Epidemiology and Genetic Variability of Circulating Influenza B Viruses in Uruguay, 2012–2019

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Influenza Surveillance and Sample Collection
2.2. Ethical Considerations
2.3. Statistical Methods
2.4. Molecular Detection of Influenza B Viruses
2.4.1. Extraction and Real Time RT-PCR Assays for Differentiation of Influenza B Lineages
2.4.2. PCR and Sequencing
2.5. Sequence Data
2.6. Phylogenetic Analysis
3. Results
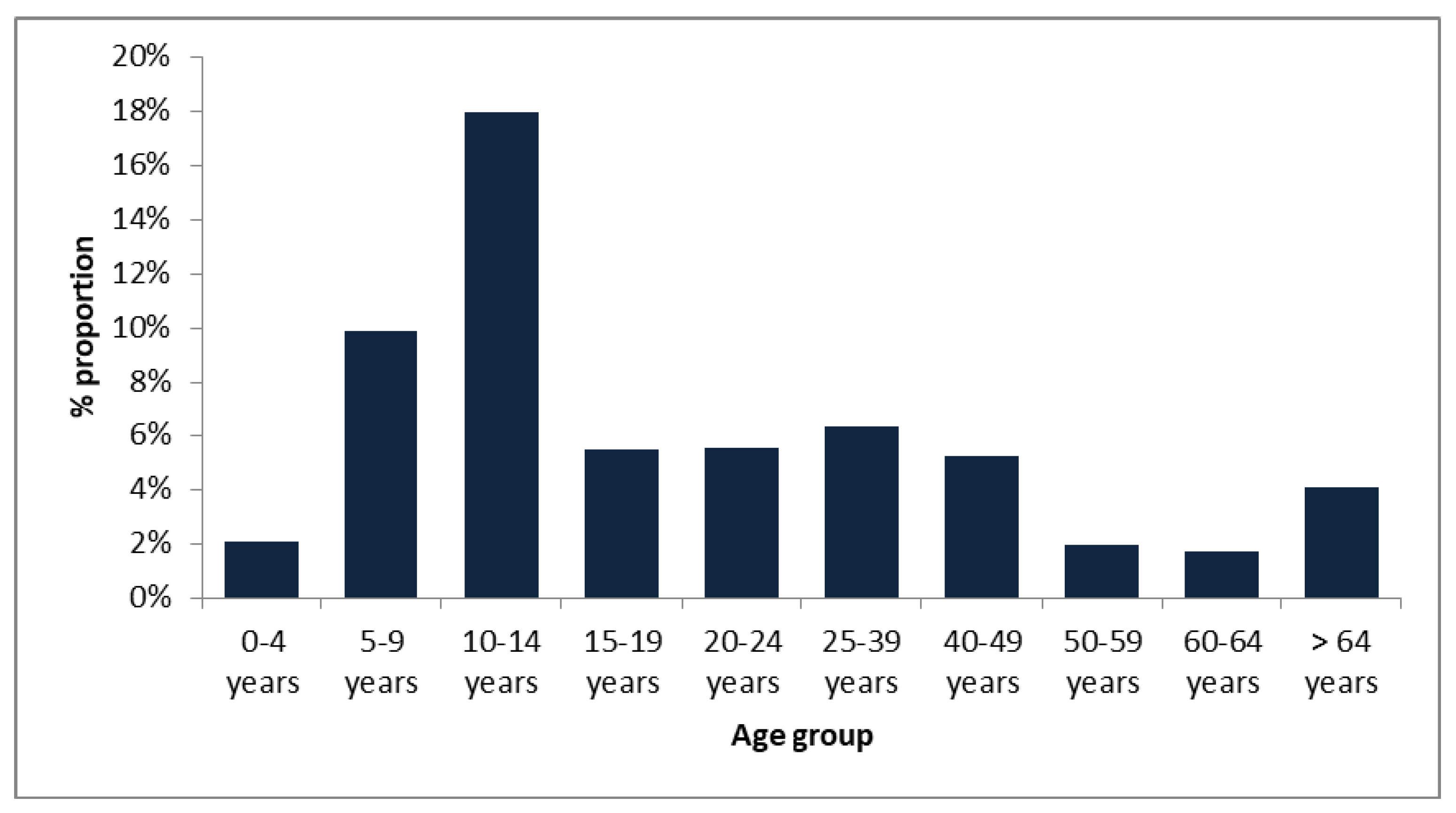
3.1. Age Distribution among Type of Surveillance
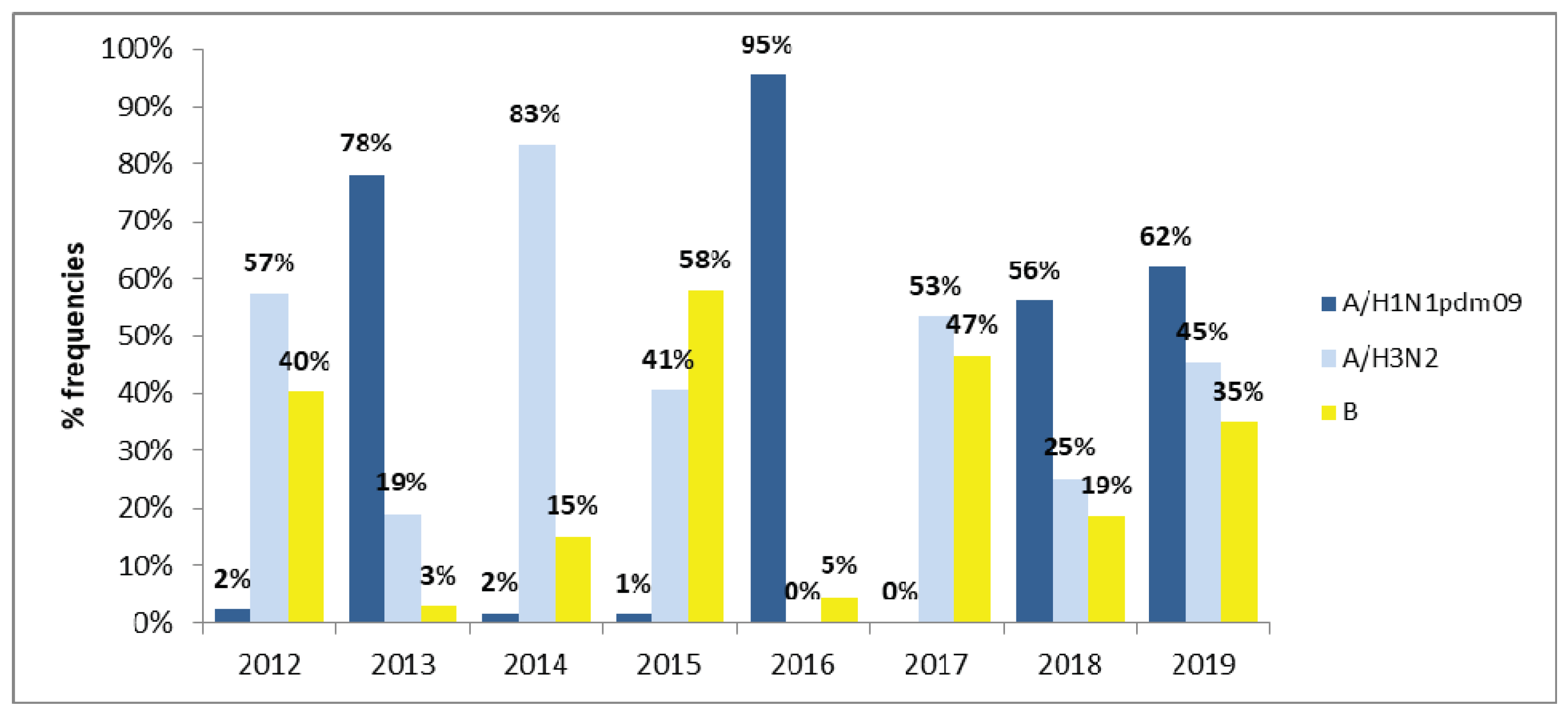
3.2. Prevalence of Influenza B Viruses
3.3. Interactions among Influenza A and B Viruses
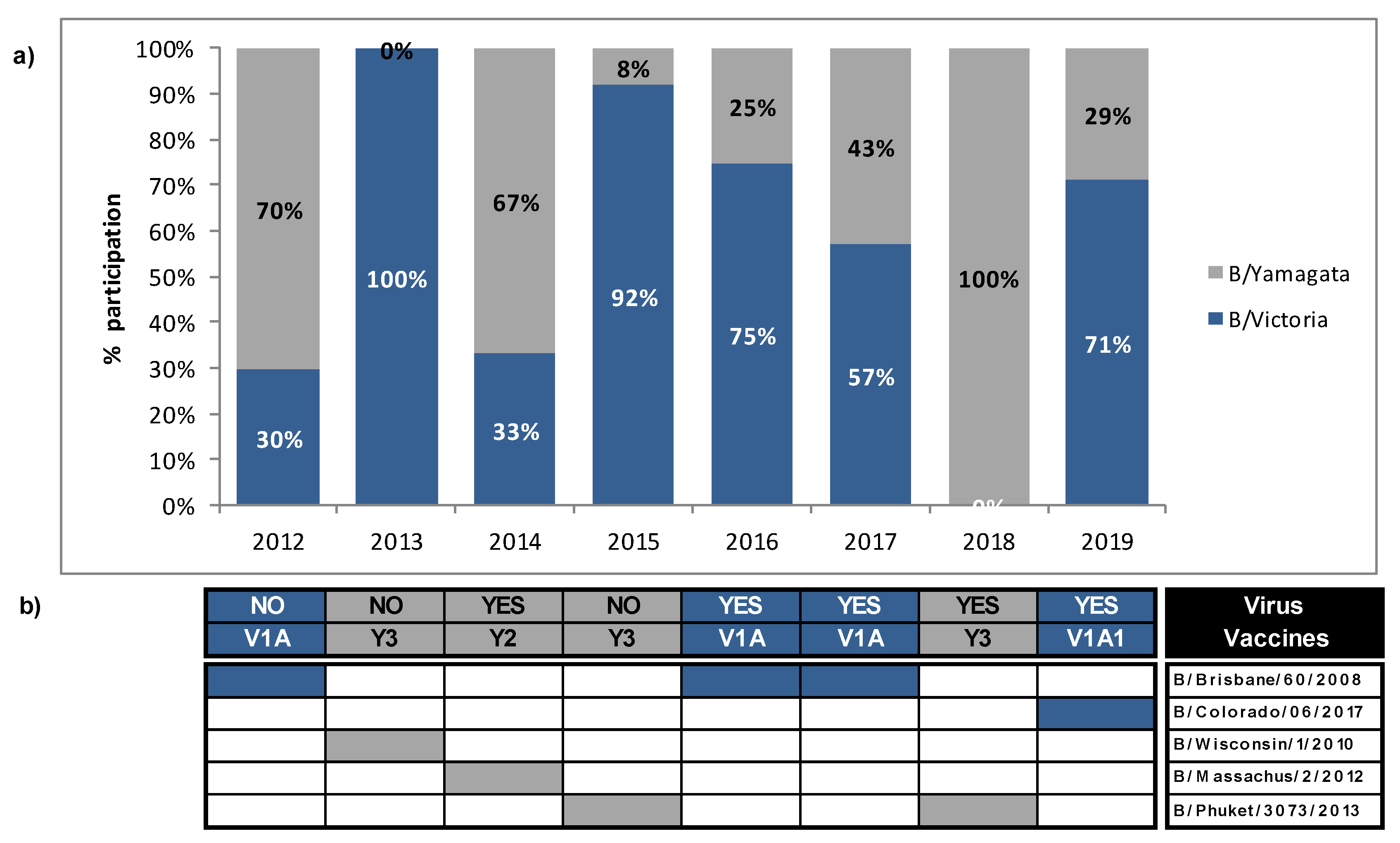
3.4. Mismatches between Circulating Strains of Influenza B and Vaccine Strains
3.5. Molecular Detection of Influenza B Subtypes
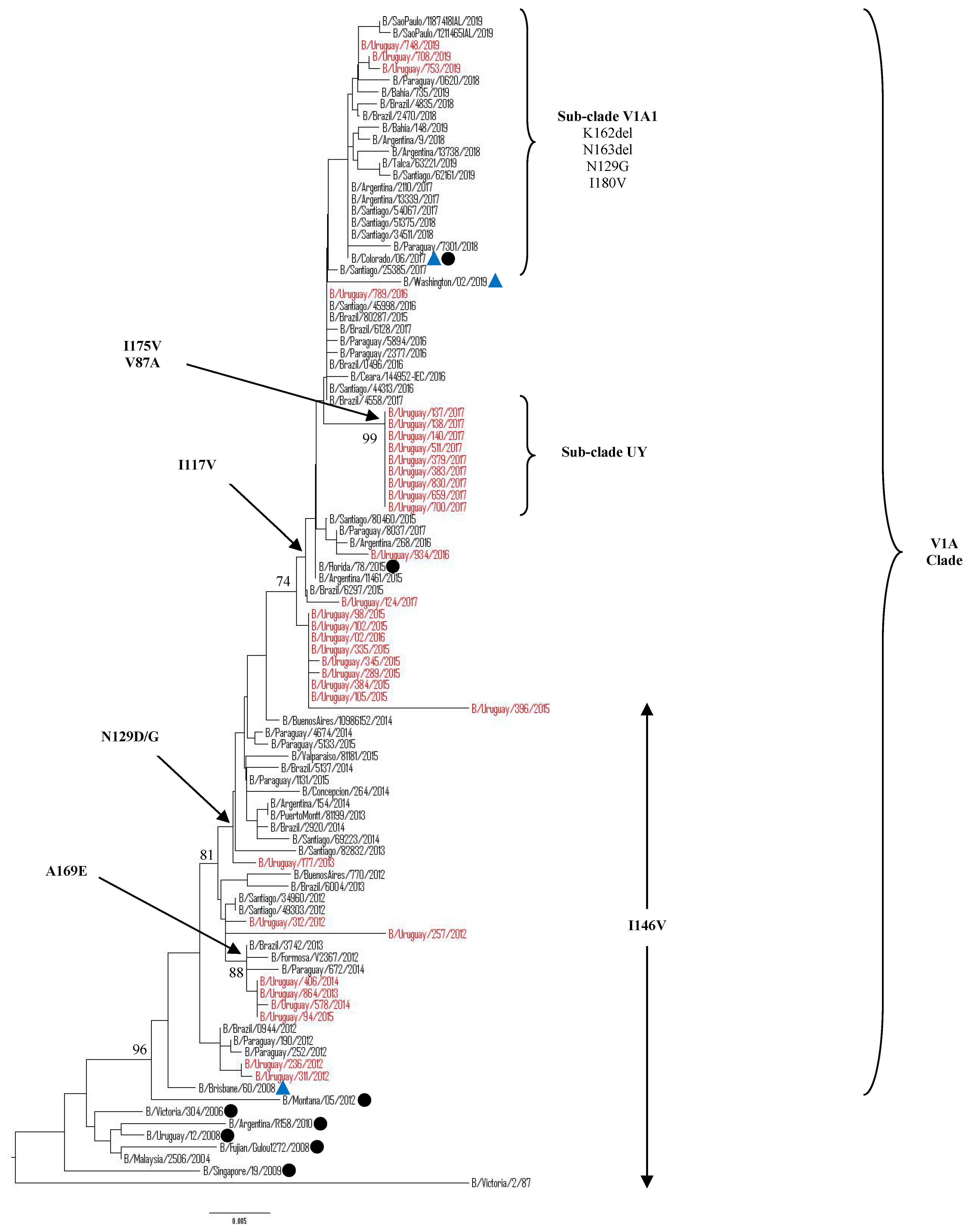
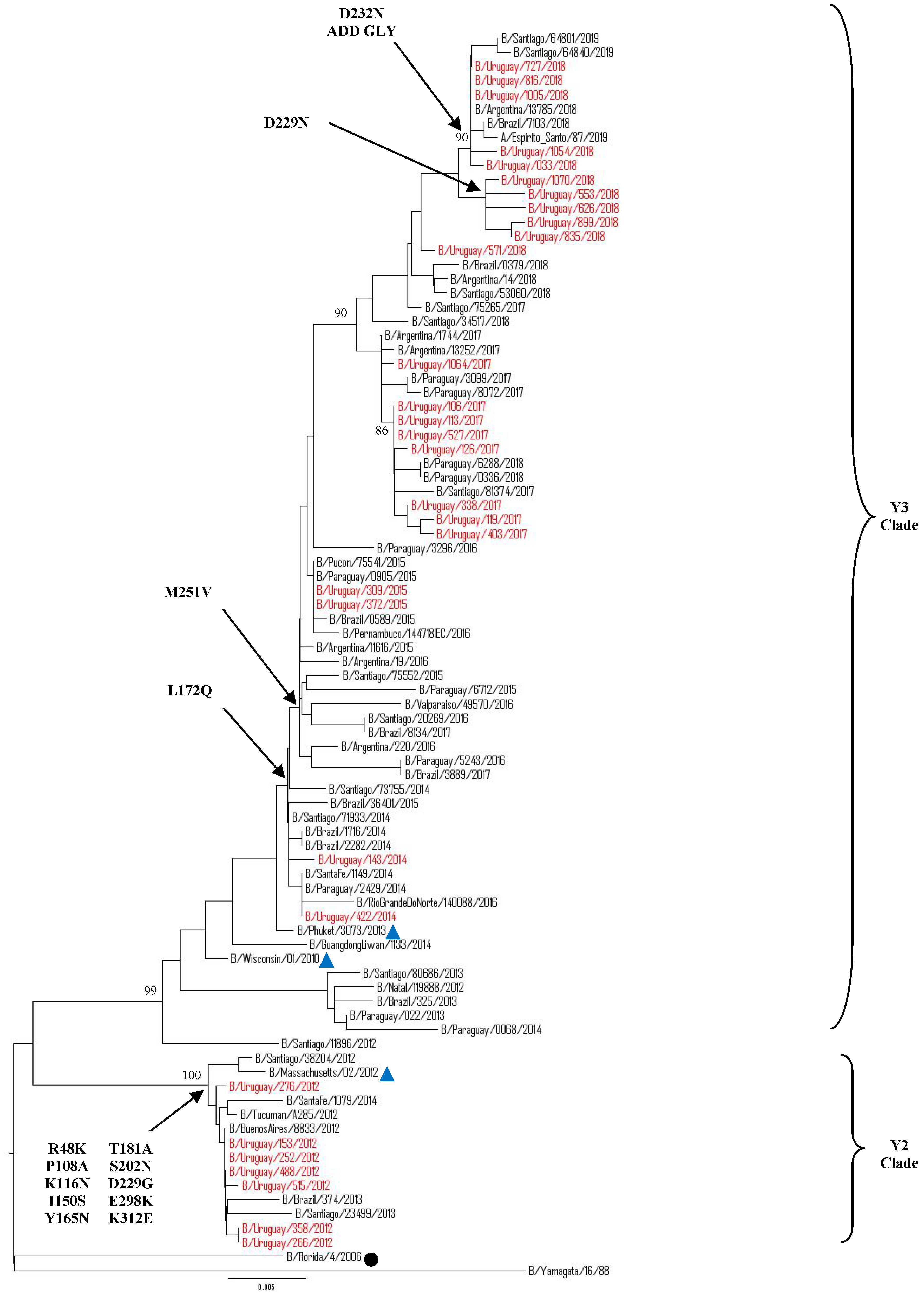
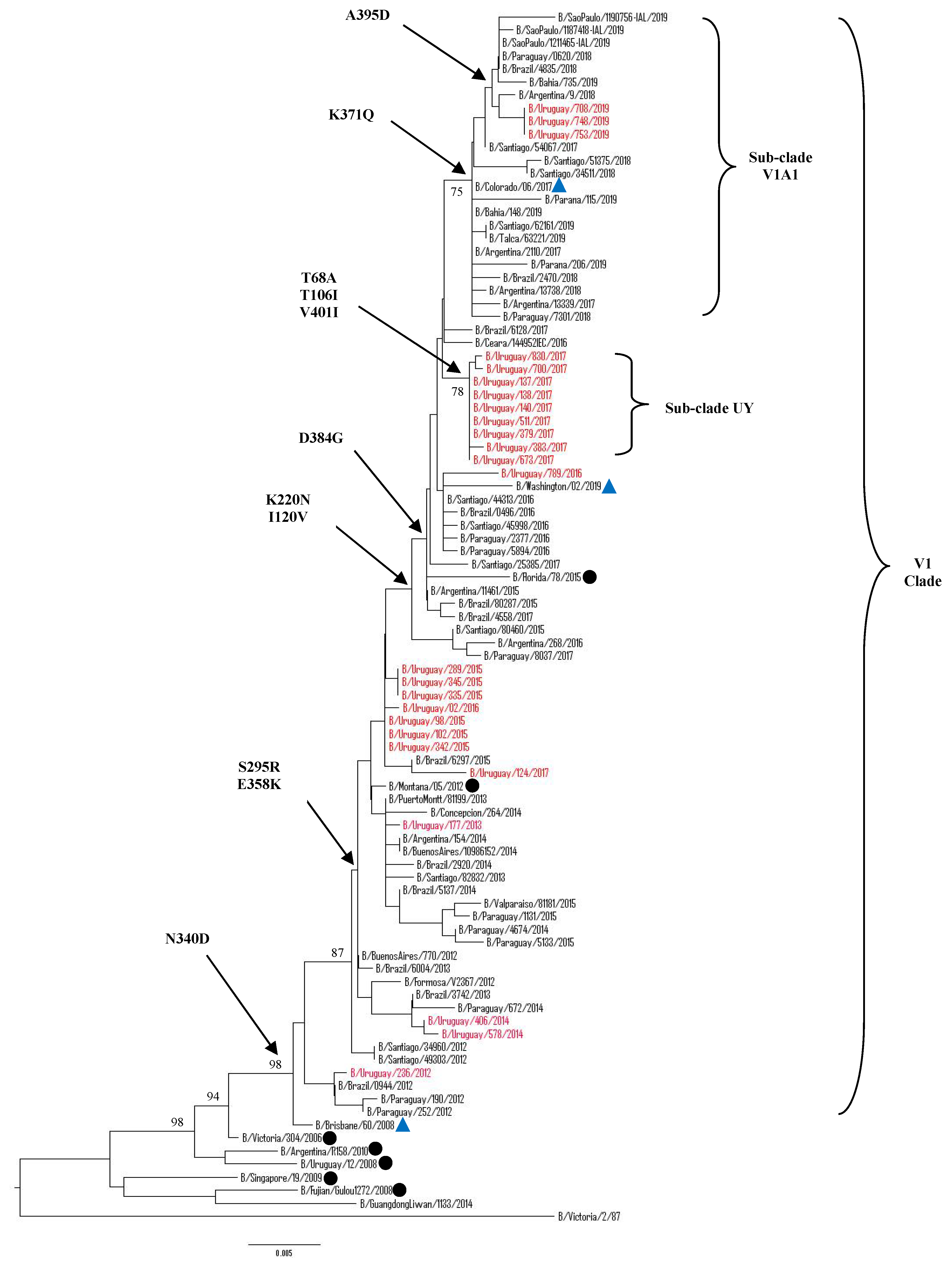
3.5.1. Sequence and Phylogenetic Analysis of the HA Gene of Uruguayan Influenza B Viruses
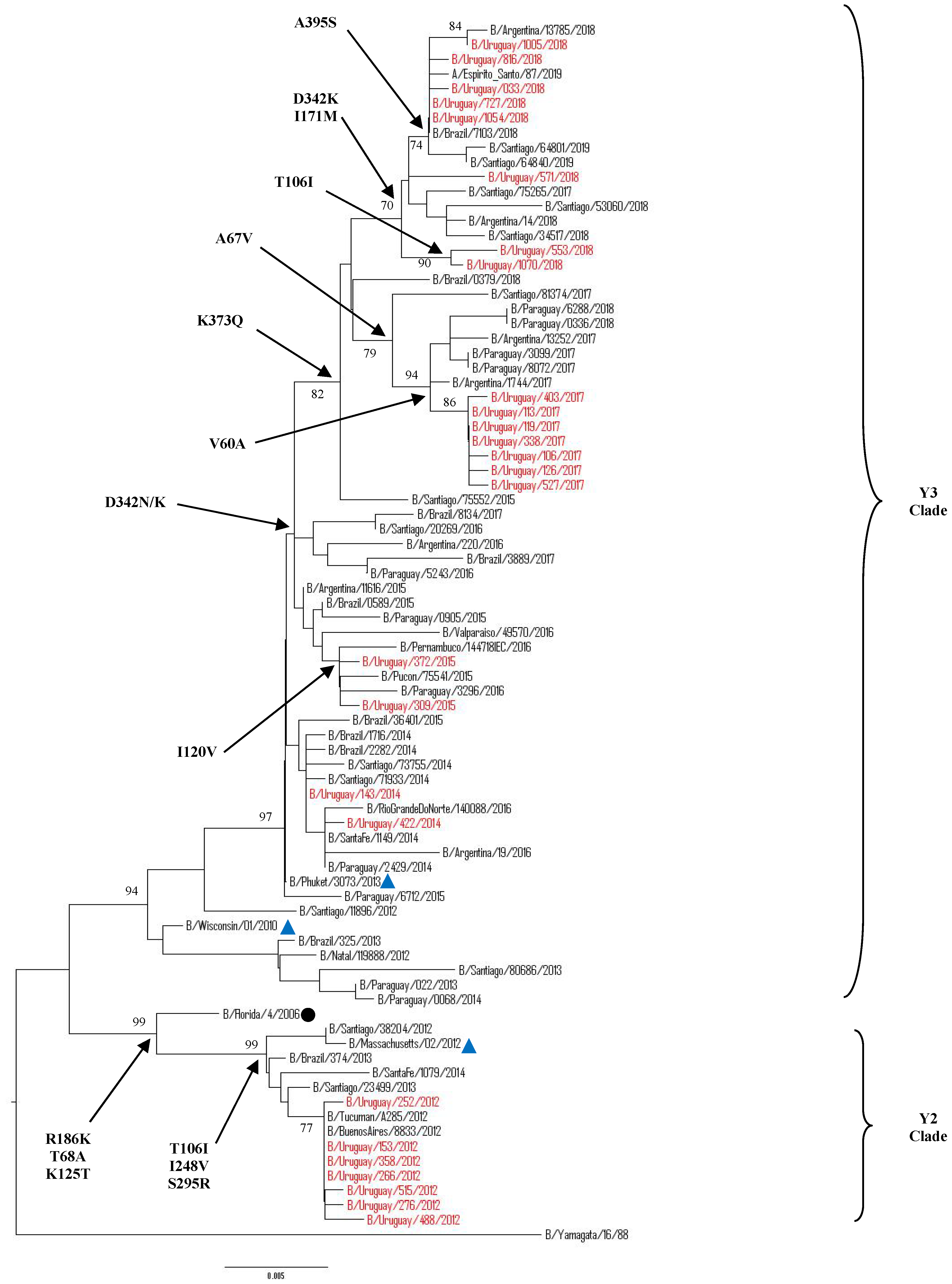
3.5.2. Sequence and Phylogenetic Analysis of the NA Gene of Uruguayan Influenza B Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Coen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modeling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Palekar, R.; Rolfes, M.; Arriola, C.; Acosta, B.; Alberto, P.; Badilla, X.; Bancej, C.; Barboza, J.; Baumeister, E.; Bruno, A.; et al. Burden of influenza-associated respiratory hospitalizations in the Americas, 2010-2015. PLoS ONE 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caini, S.; Alons, W.J.; Balmaseda, A.; Bruno, A.; Bustos, P.; Castillo, L.; de Lozano, C.; de Mora, D.; Fasce, R.; Ferrerira, W.; et al. Characteristics of seasonal influenza A and B in Latin America: Influenza surveillance data from ten countries. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Paul, G.W.; Schmier, J.K.; Kuehn, C.M.; Ryan, K.J.; Oxford, J. The burden of influenza B: A structured literature review. Am. J. Public Health. 2013. [Google Scholar] [CrossRef]
- Jennings, L.; Huang, Q.; Barr, I.; Lee, P.; Kim, W.J.; Buchy, P.; Sanicas, M.; Mungaly, B.; Chen, J. Literature review of the epidemiology of influenza B disease in 15 countries in the Asia-Pacific región. Influenza Other Respir. Viruses 2018, 12, 383–411. [Google Scholar] [CrossRef]
- Su, S.; Chaves, S.S.; Perez, A.; D´Mello, T.; Kirley, P.D.; YOusey-Hindes, K.; Farley, M.M.; Harris, M.; Sharangpani, R.; Lynfield, R.; et al. Comparing clinical characteristics between hospitalized adults with laboratory-confirmed influenza A and B infection. Clin. Infect. Dis. 2014, 59, 252–255. [Google Scholar] [CrossRef] [Green Version]
- Francis , T., Jr. A new type of virus from epidemic influenza. Science 1940, 92, 405–408. [Google Scholar] [CrossRef]
- Bedford, T.; Suchard, M.A.; Lemey, P.; Dudas, G.; Gregory, V.; Hay, A.J.; McCaulley, J.W.; Russell, C.A.; Smith, D.J.; Rambaut, A. Integrating influenza antigenic dynamics with molecular evolution. Elife 2014. [Google Scholar] [CrossRef]
- Rota, P.A.; Wallis, T.R.; Harmon, M.W.; Rota, J.S.; Kendal, A.P.; Nerome, K. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 1990, 175, 59–68. [Google Scholar] [CrossRef]
- Lo, Y.-C.; Chuang, J.-H.; Kuo, H.-W.; Huang, W.T.; Hsu, Y.F.; Liu, M.T.; Chen, C.H.; Huang, H.H.; Chang, C.H.; Chou, J.H.; et al. Surveillance and vaccine effectiveness of an influenza epidemic predominated by vaccine-mismatched influenza B/Yamagata-lineage viruses in Taiwan, 2011–2012 season. PLoS ONE 2013, 8, e58222. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.W.; Shih, S.R.; Hsiao, M.R.; Chang, S.C.; Lin, S.H.; Sun, C.F.; Tsao, K.C. Multiple genotypes of influenza B viruses co-circulated in Taiwan in 2004 and 2005. J. Clin. Microbiol. 2007, 45, 1515–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijaykrishna, D.; Holmes, E.C.; Joseph, U.; Fourment, M.; Su, Y.C.; Halpin, R.; Lee, R.T.; Deng, Y.M.; Gunalan, V.; Lin, X.; et al. The contrasting phylodynamics of human influenza B viruses. Elife 2015, 4, e05055. [Google Scholar] [CrossRef] [PubMed]
- Beran, J.; Wertzova, V.; Honegr, K.; Kaliskova, E.; Havlickova, M.; Havlik, J.; Jirincova, H.; Van Belle, P.; Jain, V.; Innis, B.; et al. Challenge of conducting a placebo-controlled randomized efficacy study for influenza vaccine in a season with low attack rate and a mismatched vaccine B strain: A concrete example. BMC Infect. Dis. 2009. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization Global Influenza Surveillance Network-WHO GISN. Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza; WHO Press: Geneva, Switzerland, 2011; Available online: https://apps.who.int/iris/bitstream/handle/10665/44518/9789241548090_eng.pdf;jsessionid=F9E0EF9A45D987DFB11EABCD433B5223?sequence=1 (accessed on 20 January 2020).
- World Health Organization. WHO information for the molecular detection of influenza viruses. Updated July, 2017. 2017. Available online: https://www.who.int/influenza/gisrs_laboratory/WHO_information_for_the_molecular_detection_of_influenza_viruses_20171023_Final.pdf (accessed on 20 January 2020).
- World Health Organization. WHO recommendations on the composition of influenza virus vaccines. Available online: https://www.who.int/influenza/vaccines/virus/recommendations/en/ (accessed on 20 January 2020).
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA 6 Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Saitoi, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 403–425. [Google Scholar]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Wang, Q.; Cheng, F.; Lu, M.; Tian, X.; Ma, J. Crystal structure of unliganded influenza B virus hemagglutinin. J. Virol. 2008, 82, 3011–3020. [Google Scholar] [CrossRef] [Green Version]
- Colman, P.M.; Varghese, J.N.; Laver, W.G. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 1983, 303, 41–44. [Google Scholar] [CrossRef]
- Tewawong, N.; Suwannakarn, K.; Prachayangprecha, S.; Korkong, S.; Vichiwattana, P.; Vongpunsawad, S.; Poovorawan, Y. Molecular epidemiology and phylogenetic analyses of influenza B virus in Thailand during 2010 to 2014. PLoS ONE 2015, 10, e0116302. [Google Scholar] [CrossRef] [Green Version]
- Gubareva, L.; Matrosovich, M.N.; Brenner, M.K.; Bethell, R.C.; Webster, R.G. Evidence for zanamivir resistance in an immunocompromised child infected with influenza B virus. J. Infect. Dis. 1998, 178, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- WHO. Laboratory methodologies for testing the antiviral susceptibility of influenza viruses: Neuraminidase inhibitor (NAI). A summary of amino acid substitutions in the influenza neuraminidase associated with resistance or reduced susceptibility to NAIs. Available online: https://www.who.int/influenza/gisrs_laboratory/antiviral_susceptibility/nai_overview/en/ (accessed on 20 January 2020).
- Goñi, N.; Baz, M.; Ruchansky, D.; Coppola, L.; Russi, J.C.; Cristina, J. Influenza B viruses isolated in Uruguay during the 2002-2005 seasons: Genetic relations and vaccine strain match. Virus Res. 2007, 123, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Hortal, M.; Russi, J.C.; Arbiza, J.R.; Canepa, E.; Chiparelli, H.; Ilarramendi, A. Identification of viruses in a study of acute respiratory tract infection in children from Uruguay. Rev. Infect. Dis. 1990, 12, S995–S997. [Google Scholar] [CrossRef] [PubMed]
- Palekar, R.; Rodriguez, A.; Avila, C.; Barrera, G.; Barrera, M.; Brenes, H.; Bruno, A.; El Omeiri, N.; Fasce, R.; Ferreira de Almeida, W.; et al. Patterns of influenza B circulation in Latin America and the Caribbean, 2010–2017. PLoS ONE 2019, 14, e0219595. [Google Scholar] [CrossRef] [Green Version]
- Tran, D.; Vaudry, W.; Moore, D.; Bettinger, J.A.; Halperin, S.A.; Scheifele, D.W.; Jadvji, T.; Lee, L.; Mersereau, T. Hospitalization for influenza A. versus B. Pediatrics 2016. [Google Scholar] [CrossRef] [Green Version]
- Seleka, M.; Treurnicht, F.K.; Tempia, S.; Hellferscee, O.; Mtshali, S.; Cohen, A.L.; Buys, A.; McAnerney, J.M.; Besselaar, T.G.; Pretorius, M.; et al. Epidemiology of influenza B/Yamagata and B/Victoria lineages in south africa, 2005-2014. PLoS ONE 2017. [Google Scholar] [CrossRef]
- Caini, S.; Huang, Q.S.; Ciblak, M.A.; Kusznierz, G.; Owen, R.; Wangchuk, S.; Henriques, C.M.; Njouom, R.; Fasce, R.A.; Yu, H.; et al. Epidemiological and virological characteristics of influenza B: Results of the Global Influenza B Study. Influenza Other Respir. Viruses 2015, 9, 3–12. [Google Scholar] [CrossRef]
- Barr, I.G.; Vijaykrishna, D.; Sullivan, S.G. Differential age susceptibility to influenza B/Victoria lineage viruses in the 2015 Australian influenza season. Euro Surveill. 2016. [Google Scholar] [CrossRef]
- Glezen, W.P. Changing Epidemiology of Influenza B Virus. Clin. Infect. Dis. 2014, 59, 1525–1526. [Google Scholar] [CrossRef] [Green Version]
- McCullers, J.A.; Wang, G.C.; He, S.; Webster, R.G. Reassortment and insertion-deletion are strategies for the evolution of influenza B viruses in nature. J. Virol. 1999, 73, 7343–7348. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.F.; Nogales, A.; Finch, C.; Tuffy, K.M.; Domm, W.; Perez, D.R.; Topham, D.J.; Martínez-Sobrido, L. Influenza A and B virus intertypic reassortment through compatible viral packaging signals. J. Virol. 2014, 88, 10778–10791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudas, G.; Bedford, T.; Lycett, S.; Rambaut, A. Reassortment between influenza B lineages and the emergence of a co adapted PB1-PB2-HA gene complex. Mol. Biol. Evol. 2014, 32, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.R.; Huang, Y.P.; Chang, F.Y.; Hsu, L.C.; Lin, Y.C.; Huang, H.Y.; Wu, F.T.; Wu, H.S.; Liu, M.T. Phylogenetic and evolutionary history of influenza B viruses, which caused a large epidemic in 2011-2012, Taiwan. PLoS ONE 2012, 7, e47179. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Holmes, E.C. The evolutionary dynamics of human influenza B virus. J. Mol. Evol. 2008, 66, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Langat, P.; Raghwani, J.; Dudas, G.; Bowden, T.A.; Edwards, S.; Gall, A.; Bedford, T.; Rambaut, A.; Daniels, R.S.; Russell, C.; et al. Genome-wide evolutionary dynamics of influenza B viruses on a global scale. PLoS Pathog. 2017. [Google Scholar] [CrossRef] [Green Version]
- Tsedenbal, N.; Tsend-Ayush, A.; Badarch, D.; Jav, S.; Pagbajab, N. Influenza B viruses circulated during last 5 years in Mongolia. PLoS ONE 2018. [Google Scholar] [CrossRef] [Green Version]
- Horthongkham, N.; Athipanyasilp, N.; Pattama, A.; Kaewnapan, B.; Sornprasert, S.; Srisurapanont, S.; Kantakamalakul, W.; Amaranond, P.; Sutthent, R. Epidemiological, Clinical and Virological characteristics of influenza B virus from patients at the hospital Tertary care units in Bangkok during 2011–2014. PLoS ONE 2016. [Google Scholar] [CrossRef] [Green Version]
- Trucchi, C.; Alicino, C.; Orsi, A.; Paganino, C.; Barberis, I.; Grammatico, F.; Canepa, P.; Rappazzo, E.; Bruzzone, B.; Sticchi, L.; et al. Fifteen years of epidemiologic, virologic and syndromic influenza surveillance: A focus on type B virus and the effects of vaccine mismatch in Liguria region, Italy. Hum.Vaccin. Immunother. 2017, 13, 456–463. [Google Scholar] [CrossRef]
- Tan, Y.; Guan, W.; Lam, T.T.; Pan, S.; Wu, S.; Zhan, Y.; Viboud, C.; Holmes, E.C.; Yang, Z. Differing epidemiological dynamics of influenza B virus lineages in Guangzhou, Southern China,2009-2010. J. Virol. 2013, 87, 12447–12456. [Google Scholar] [CrossRef] [Green Version]
- Caini, S.; Kusznierz, G.; Garate, V.V.; Wangchuk, S.; Thapa, B.; de Paula Júnior, F.J.; Ferreira de Almeida, W.A.; Njouom, R.; Fasce, R.A.; Bustos, P.; et al. The epidemiological signature of influenza B virus and its B/Victoria and B/Yamagata lineages in the 21 st century. PLoS ONE 2019. [Google Scholar] [CrossRef] [Green Version]
- Orsi, A.; Colomba, G.M.E.; Pojero, F.; Calamusa, G.; Alicino, C.; Trucchi, C.; Canepa, P.; Ansaldi, F.; Vitale, F.; Tramuto, F. Trends of impact of influenza B during the 2010-2016 seasons in 2 regions of north and south italy: The impact of the vaccine mismatch on influenza immunization strategy. Hum. Vaccin. Immunother. 2018, 14, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yu, Z.; Liu, S.; Zhang, X.; Wang, X.; Cai, J.; Ling, F.; Chen, E. Comparison of influenza epidemiological and virological characteristics between outpatients and inpatients in Zhejiang province, China, March 2011-June 2015. Int. J. Env. Res. Public Health 2017, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmaa, A.; Kamigaki, T.; Darmaa, B.; Nymadawa, P.; Oshitani, H. Epidemiology and impact of influenza in Mongolia 2007–2012. Influenza Other Respir. Viruses 2014, 8, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Tallo, V.L.; Kamigaki, T.; Tan, A.G. Estimating influenza outpatient´s and impatients incidences from 2009 to 2011 in a tropical urban setting in the Philippines. Influenza Other Respir. Viruses 2014, 8, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Pan, L.; Sun, Q.; Zhu, W.; Zhu, L.; Ye, C.; Xue, C.; Wang, Y.; Liu, Q.; Ma, P.; et al. The clinical and etiological characteristics of influenza-like illness (ILI) in outpatients in Shangai, China, 2011 to 2013. PLoS ONE 2015. [Google Scholar] [CrossRef]
- Noh, J.; Song, J.Y.; Cheong, H.J.; Choi, W.S.; Lee, J.; Lee, J.S.; Wie, S.H.; Jeong, H.W.; Kim, Y.K.; Choi, S.H.; et al. Laboratory surveillance of influenza –like illness in seven teaching hospitals, South Koreal: 2011–2012 season. PLoS ONE 2013, 8, e64295. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, N.; Xie, Z.; Lu, R.; He, B.; Liu, C.; Ma, X.; Tan, W. Viral etiology and clinical profiles of children with severe acute respiratory infections in China. PLoS ONE 2013, 8, e72606. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Recommended composition of influenza virus vaccines for use in the 2018–2019 northern hemisphere influenza season. Wkly Epidemiol. Rec. 2018, 93, 133–152. [Google Scholar]
- Kuo, S.; Chen, G.W.; Velu, A.B.; Dash, S.; Han, Y.J.; Tsao, K.C.; Shih, S.R. Circulating pattern and genomic characteristics of influenza B viruses in Taiwan from 2003 to 2014. J. Formos. Med. Assoc. 2016, 115, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Kirk, B.D.; Ma, J.; Wang, Q. Diversifying selective pressure on influenza B virus hemagglutinin. J. Med. Virol. 2009, 81, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Tramuto, F.; Orsi, A.; Maida, C.M.; Costantino, C.; Trucchi, C.; Alicino, C.; Vitale, F.; Ansaldi, F. The molecular epidemiology and evolutionary dynamics of influenza B virus in two italian regions during 2010-2015: The experience of Sicily and Liguria. Int. J. Mol. Sci. 2016, 17, 549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nerome, R.; Hiromoto, Y.; Sugita, S.; Tanabe, N.; Ishida, M.; Matsumoto, M.; Lindstrom, S.E.; Takahashi, T.; Nerome, K. Evolutionary characteristics of influenza B virus since its first isolation in 1940: Dynamic circulation of deletion and insertion mechanism. Arch. Virol. 1998, 143, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Suptawiwat, O.; Ninpan, K.; Boonarkart, C.; Ruangrung, K.; Auewarakul, P. Evolutionary dynamic of antigenic residues on influenza B hemagglutinin. Virology 2017, 502, 84–96. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.; Gilbertson, B.; Trifkovic, S.; Brown, L.; McKimm-Breschkin, J.L. Influenza virus neuraminidase structure and functions. Front. Microbiol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.P.; Gregory, V.; Collins, P.; Kloess, J.; Wharton, S.; Cattle, N.; Lackenby, A.; Daniels, R.; Hay, A. Neuraminidase receptor binding variants of human influenza A(H3N2) viruses resulting from substitution of aspartic acid 151 in the catalytic site: A role in virus attachment? J. Virol. 2010, 84, 6769–6781. [Google Scholar] [CrossRef] [Green Version]
- Mohr, P.G.; Deng, Y.M.; McKimm-Breschkin, J.L. The neuraminidases of MDCK grown human influenza A (H3N2) viruses isolated since 1994 can demonstrate receptor binding. Virol. J. 2015. [Google Scholar] [CrossRef] [Green Version]
- Tivane, A.; Daniels, R.; Nguenha, N.; Machalele, L.; Nacoto, A.; Pale, M.; Mateonane, E.; Mavale, S.; Chilundo, J.; Muteto, D. Antigenic and genetic characterization of influenza viruses isolated in Mozambique during the 2015 season. PLoS ONE 2018. [Google Scholar] [CrossRef]
- Norwegian Institute of Public Health. Influenza Epidemiological Information prepared for the WHO Consultation on the Composition of Influenza Virus Vaccines for Use in the 2018 Southern Hemisphere Influenza Season. 2017. Available online: https://www.fhi.no/globalassets/dokumenterfiler/influensa/influensaovervaking-gml/norsk-rapport-for-whos-influenza-vaksinemote-september-2017.pdf (accessed on 5 November 2019).
- Mosterin Hopping, A.; Fonville, J.M.; Russell, C.A.; James, S.; Smith, D.J. Influenza B vaccine lineage selection-an optimized trivalent vaccine. Vaccine 2016, 34, 1617–1622. [Google Scholar] [CrossRef]
- Demicheli, V.; Jefferson, T.; Ferroni, E.; Rivett, A.; Di Pietrantonj, C. Vaccines for preventing influenza in healthy adults. Cochrane Database Syst. Rev. 2018. [Google Scholar] [CrossRef]
- Qin, Y.; Zhang, Y.; Wu, P. Influenza vaccine effectiveness in preventing hospitalization among Beijing residents in China, 2013–2015. Vaccine 2016, 34, 2329–2333. [Google Scholar] [CrossRef] [Green Version]
- Moa, A.M.; Muscatello, D.J.; Turner, R.M.; MacIntyre, C.R. Epidemiology of influenza B in Australia: 2001-2014. Influenza Other Respir. Viruses 2017, 11, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Tinoco, Y.O.; Azziz-Baumgartner, E.; Uyek, T.M. Burden of Influenza in 4 Ecologically Distinct Regions of Peru: Household active surveillance of a community cohort, 2009-2015. Clin. Infect. Dis. 2017, 65, 1532–1541. [Google Scholar] [CrossRef]
- Reed, C.; Chaves, S.S.; Kirley, P.D.; Emerson, R.; Aragon, D.; Hancock, E.B.; Butler, L.; Hollick, G.; Baumbach, J.; Hollick, G.; et al. Estimating influenza disease burden from population-based surveillance data in the United States. PLoS ONE 2015, 10, e0118369. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control (ECDC). Immunization Schedules by Target Disease. Available online: http://vaccine-schedule.ecdc.europa.eu/Pages/Scheduler.aspx (accessed on 27 February 2019).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples Processed | ILI Total | SARI Total | IBV Total | B/Vic | B/Yam | IBV Untyped | * p-Value | ILI (+IBV) | SARI (+IBV) | * p-Value | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Demographic feature | |||||||||||
| Mean age in years | 20.5 | 20.5 | 20.1 | 22 | 15.4 | 29.4 | 14.3 | 24.0 | |||
| Median age in years | 4 | 10 | 4 | 8 | 6.5 | 13.5 | <0.05 | 9.5 | 9 | >0.05 | |
| Number of patients between | |||||||||||
| 0–4 years | 3210 | 166 | 3044 | 67 | 39 | 27 | 1 | 9 | 57 | ||
| 5–9 years | 445 | 52 | 393 | 44 | 30 | 14 | 0 | 13 | 31 | ||
| 10–14 years | 145 | 30 | 115 | 26 | 16 | 9 | 1 | 8 | 18 | ||
| 15–19 years | 55 | 10 | 45 | 3 | 2 | 1 | 0 | 2 | 1 | ||
| 20–24 years | 90 | 25 | 65 | 5 | 2 | 2 | 1 | 3 | 2 | ||
| 25–39 years | 284 | 74 | 210 | 18 | 7 | 10 | 1 | 7 | 11 | ||
| 40–49 years | 210 | 49 | 161 | 11 | 2 | 9 | 0 | 1 | 10 | ||
| 50–59 years | 304 | 21 | 283 | 6 | 1 | 5 | 0 | 1 | 5 | ||
| 60–64 years | 177 | 8 | 169 | 3 | 1 | 2 | 0 | 0 | 4 | ||
| >64 years | 834 | 20 | 814 | 34 | 10 | 19 | 5 | 0 | 33 | ||
| Total | 5754 | 455 | 5299 | 217 | 110 | 98 | 9 | 44 | 172 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivas, M.J.; Alegretti, M.; Cóppola, L.; Ramas, V.; Chiparelli, H.; Goñi, N. Epidemiology and Genetic Variability of Circulating Influenza B Viruses in Uruguay, 2012–2019. Microorganisms 2020, 8, 591. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8040591
Rivas MJ, Alegretti M, Cóppola L, Ramas V, Chiparelli H, Goñi N. Epidemiology and Genetic Variability of Circulating Influenza B Viruses in Uruguay, 2012–2019. Microorganisms. 2020; 8(4):591. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8040591
Chicago/Turabian StyleRivas, María José, Miguel Alegretti, Leticia Cóppola, Viviana Ramas, Héctor Chiparelli, and Natalia Goñi. 2020. "Epidemiology and Genetic Variability of Circulating Influenza B Viruses in Uruguay, 2012–2019" Microorganisms 8, no. 4: 591. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8040591