Rapid Diagnostics of Orthopaedic-Implant-Associated Infections Using Nanopore Shotgun Metagenomic Sequencing on Tissue Biopsies

 ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
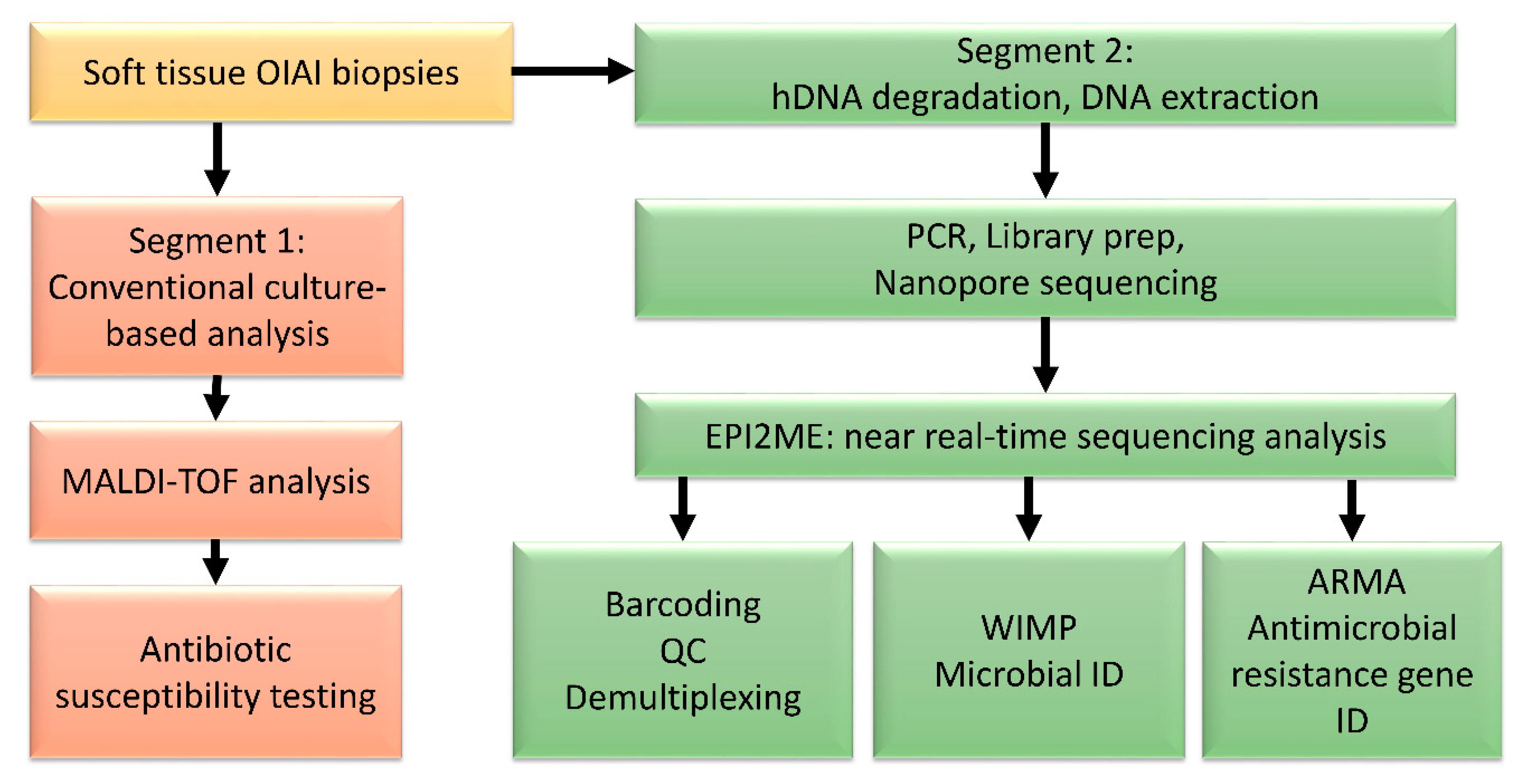
2. Materials and Methods
2.1. Patient Selection
2.2. DNA Extraction
2.3. Nanopore Sequencing and Analysis
3. Results
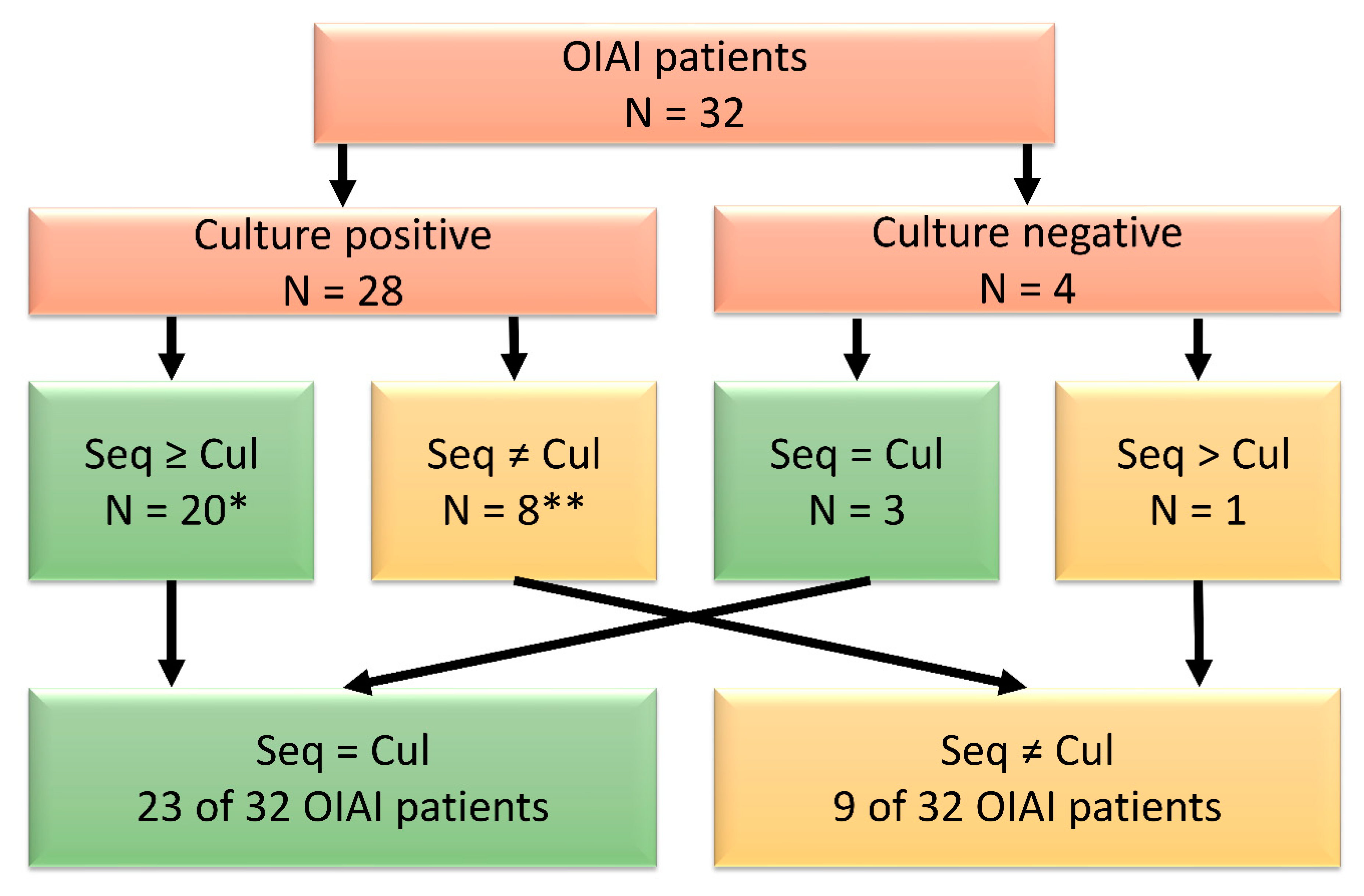
3.1. Pilot Study
3.2. Microbial Identification
3.3. Time to Results of Sequencing
3.4. Antimicrobial Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zmistowski, B.; Karam, J.A.; Durinka, J.B.; Casper, D.S.; Parvizi, J. Periprosthetic joint infection increases the risk of one-year mortality. J. Bone Jt. Surg. Ser. A 2013, 95, 2177–2184. [Google Scholar] [CrossRef] [Green Version]
- Siljander, M.P.; Sobh, A.H.; Baker, K.C.; Baker, E.A.; Kaplan, L.M. Multidrug-Resistant Organisms in the Setting of Periprosthetic Joint Infection—Diagnosis, Prevention, and Treatment. J. Arthroplast. 2018, 33, 185–194. [Google Scholar] [CrossRef]
- Karachalios, T.; Koutalos, A.; Komnos, G. Management strategies for infected total hip arthroplasty. A critical appreciation of problems and techniques. HIP Int. 2014, 24, S44–S47. [Google Scholar] [CrossRef]
- Lagier, J.C.; Hugon, P.; Khelaifia, S.; Fournier, P.E.; La Scola, B.; Raoult, D. The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef] [Green Version]
- Aamot, H.V.; Noone, J.C.; Skråmm, I.; Truls, M. Are conventional microbiological diagnostics sufficiently expedient in the era of rapid diagnostics? Evaluation of conventional microbiological diagnostics of orthopedic implant-associated infections (OIAI) Are conventional microbiological diagnostics. Acta Orthop. 2020, 1–6. [Google Scholar] [CrossRef]
- Kawamura, M.; Kobayashi, N.; Inaba, Y.; Choe, H.; Tezuka, T.; Kubota, S.; Saito, T. A new multiplex real-time polymerase chain reaction assay for the diagnosis of periprosthetic joint infection. Mod. Rheumatol. 2017, 27, 1072–1078. [Google Scholar] [CrossRef]
- Sambri, A.; Pignatti, G.; Romagnoli, M.; Donati, D.; Marcacci, M.; Cadossi, M. Intraoperative diagnosis of Staphylococcus aureus and coagulase-negative Staphylococcus using Xpert MRSA/SA SSTI assay in prosthetic joint infection. New Microbiol. 2017, 40, 130–134. [Google Scholar] [PubMed]
- Aamot, H.V.; Johnsen, B.O.; Skråmm, I. Rapid diagnostics of orthopedic implant-associated infections using Unyvero ITI implant and tissue infection application is not optimal for Staphylococcus species identification. BMC Res. Notes 2019, 12, 725. [Google Scholar] [CrossRef] [PubMed]
- Zimmerli, W.; Lew, P.D.; Waldvogel, F.A. Pathogenesis of foreign body infection. Evidence for a local granulocyte defect. J. Clin. Investig. 1984, 73, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Southwood, R.T.; Rice, J.L.; McDonald, P.J.; Hakendorf, P.H.; Rozenbilds, M.A. Infection in experimental arthroplasties. Clin. Orthop. Relat. Res. 1987, 67, 33–36. [Google Scholar] [CrossRef]
- Helmersen, K.; Aamot, H.V. DNA extraction of microbial DNA directly from infected tissue: An optimized protocol for use in nanopore sequencing. Sci. Rep. 2020, 10, 2985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvizi, J.; Zmistowski, B.; Berbari, E.F.; Bauer, T.W.; Springer, B.D.; Della Valle, C.J.; Garvin, K.L.; Mont, M.A.; Wongworawat, M.D.; Zalavras, C.G. New definition for periprosthetic joint infection: From the workgroup of the musculoskeletal infection society. Clin. Orthop. Relat. Res. 2011, 469, 2992–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Society of Clinical Microbiology and Infectious Diseases. Antimicrob. Susceptibility Test. EUCAST Disk Diffus. Method, Version 6.0. (2017); EUCAST. Break. Tables Interpret. MICs Zo. Diameters, Version 7.1. 2017. Available online: http//www.eucast.org (accessed on 31 December 2020).
- Schmidt, K.; Mwaigwisya, S.; Crossman, L.C.; Doumith, M.; Munroe, D.; Pires, C.; Khan, A.M.; Woodford, N.; Saunders, N.J.; Wain, J.; et al. Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J. Antimicrob. Chemother. 2017, 72, 104–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Petkovšek, Ž.; Eleršič, K.; Gubina, M.; Žgur-Bertok, D.; Erjavec, M.S. Virulence potential of Escherichia coli isolates from skin and soft tissue infections. J. Clin. Microbiol. 2009, 47, 1811–1817. [Google Scholar] [CrossRef]
- McDowell, A.; Nagy, I.; Magyari, M.; Barnard, E.; Patrick, S. The Opportunistic Pathogen Propionibacterium acnes: Insights into Typing, Human Disease, Clonal Diversification and CAMP Factor Evolution. PLoS ONE 2013, 8, e70897. [Google Scholar] [CrossRef] [Green Version]
- Mühl, H.; Kochem, A.J.; Disqué, C.; Sakka, S.G. Activity and DNA contamination of commercial polymerase chain reaction reagents for the universal 16S rDNA real-time polymerase chain reaction detection of bacterial pathogens in blood. Diagn. Microbiol. Infect. Dis. 2010, 66, 41–49. [Google Scholar] [CrossRef]
- Bengoechea, J.A.; Sa Pessoa, J. Klebsiella pneumoniae infection biology: Living to counteract host defences. FEMS Microbiol. Rev. 2019, 43, 123–144. [Google Scholar] [CrossRef] [Green Version]
- New England Biolabs Certificate of Analysis_LongAmp Taq Polymerase. Available online: https://www.neb.com/-/media/catalog/certificates-of-analysis/m0323l_v1_10048516.pdf?rev=8cc307a9142f4a42af1821421ac095b8&hash=75522E2DF011C79A55F45EF7401C223DFCC745BA (accessed on 2 September 2020).
- Bard, J.D.; Lewinski, M.; Summanen, P.H.; Deville, J.G. Sepsis with prolonged hypotension due to Moraxella osloensis in a non-immunocompromised child. J. Med. Microbiol. 2011, 60, 138–141. [Google Scholar] [CrossRef] [Green Version]
- Lusk, R.W. Diverse and widespread contamination evident in the unmapped depths of high throughput sequencing data. PLoS ONE 2014, 9, e110808. [Google Scholar] [CrossRef] [Green Version]
- Wally, N.; Schneider, M.; Thannesberger, J.; Kastner, M.T.; Bakonyi, T.; Indik, S.; Rattei, T.; Bedarf, J.; Hildebrand, F.; Law, J.; et al. Plasmid DNA contaminant in molecular reagents. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Charalampous, T.; Kay, G.L.; Richardson, H.; Aydin, A.; Baldan, R.; Jeanes, C.; Rae, D.; Grundy, S.; Turner, D.J.; Wain, J.; et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 2019, 37, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Putman, M.; Van Veen, H.W.; Degener, J.E.; Konings, W.N. The lactococcal secondary multidrug transporter LmrP confers resistance to lincosamides, macrolides, streptogramins and tetracyclines. Microbiology 2001, 147, 2873–2880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campion, J.J.; McNamara, P.J.; Evans, M.E. Evolution of ciprofloxacin-resistant Staphylococcus aureus in in vitro pharmacokinetic environments. Antimicrob. Agents Chemother. 2004, 48, 4733–4744. [Google Scholar] [CrossRef] [Green Version]
- Su, M.; Satola, S.W.; Read, T.D. Genome-based prediction of bacterial antibiotic resistance. J. Clin. Microbiol. 2019, 57, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Eyre, D.W.; Silva, D.D.; Cole, K.; Peters, J.; Cole, M.J.; Grad, Y.H.; Demczuk, W.; Martin, I.; Mulvey, M.R.; Crook, D.W.; et al. WGS to predict antibiotic MICs for Neisseria gonorrhoeae. J. Antimicrob. Chemother. 2017, 72, 1937–1947. [Google Scholar] [CrossRef] [Green Version]
- Kime, L.; Randall, C.P.; Banda, F.I.; Coll, F.; Wright, J.; Richardson, J.; Empel, J.; Parkhill, J.; O’Neill, A.J. Transient silencing of antibiotic resistance by mutation represents a significant potential source of unanticipated therapeutic failure. MBio 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Paul, G.C.; Gerbraud, G.; Bure, A.; Philippon, A.M.; Pangon, B.; Courvalin, P. TEM-4, a new plasmid-mediated β-lactamase that hydrolyzes broad-spectrum cephalosporins in a clinical isolate of Escherichia coli. Antimicrob. Agents Chemother. 1989, 33, 1958–1963. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Deng, W.; Liu, S.; Yu, X.; Mustafa, G.R.; Chen, S.; He, L.; Ao, X.; Yang, Y.; Zhou, K.; et al. Presence of heavy metal resistance genes in Escherichia coli and Salmonella isolates and analysis of resistance gene structure in E. coli E308. J. Glob. Antimicrob. Resist. 2020, 21, 420–426. [Google Scholar] [CrossRef]
- Fernández-Martínez, M.; Del Ruiz Castillo, B.; Lecea-Cuello, M.J.; Rodríguez-Baño, J.; Pascual, Á.; Martínez-Martínez, L. Prevalence of Aminoglycoside-Modifying Enzymes in Escherichia coli and Klebsiella pneumoniae Producing Extended Spectrum β-Lactamases Collected in Two Multicenter Studies in Spain. Microb. Drug Resist. 2018, 24, 367–376. [Google Scholar] [CrossRef]
- Street, T.L.; Sanderson, N.D.; Atkins, B.L.; Brent, A.J.; Cole, K.; Foster, D.; McNally, M.A.; Oakley, S.; Peto, L.; Taylor, A.; et al. Molecular Diagnosis of Orthopedic-Device-Related Infection Directly from Sonication Fluid by Metagenomic Sequencing. J. Clin. Microbiol. 2017, 55, 2334–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, N.D.; Street, T.L.; Foster, D.; Swann, J.; Atkins, B.L.; Brent, A.J.; McNally, M.A.; Oakley, S.; Taylor, A.; Peto, T.E.A.; et al. Real-time analysis of nanopore-based metagenomic sequencing from infected orthopaedic devices. BMC Genom. 2018, 19, 714. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noone, J.C.; Helmersen, K.; Leegaard, T.M.; Skråmm, I.; Aamot, H.V. Rapid Diagnostics of Orthopaedic-Implant-Associated Infections Using Nanopore Shotgun Metagenomic Sequencing on Tissue Biopsies. Microorganisms 2021, 9, 97. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9010097
Noone JC, Helmersen K, Leegaard TM, Skråmm I, Aamot HV. Rapid Diagnostics of Orthopaedic-Implant-Associated Infections Using Nanopore Shotgun Metagenomic Sequencing on Tissue Biopsies. Microorganisms. 2021; 9(1):97. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9010097
Chicago/Turabian StyleNoone, J. Christopher, Karin Helmersen, Truls Michael Leegaard, Inge Skråmm, and Hege Vangstein Aamot. 2021. "Rapid Diagnostics of Orthopaedic-Implant-Associated Infections Using Nanopore Shotgun Metagenomic Sequencing on Tissue Biopsies" Microorganisms 9, no. 1: 97. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9010097