Transcriptional Profiling of Immune and Inflammatory Responses in the Context of SARS-CoV-2 Fungal Superinfection in a Human Airway Epithelial Model

 , , , , , ,
, , , , , ,
Abstract
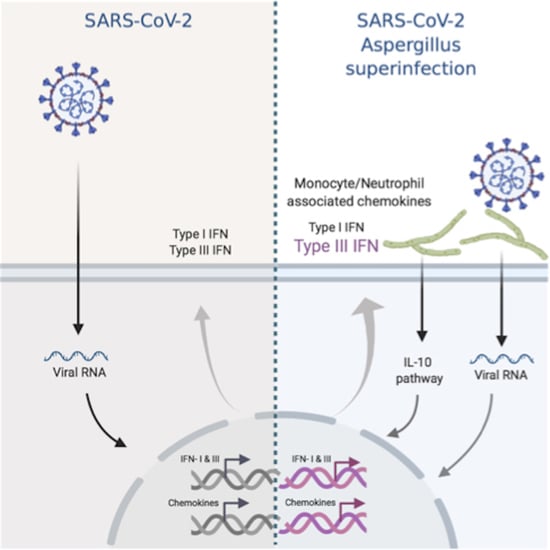
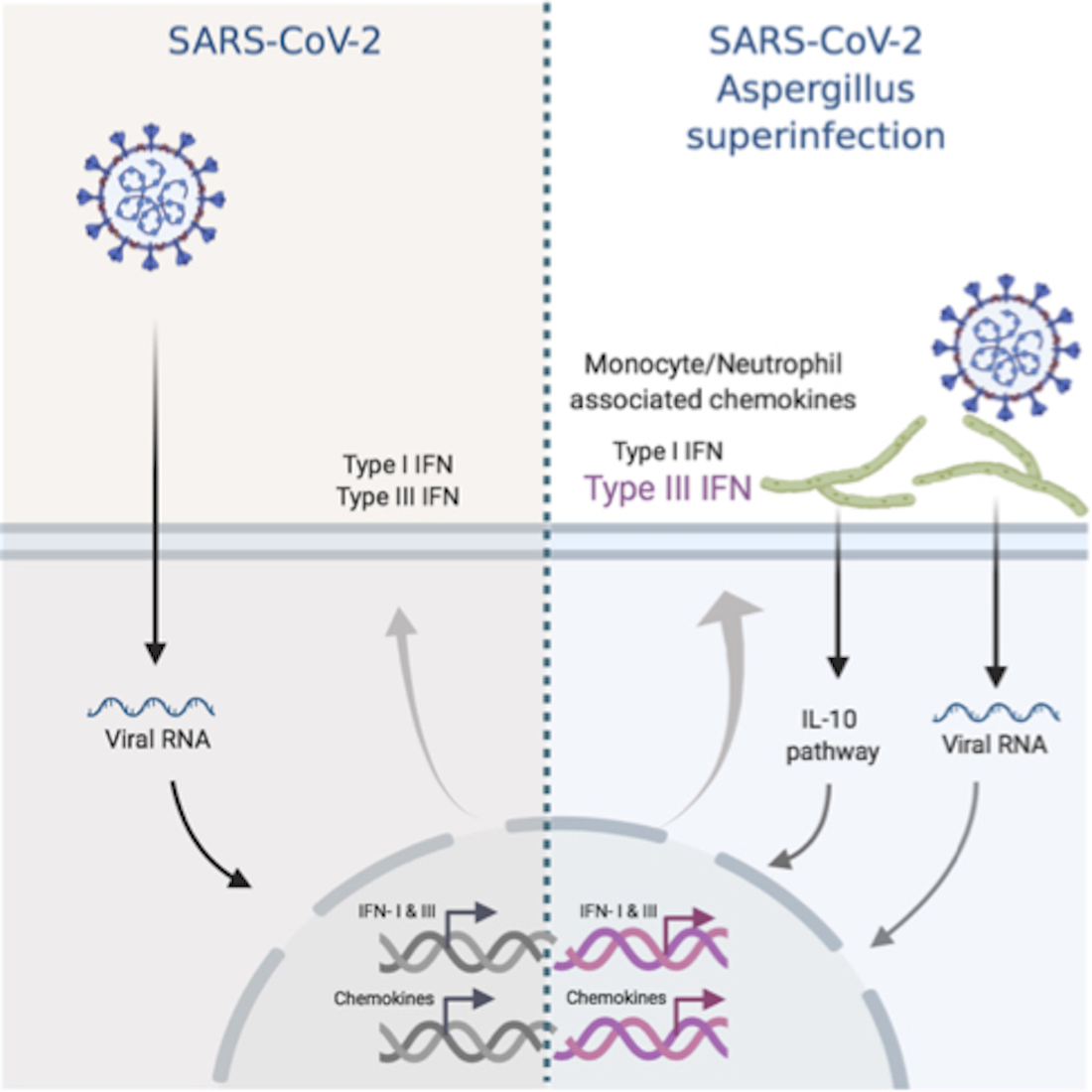
:
{kind=link}
{kind=link}
{kind=link}
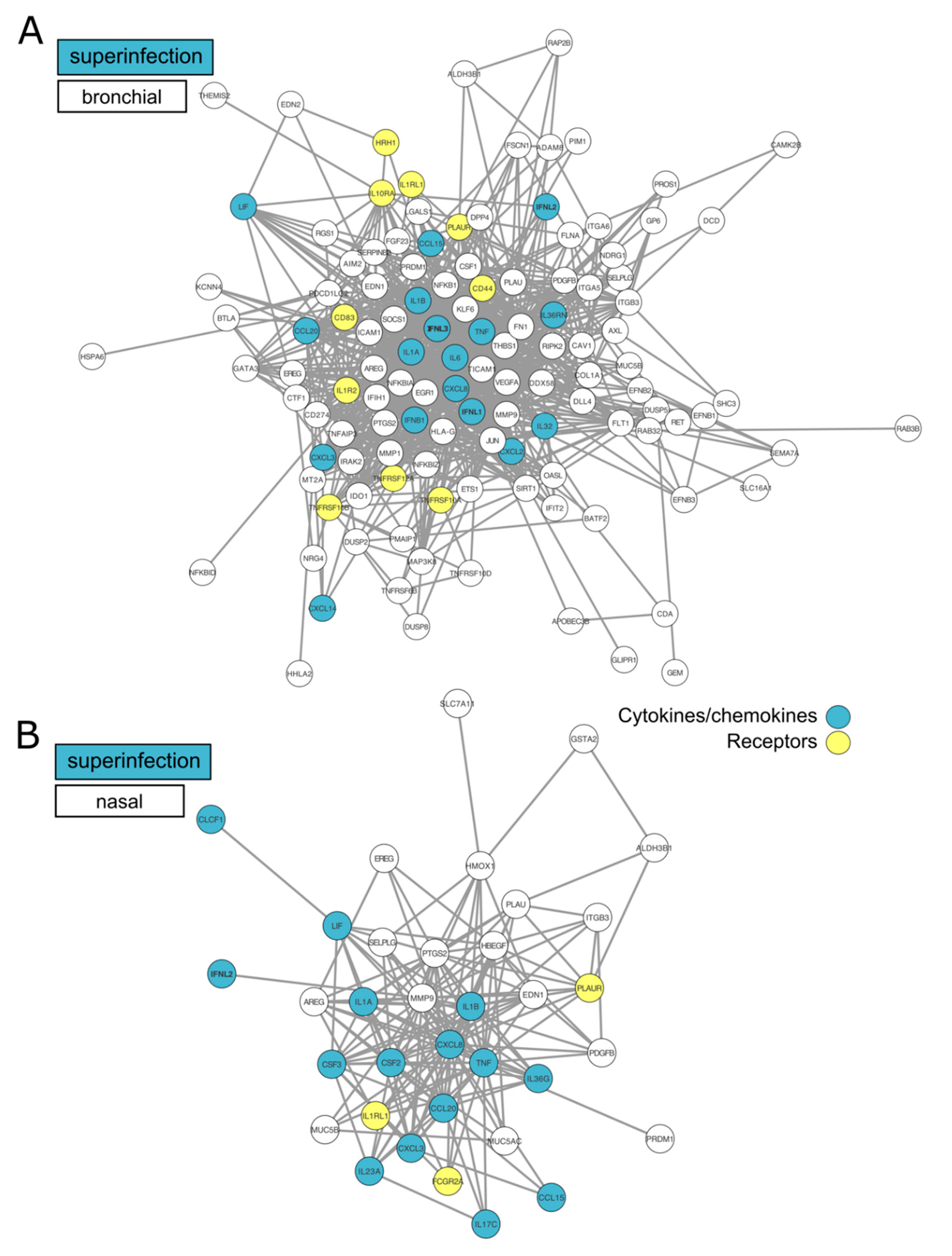{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reconstituted Human Airway Epithelial Model
2.2. Pathogens
2.3. mRNA Sequencing
2.4. Differential Expression Analysis
2.5. In silico Functional Analysis
3. Results
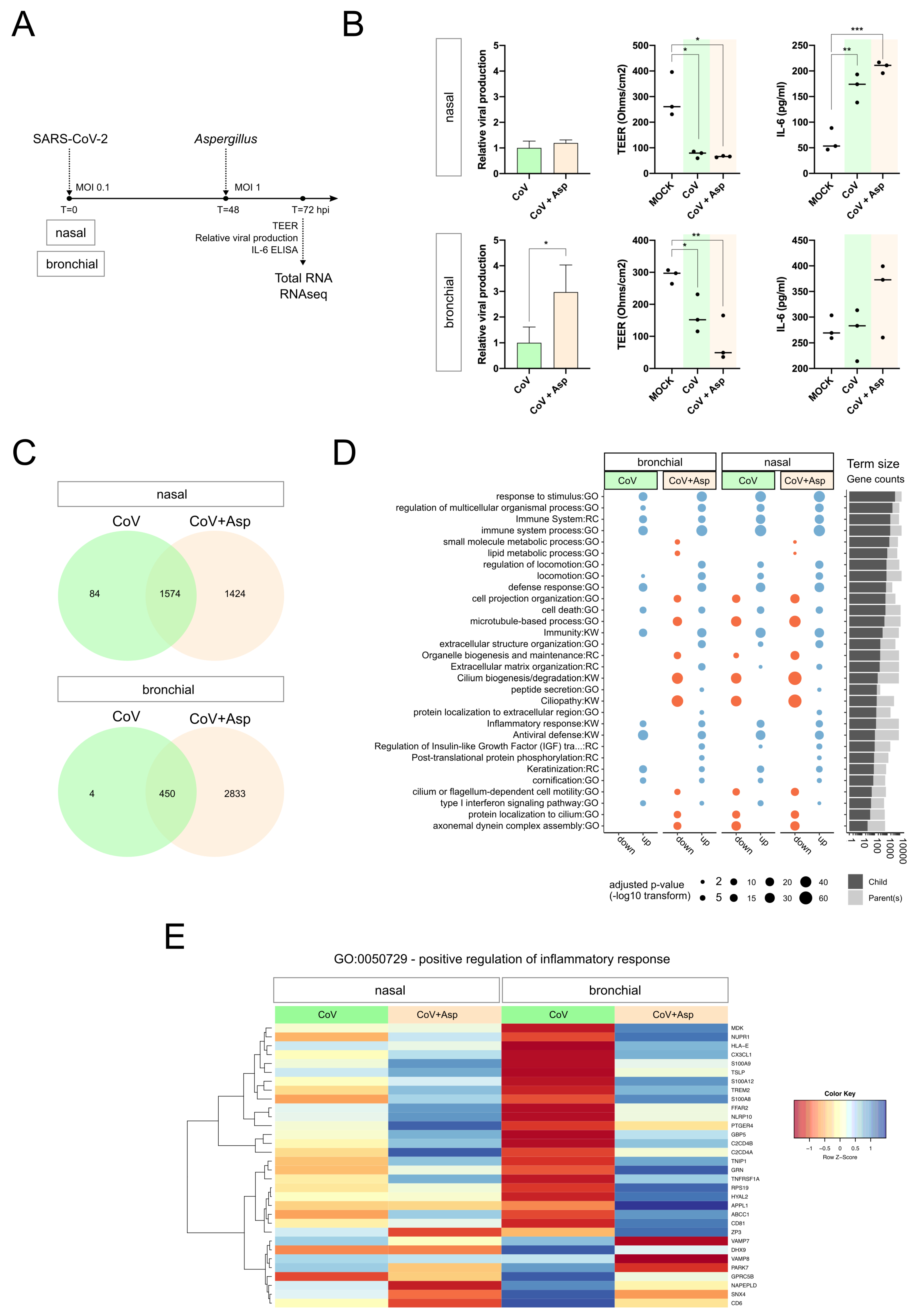
3.1. Simple Infection and Superinfection Induce a Strong Deregulation of Core Components of Innate Immune and Inflammatory Responses
3.2. CoV+Asp Superinfection Transcriptomic Signature Recapitulates the Signature CoV Infection, but with a Distinct Regulation of the Inflammatory Response
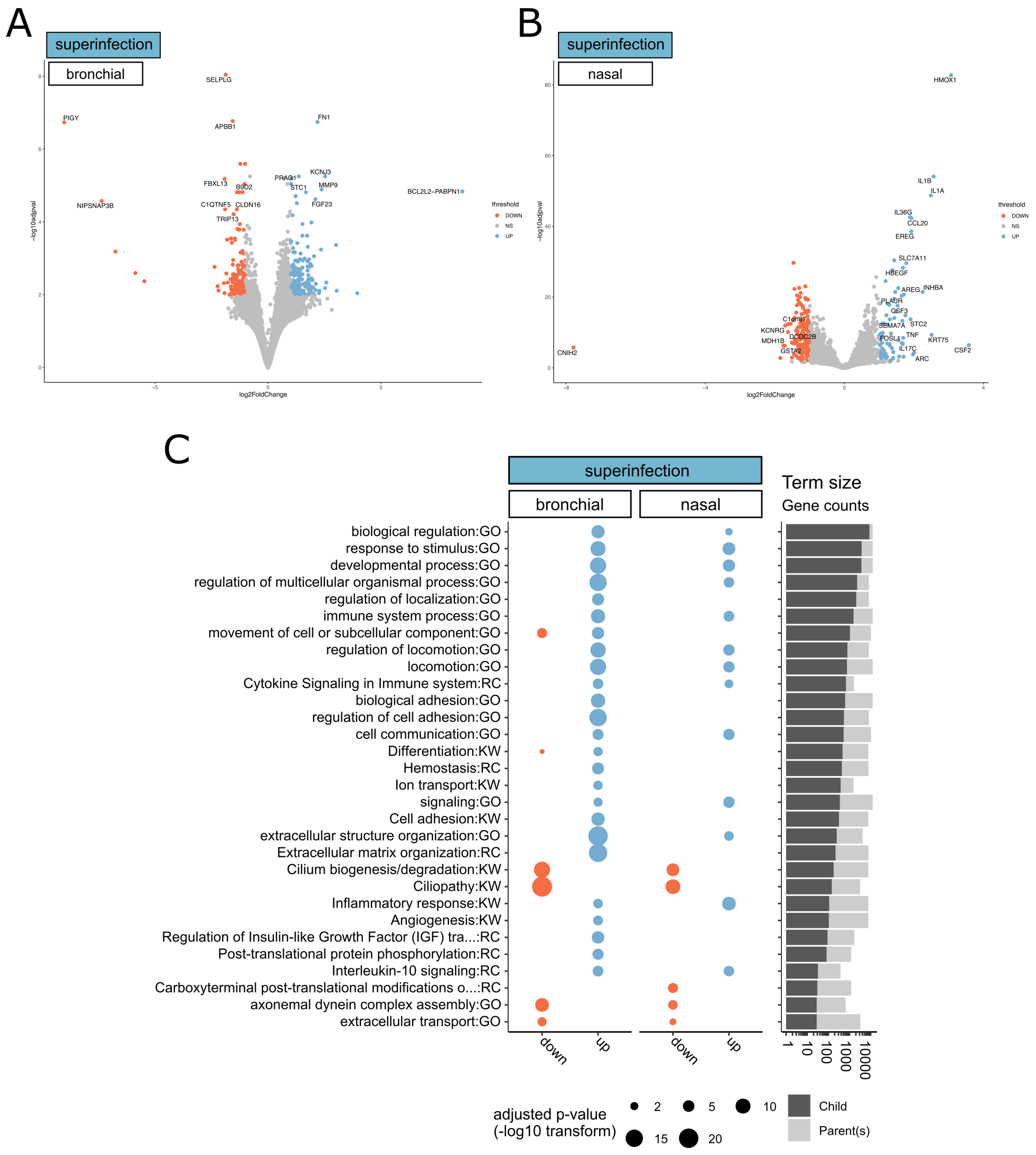
3.3. Transcriptional Footprints of SARS-CoV-2 Aspergillus Superinfection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Messina, F.; COVID 19 INMI Network Medicine for IDs Study Group; Giombini, E.; Agrati, C.; Vairo, F.; Bartoli, T.A.; Al Moghazi, S.; Piacentini, M.; Locatelli, F.; Kobinger, G.; et al. COVID-19: Viral–host interactome analyzed by network based-approach model to study pathogenesis of SARS-CoV-2 infection. J. Transl. Med. 2020, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Bosch, A.A.T.M.; Biesbroek, G.; Trzcinski, K.; Sanders, E.A.M.; Bogaert, D. Viral and Bacterial Interactions in the Upper Respiratory Tract. PLoS Pathog. 2013, 9, e1003057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullers, J.A. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Genet. 2014, 12, 252–262. [Google Scholar] [CrossRef]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Predominant Role of Bacterial Pneumonia as a Cause of Death in Pandemic Influenza: Implications for Pandemic Influenza Preparedness. J. Infect. Dis. 2008, 198, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Paget, C.; Trottein, F. Mechanisms of Bacterial Superinfection Post-influenza: A Role for Unconventional T Cells. Front. Immunol. 2019, 10, 336. [Google Scholar] [CrossRef]
- Bengoechea, J.A.; Bamford, C.G. SARS -CoV-2, bacterial co-infections, and AMR: The deadly trio in COVID -19? EMBO Mol. Med. 2020, 12. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Lescure, F.-X.; Bouadma, L.; Nguyen, D.; Parisey, M.; Wicky, P.-H.; Behillil, S.; Gaymard, A.; Bouscambert-Duchamp, M.; Donati, F.; Le Hingrat, Q.; et al. Clinical and virological data of the first cases of COVID-19 in Europe: A case series. Lancet Infect. Dis. 2020, 20, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Alanio, A.; Dellière, S.; Fodil, S.; Bretagne, S.; Mégarbane, B. Prevalence of putative invasive pulmonary aspergillosis in critically ill patients with COVID-19. Lancet Respir. Med. 2020, 8, e48–e49. [Google Scholar] [CrossRef]
- De Lamballerie, C.N.; Pizzorno, A.; Dubois, J.; Julien, T.; Padey, B.; Bouveret, M.; Traversier, A.; Legras-Lachuer, C.; Lina, B.; Boivin, G.; et al. Characterization of cellular transcriptomic signatures induced by different respiratory viruses in human reconstituted airway epithelia. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pizzorno, A.; Terrier, O.; De Lamballerie, C.N.; Julien, T.; Padey, B.; Traversier, A.; Roche, M.; Hamelin, M.-E.; Rhéaume, C.; Croze, S.; et al. Repurposing of Drugs as Novel Influenza Inhibitors From Clinical Gene Expression Infection Signatures. Front. Immunol. 2019, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Pizzorno, A.; Padey, B.; Julien, T.; Trouillet-Assant, S.; Traversier, A.; Errazuriz-Cerda, E.; Fouret, J.; Dubois, J.; Gaymard, A.; Lescure, F.-X.; et al. Characterization and Treatment of SARS-CoV-2 in Nasal and Bronchial Human Airway Epithelia. Cell Rep. Med. 2020, 1, 100059. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Maglott, D.R.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res. 2004, 33, D54–D58. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.I.; Love, M.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2016, 4, 1521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Breuza, L.; Poux, S.; Estreicher, A.; Famiglietti, M.L.; Magrane, M.; Tognolli, M.; Bridge, A.; Baratin, D.; Redaschi, N. The UniProt Consortium The UniProtKB guide to the human proteome. Database 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, S.; Bauer, S.; Robinson, P.N.; Vingron, M. Improved detection of overrepresentation of Gene-Ontology annotations with parent child analysis. Bioinformatics 2007, 23, 3024–3031. [Google Scholar] [CrossRef]
- Clancy, C.J.; Nguyen, M.H. Coronavirus Disease 2019, Superinfections, and Antimicrobial Development: What Can We Expect? Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Verweij, P.E.; Gangneux, J.-P.; Bassetti, M.; Brüggemann, R.J.M.A.; Cornely, O.; Koehler, P.; Lass-Flörl, C.; Van De Veerdonk, F.L.; Chakrabarti, A.; Hoenigl, M. Diagnosing COVID-19-associated pulmonary aspergillosis. Lancet Microbe 2020, 1, e53–e55. [Google Scholar] [CrossRef]
- Ziegler, C.G.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Kulkarni, H.; Robert, H.; Williams, G.; Andrew, P.; Easton, A.; O’Callaghan, C. Influenza virus infection of human ciliated respiratory epithelial cells in culture. Eur. Respir. J. 2013, 42 (Suppl. S57), 4406. [Google Scholar]
- Wu, N.-H.; Yang, W.; Beineke, A.; Dijkman, R.; Matrosovich, M.; Baumgärtner, W.; Thiel, V.E.; Valentin-Weigand, P.; Meng, F.; Herrler, G. The differentiated airway epithelium infected by influenza viruses maintains the barrier function despite a dramatic loss of ciliated cells. Sci. Rep. 2016, 6, 39668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, V.; Dutta, O.; Mc Elrath, C.; Du, P.; Chang, Y.-J.; Cicciarelli, B.; Pitler, A.; Whitehead, I.; Obar, J.J.; Durbin, J.E.; et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci. Immunol. 2017, 2, eaan5357. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Clemons, K.V.; Grunig, G.; Sobel, R.A.; Mirels, L.F.; Rennick, D.M.; Stevens, D.A. Role of IL-10 in invasive aspergillosis: Increased resistance of IL-10 gene knockout mice to lethal systemic aspergillosis. Clin. Exp. Immunol. 2000, 122, 186–191. [Google Scholar] [CrossRef]
- Santacroce, L.; Charitos, I.A.; Ballini, A.; Inchingolo, F.; Luperto, P.; De Nitto, E.; Topi, S. The Human Respiratory System and its Microbiome at a Glimpse. Biology 2020, 9, 318. [Google Scholar] [CrossRef]
- Bao, L.; Zhang, C.; Dong, J.; Zhao, L.; Li, Y.; Sun, J. Oral Microbiome and SARS-CoV-2: Beware of Lung Co-infection. Front. Microbiol. 2020, 11, 1840. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolas de Lamballerie, C.; Pizzorno, A.; Fouret, J.; Szpiro, L.; Padey, B.; Dubois, J.; Julien, T.; Traversier, A.; Dulière, V.; Brun, P.; et al. Transcriptional Profiling of Immune and Inflammatory Responses in the Context of SARS-CoV-2 Fungal Superinfection in a Human Airway Epithelial Model. Microorganisms 2020, 8, 1974. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121974
Nicolas de Lamballerie C, Pizzorno A, Fouret J, Szpiro L, Padey B, Dubois J, Julien T, Traversier A, Dulière V, Brun P, et al. Transcriptional Profiling of Immune and Inflammatory Responses in the Context of SARS-CoV-2 Fungal Superinfection in a Human Airway Epithelial Model. Microorganisms. 2020; 8(12):1974. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121974
Chicago/Turabian StyleNicolas de Lamballerie, Claire, Andrés Pizzorno, Julien Fouret, Lea Szpiro, Blandine Padey, Julia Dubois, Thomas Julien, Aurélien Traversier, Victoria Dulière, Pauline Brun, and et al. 2020. "Transcriptional Profiling of Immune and Inflammatory Responses in the Context of SARS-CoV-2 Fungal Superinfection in a Human Airway Epithelial Model" Microorganisms 8, no. 12: 1974. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121974