± 0.10 | ND | 5.34 ± 0.00 | ND | 6.92 ± 0.04 | 2.82 |
| R29 | #26 | 27.04.2017 | ND | <LOD | ND | <LOD | G2P[8] | 4.59 | 4.31 |
| R30 | #27 | 04.05.2017 | ND | <LOD | ND | <LOD | ND | 6.31 ± 0.04 | 4.75 |
| R31 | #28 | 11.05.2017 | ND | <LOD | ND | <LOD | G3P[8] | <LOD | 11.03 |
| R32 | #29 | 18.05.2017 | ND | <LOD | GII.6 | <LOD | GxP[8] | 6.13 ± 0.01 | 9.05 |
| R33 | #30 | 25.02.2017 | ND | <LOD | ND | <LOD | GxP[8] | 6.80 | 9.34 |
| R34 | #31 | 01.06.2017 | ND | <LOD | ND | 5.17 | ND | <LOD | 6.73 |
| R35 | #32 | 08.06.2017 | ND | <LOD | ND | <LOD | ND | 4.99 | 2.02 |
| R36 | #33 | 15.06.2017 | ND | 4.33 ± 0.45 | ND | <LOD | ND | 5.28 | 7.10 |
| R37 | #34 | 22.06.2017 | ND | 4.81 ± 0.57 | ND | 4.69 ± 0.00 | ND | <LOD | 9.40 |
| R38 | #35 | 28.06.2017 | ND | 5.41 ± 0.01 | GII.17 | 5.04 ± 0.27 | ND | 5.98 | 2.48 |
| R39 | #36 | 05.07.2017 | ND | 5.45 ± 0.26 | GII.6 | 5.43 ± 1.58 | ND | 5.65 | 1.53 |
| R40 | #37 | 06.07.2017 | ND | 5.03 ± 0.02 | GII.17 | <LOD | ND | 4.95 | 3.53 |
| R41 | #38 | 13.07.2017 | GI.4 | 5.73 ± 0.54 | ND | 5.50 ± 0.15 | ND | <LOD | 8.04 |
| R42 | #39 | 20.07.2017 | ND | 5.75 ± 0.31 | ND | 5.25 ± 0.00 | ND | 4.73 ± 0.10 | 2.34 |
| R43 | #40 | 27.07.2017 | ND | 5.49 ± 0.22 | ND | 5.58 ± 0.13 | ND | <LOD | 26.30 |
| R44 | #41 | 10.08.2017 | ND | 4.75 ± 0.37 | ND | 5.23 ± 0.37 | ND | <LOD | 13.44 |
| R45 | #42 | 17.08.2017 | ND | 5.51 ± 0.00 | ND | 5.65 ± 0.11 | G1G2P[x] | <LOD | 25.72 |
| R46 | #43 | 30.08.2017 | ND | <LOD | ND | 5.13 ± 0.18 | ND | 5.20 ± 0.07 | 10.05 |
| R48 | #44 | 7.09.2017 | ND | 5.02 ± 0.17 | ND | 5.16 ± 0.00 | ND | <LOD | 9.86 |
| R49 | #45 | 13.09.2017 | ND | 5.67 ± 0.06 | ND | 5.64 ± 0.09 | ND | <LOD | 4.24 |
| R50 | #46 | 20.09.2017 | ND | 5.28 ± 0.08 | ND | 6.19 ± 0.09 | ND | 4.72 ± 0.13 | 13.27 |
| Norovirus | Rotavirus | ||||
|---|---|---|---|---|---|
| Genotype | GI (%) | GII (%) | Genotype | VP7 (%) | VP4 (%) |
| ND | 87.5 (28/32) | 45.7 (16/35) | ND | 63.5 (21/33) | 42.3 (14/33) |
| GI.2 | 6.25 (2/32) | - | P[8] | - | 57.7 (19/33) |
| GI.4 | 6.25 (2/32) | - | G1+G2 | 3 (1/33) | - |
| GII.2 | - | 40 (14/35) | G2 | 15.2 (5/33) | - |
| GII.6 | - | 8.6 (3/35) | G3 | 6.1 (2/33) | - |
| GII.17 | - | 5.7 (2/35) | G9 | 6.1 (2/33) | - |
| G12 | 6.1 (2/33) | - | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiso-Bellón, C.; Randazzo, W.; Pérez-Cataluña, A.; Vila-Vicent, S.; Gozalbo-Rovira, R.; Muñoz, C.; Buesa, J.; Sanchez, G.; Rodríguez Díaz, J. Epidemiological Surveillance of Norovirus and Rotavirus in Sewage (2016–2017) in Valencia (Spain). Microorganisms 2020, 8, 458. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8030458
Santiso-Bellón C, Randazzo W, Pérez-Cataluña A, Vila-Vicent S, Gozalbo-Rovira R, Muñoz C, Buesa J, Sanchez G, Rodríguez Díaz J. Epidemiological Surveillance of Norovirus and Rotavirus in Sewage (2016–2017) in Valencia (Spain). Microorganisms. 2020; 8(3):458. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8030458
Chicago/Turabian StyleSantiso-Bellón, Cristina, Walter Randazzo, Alba Pérez-Cataluña, Susana Vila-Vicent, Roberto Gozalbo-Rovira, Carlos Muñoz, Javier Buesa, Gloria Sanchez, and Jesús Rodríguez Díaz. 2020. "Epidemiological Surveillance of Norovirus and Rotavirus in Sewage (2016–2017) in Valencia (Spain)" Microorganisms 8, no. 3: 458. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8030458