
Systematic Analysis of Functionally Related Gene Clusters in the Opportunistic Pathogen, Candida albicans

Abstract
:1. Introduction
2. Materials and Methods
2.1. The Identification and Significance of Functional Clustering in Candida albicans
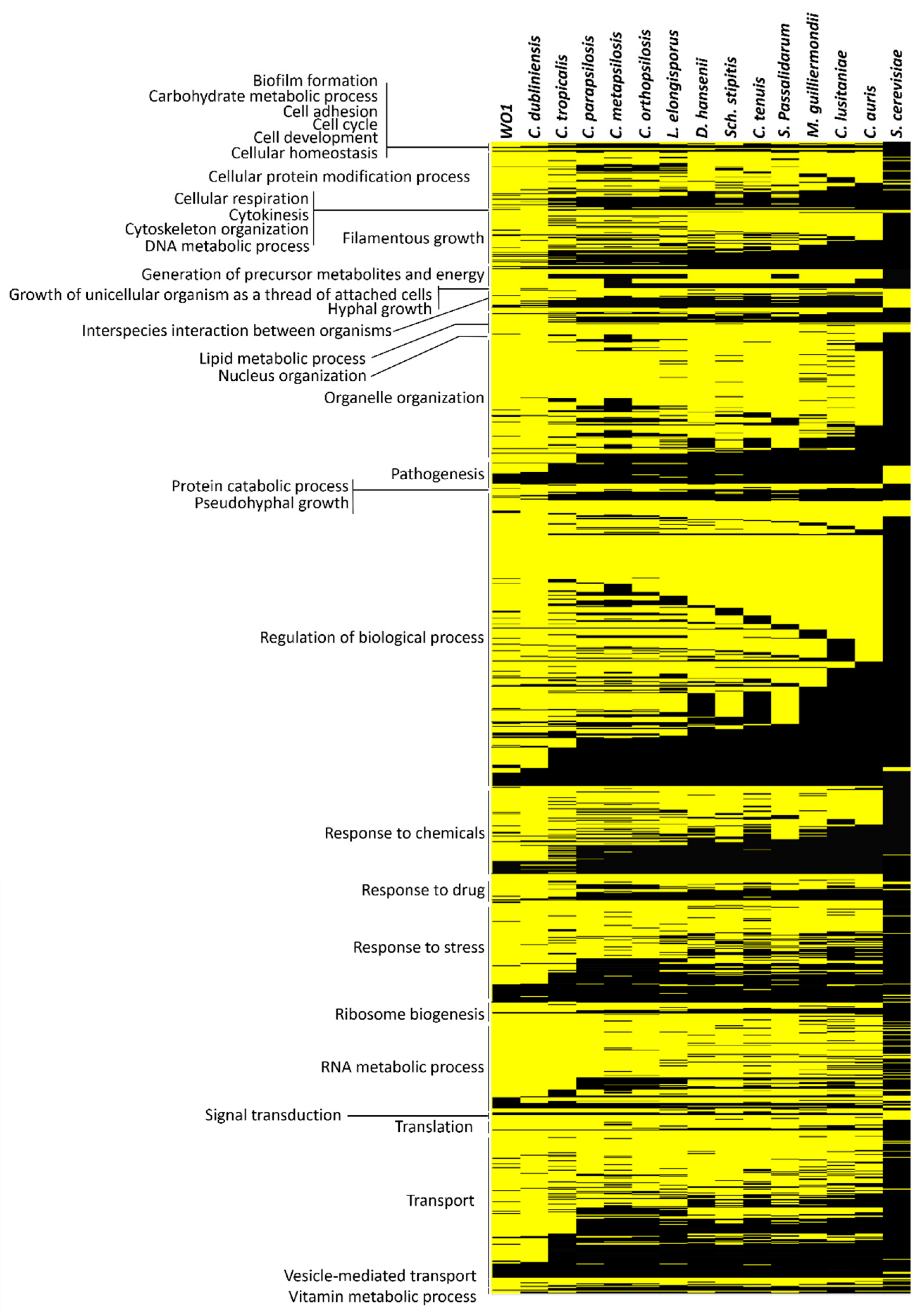
2.2. The Conservation of the Functional Clusters across Divergent Candida Lineages
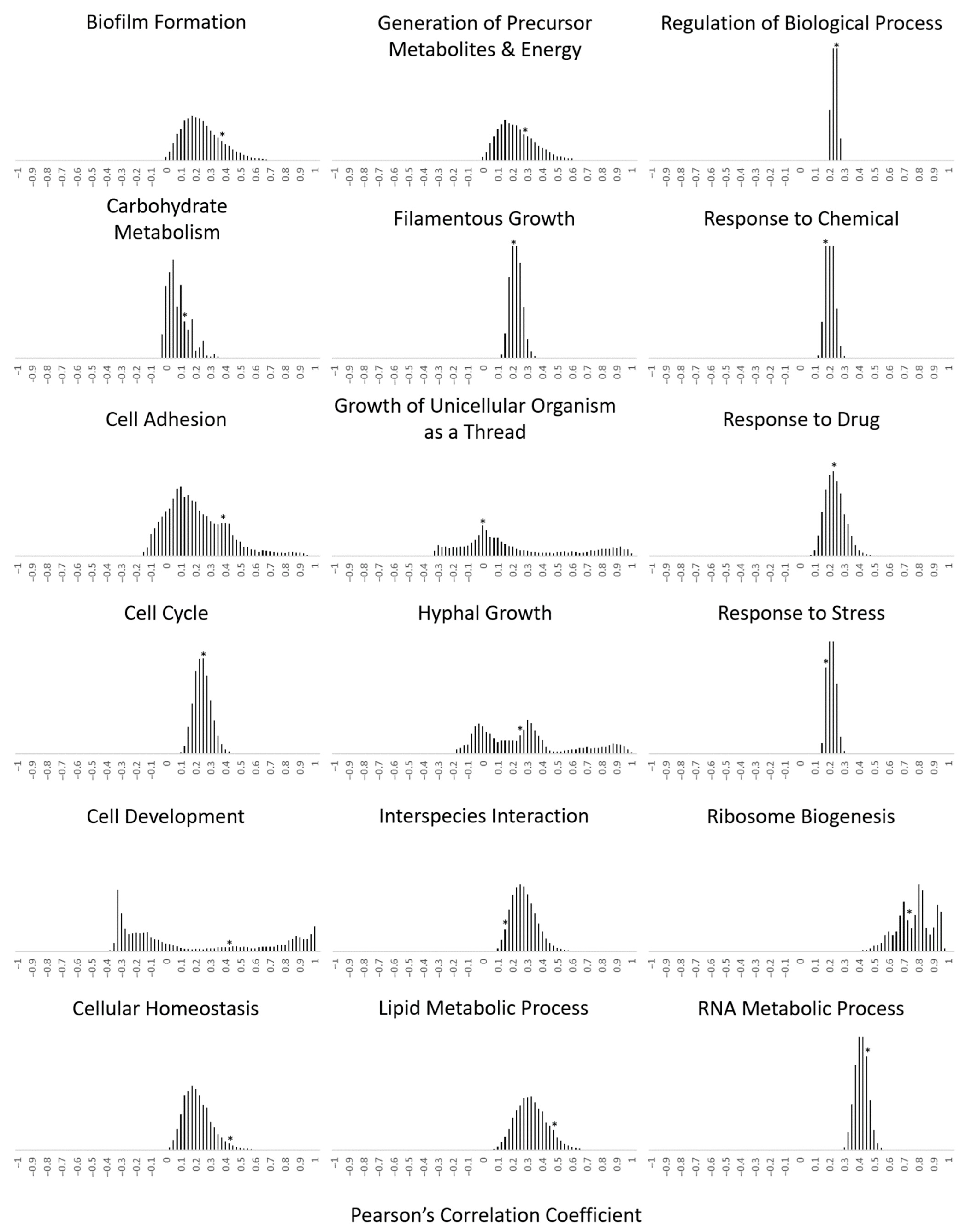
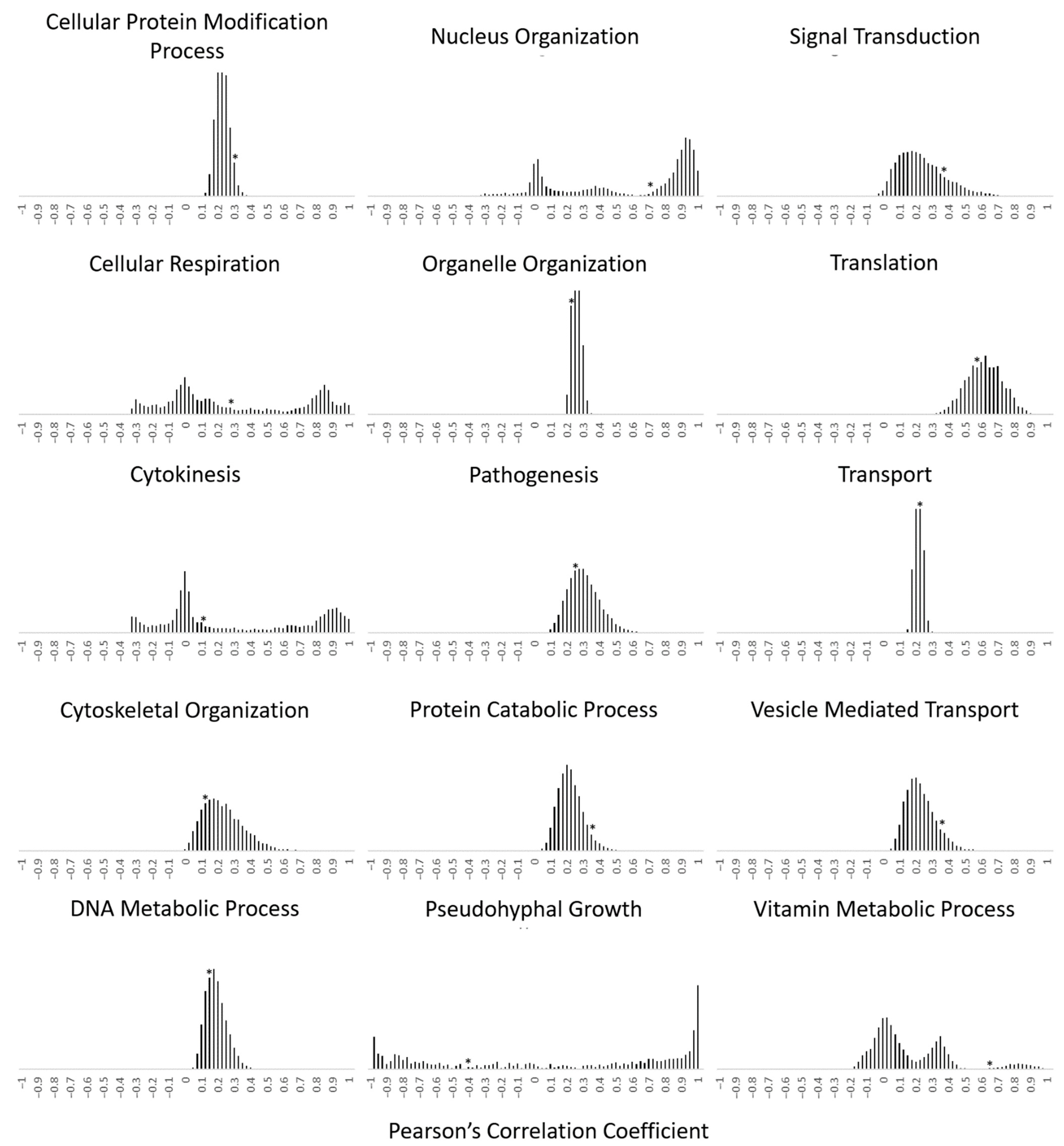
2.3. Calculation of the Transcriptional Relationship within Gene Families with the Pearson’s Correlation Coefficient
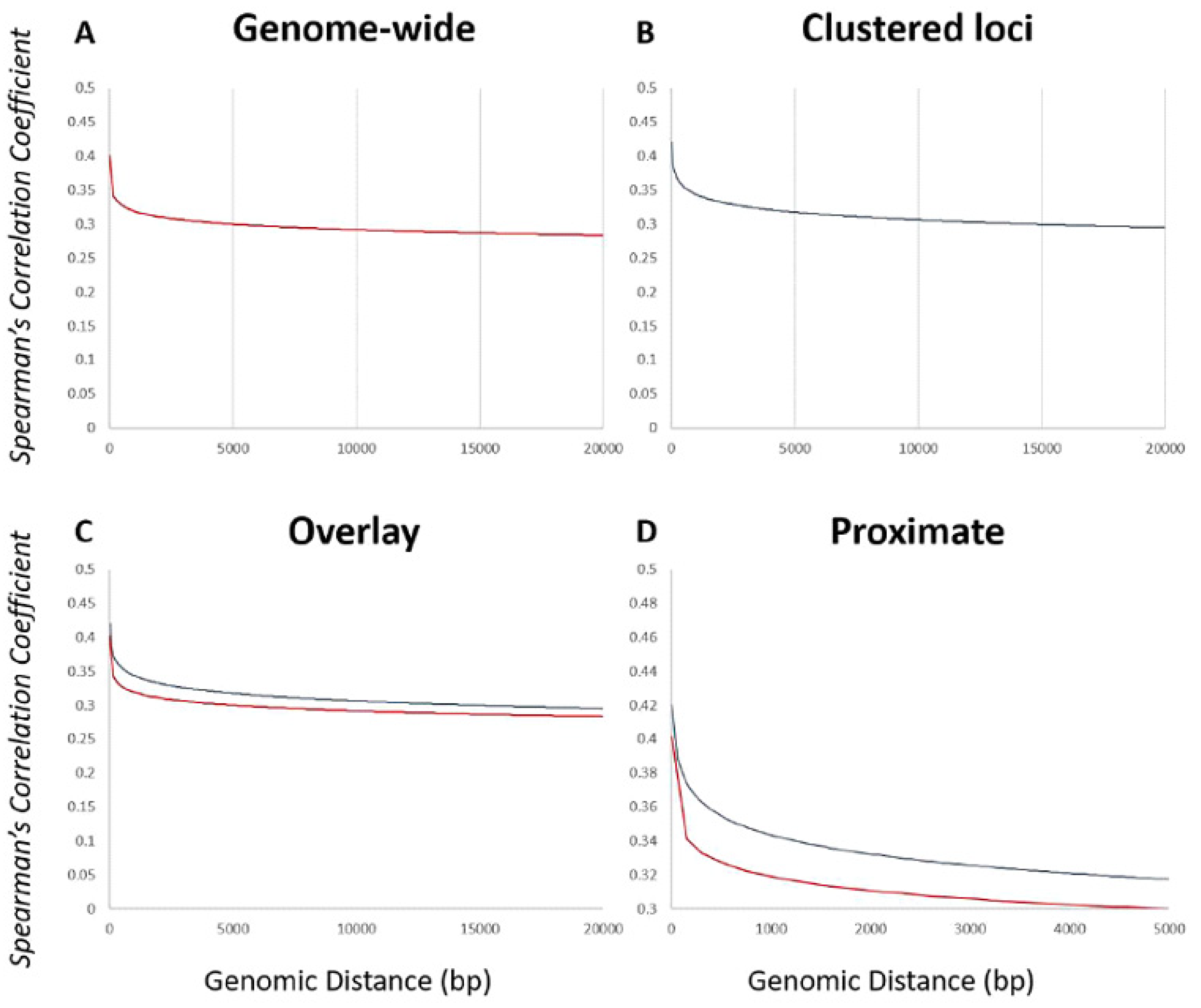
2.4. Calculation of the Transcriptional Relationship within a Genomic Neighborhood Using the Spearman’s Correlation Coefficient
2.5. Microarray and RNA-Sequencing Data Sets Used for Analysis
3. Results
3.1. Functionally Related Genes Cluster throughout the Candida albicans Genome
3.2. Functionally Clustered Genes Exhibit Tighter Transcriptional Coregulation within Their Gene Family
3.3. Clustered Genes Do Not Exhibit a Bias for a Divergent Orientation
3.4. Clustered Grouping Are Highly Conserved among Closely Related Candida Species and Deteriorate with Greater Evolutionary Distance
3.5. Functionally Clustered Gene Sets Display a Higher Transcriptional Coregulation than Would Be Expected by Chance Groupings of Functionally Related Genes
3.6. Functionally Related Genes Congregate to Transcriptionally Permissive Regions throughout the Genome
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pappas, P.G.; Kauffman, C.A.; Andes, D.R.; Clancy, C.J.; Marr, K.A.; Ostrosky-Zeichner, L.; Reboli, A.C.; Schuster, M.G.; Vazquez, J.A.; Walsh, T.J. Clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2016, 62, e1–e50. [Google Scholar] [CrossRef]
- De Oliveira Santos, G.C.; Vasconcelos, C.C.; Lopes, A.J.; de Sousa Cartágenes, M.d.S.; do Nascimento, F.R.; Ramos, R.M.; Pires, E.R.; de Andrade, M.S.; Rocha, F.M.; de Andrade Monteiro, C. Candida infections and therapeutic strategies: Mechanisms of action for traditional and alternative agents. Front. Microbiol. 2018, 9, 1351. [Google Scholar] [CrossRef] [PubMed]
- Nett, J.E.; Andes, D.R. Contributions of the biofilm matrix to Candida pathogenesis. J. Fungi 2020, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, J.F.; Kavanagh, K.; Sobel, J.D.; Kauffman, C.A.; Newman, C.A. Candida urinary tract infection: Pathogenesis. Clin. Infect. Dis. 2011, 52, S437–S451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassone, A. Vulvovaginal Candida albicans infections: Pathogenesis, immunity and vaccine prospects. Int. J. Obstet. Gynaecol. 2015, 122, 785–794. [Google Scholar] [CrossRef]
- Cheng, S.; Clancy, C.J.; Checkley, M.A.; Handfield, M.; Hillman, J.D.; Progulske-Fox, A.; Lewin, A.S.; Fidel, P.L.; Nguyen, M.H. Identification of Candida albicans genes induced during thrush offers insight into pathogenesis. Mol. Microbiol. 2003, 48, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Pappas, P.G.; Lionakis, M.S.; Arendrup, M.C.; Ostrosky-Zeichner, L.; Kullberg, B.J. Invasive candidiasis. Nat. Rev. Dis. Primers 2018, 4, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Perlin, D.S. Mechanisms of echinocandin antifungal drug resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 1. [Google Scholar] [CrossRef]
- Healey, K.R.; Perlin, D.S. Fungal resistance to echinocandins and the MDR phenomenon in Candida glabrata. J. Fungi 2018, 4, 105. [Google Scholar]
- Healey, K.R.; Zhao, Y.; Perez, W.B.; Lockhart, S.R.; Sobel, J.D.; Farmakiotis, D.; Kontoyiannis, D.P.; Sanglard, D.; Taj-Aldeen, S.J.; Alexander, B.D. Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat. Commnu. 2016, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kordalewska, M.; Perlin, D.S. Identification of drug resistant Candida auris. Front. Microbiol. 2019, 10, 1918. [Google Scholar] [CrossRef] [PubMed]
- Revie, N.M.; Iyer, K.R.; Robbins, N.; Cowen, L.E. Antifungal drug resistance: Evolution, mechanisms and impact. Curr. Opin. Microbiol. 2018, 45, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Nantel, A.; Dignard, D.; Bachewich, C.; Harcus, D.; Marcil, A.; Bouin, A.-P.; Sensen, C.W.; Hogues, H.; van het Hoog, M.; Gordon, P. Transcription profiling of Candida albicans cells undergoing the yeast-to-hyphal transition. Mol. Biol. Cell 2002, 13, 3452–3465. [Google Scholar] [CrossRef] [Green Version]
- Whiteway, M.; Bachewich, C. Morphogenesis in Candida albicans. Annu. Rev. Microbiol. 2007, 61, 529–553. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Tan, Y.; Cahan, P. Understanding development and stem cells using single cell-based analyses of gene expression. Development 2017, 144, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Rué, P.; Martinez Arias, A. Cell dynamics and gene expression control in tissue homeostasis and development. Mol. Syst. Biol. 2015, 11, 792. [Google Scholar] [CrossRef] [PubMed]
- Cardoso-Moreira, M.; Halbert, J.; Valloton, D.; Velten, B.; Chen, C.; Shao, Y.; Liechti, A.; Ascenção, K.; Rummel, C.; Ovchinnikova, S. Gene expression across mammalian organ development. Nature 2019, 571, 505–509. [Google Scholar] [CrossRef]
- Chandrangsu, P.; Rensing, C.; Helmann, J.D. Metal homeostasis and resistance in bacteria. Nat. Rev. Microbiol. 2017, 15, 338. [Google Scholar]
- Li, C.; Li, Y.; Ding, C. The role of copper homeostasis at the host-pathogen axis: From bacteria to fungi. Int. J. Mol. Sci. 2019, 20, 175. [Google Scholar] [CrossRef] [Green Version]
- Brickner, J.H.; Walter, P. Gene recruitment of the activated INO1 locus to the nuclear membrane. PLoS Biol. 2004, 2, e342. [Google Scholar] [CrossRef]
- Egecioglu, D.; Brickner, J.H. Gene positioning and expression. Curr. Opin. Cell Biol. 2011, 23, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasch, A.P.; Werner-Washburne, M. The genomics of yeast responses to environmental stress and starvation. Funct. Integr. Genom. 2002, 2, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Gasch, A.P.; Yu, F.B.; Hose, J.; Escalante, L.E.; Place, M.; Bacher, R.; Kanbar, J.; Ciobanu, D.; Sandor, L.; Grigoriev, I.V. Single-cell RNA sequencing reveals intrinsic and extrinsic regulatory heterogeneity in yeast responding to stress. PLoS Biol. 2017, 15, e2004050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Martínez, M.E.; Benet, M.; Alepuz, P.; Tordera, V. Nut1/Hos1 and Sas2/Rpd3 control the H3 acetylation of two different sets of osmotic stress-induced genes. Epigenetics 2020, 15, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, E.; Shimaji, K.; Umegawachi, T.; Tomida, S.; Yoshida, H.; Yoshimoto, N.; Izawa, S.; Kimura, H.; Yamaguchi, M. The Histone deacetylase gene Rpd3 is required for starvation stress resistance. PLoS ONE 2016, 11, e0167554. [Google Scholar] [CrossRef]
- Rocha, C.R.; Schroppel, K.; Harcus, D.; Marcil, A.; Dignard, D.; Taylor, B.N.; Thomas, D.Y.; Whiteway, M.; Leberer, E. Signaling through adenylyl cyclase is essential for hyphal growth and virulence in the pathogenic fungus Candida albicans. Mol. Biol. Cell 2001, 12, 3631–3643. [Google Scholar] [CrossRef] [Green Version]
- Harcus, D.; Nantel, A.; Marcil, A.; Rigby, T.; Whiteway, M. Transcription profiling of cyclic AMP signaling in Candida albicans. Mol. Biol. Cell 2004, 15, 4490–4499. [Google Scholar] [CrossRef] [Green Version]
- Gasch, A.P.; Spellman, P.T.; Kao, C.M.; Carmel-Harel, O.; Eisen, M.B.; Storz, G.; Botstein, D.; Brown, P.O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 2000, 11, 4241–4257. [Google Scholar] [CrossRef]
- Rangel, D.E. Stress induced cross-protection against environmental challenges on prokaryotic and eukaryotic microbes. World J. Microbiol. Biotechnol. 2011, 27, 1281–1296. [Google Scholar] [CrossRef]
- Kreuzer, K.N. DNA damage responses in prokaryotes: Regulating gene expression, modulating growth patterns, and manipulating replication forks. Cold Spring Harbor Perspect. Biol. 2013, 5, a012674. [Google Scholar] [CrossRef]
- Enjalbert, B.; Nantel, A.; Whiteway, M. Stress-induced gene expression in Candida albicans: Absence of a general stress response. Mol. Biol. Cell 2003, 14, 1460–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, M.D.; Farrer, R.A.; Tan, K.; Miao, Z.; Walker, L.A.; Cuomo, C.A.; Wheeler, R.T.; Brown, A.J.; Wong, K.H.; Cowen, L.E. Hsf1 and Hsp90 orchestrate temperature-dependent global transcriptional remodelling and chromatin architecture in Candida albicans. Nat. Commun. 2016, 7, 1–13. [Google Scholar]
- Nobile, C.J.; Fox, E.P.; Nett, J.E.; Sorrells, T.R.; Mitrovich, Q.M.; Hernday, A.D.; Tuch, B.B.; Andes, D.R.; Johnson, A.D. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 2012, 148, 126–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, E.M.; Howlett, B.J. Secondary metabolism: Regulation and role in fungal biology. Curr. Opin. Microbiol. 2008, 11, 481–487. [Google Scholar] [CrossRef]
- Eldabagh, R.S.; Mejia, N.G.; Barrett, R.L.; Monzo, C.R.; So, M.K.; Foley, J.J.; Arnone, J.T. Systematic identification, characterization, and conservation of adjacent-gene coregulation in the budding yeast Saccharomyces cerevisiae. Msphere 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Hagee, D.; Hardan, A.A.; Botero, J.; Arnone, J.T. Genomic Clustering within Functionally Related Gene Families in Ascomycota Fungi. Computat. Struct. Biotechnol. J. 2020. [Google Scholar] [CrossRef]
- Xu, H.; Liu, J.-J.; Liu, Z.; Li, Y.; Jin, Y.-S.; Zhang, J. Synchronization of stochastic expressions drives the clustering of functionally related genes. Sci. Adv. 2019, 5, eaax6525. [Google Scholar] [CrossRef] [Green Version]
- Tye, B.W.; Commins, N.; Ryazanova, L.V.; Wühr, M.; Springer, M.; Pincus, D.; Churchman, L.S. Proteotoxicity from aberrant ribosome biogenesis compromises cell fitness. Elife 2019, 8, e43002. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Consortium, G.O. The gene ontology resource: 20 years and still GOing strong. Nucl. Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Skrzypek, M.S.; Binkley, J.; Binkley, G.; Miyasato, S.R.; Simison, M.; Sherlock, G. The Candida Genome Database (CGD): Incorporation of Assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucl. Acids Res. 2016, gkw924. [Google Scholar]
- Arnone, J.T.; Robbins-Pianka, A.; Arace, J.R.; Kass-Gergi, S.; McAlear, M.A. The adjacent positioning of co-regulated gene pairs is widely conserved across eukaryotes. BMC Genom. 2012, 13, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, S.L.; OhÉigeartaigh, S.S.; Byrne, K.P.; Schröder, M.S.; O’Gaora, P.; Wolfe, K.H.; Butler, G. Comparative genome analysis and gene finding in Candida species using CGOB. Mol. Biol. Evol. 2013, 30, 1281–1291. [Google Scholar]
- Fitzpatrick, D.A.; O’Gaora, P.; Byrne, K.P.; Butler, G. Analysis of gene evolution and metabolic pathways using the Candida Gene Order Browser. BMC Genom. 2010, 11, 290. [Google Scholar] [CrossRef] [Green Version]
- Cera, A.; Holganza, M.K.; Hardan, A.A.; Gamarra, I.; Eldabagh, R.S.; Deschaine, M.; Elkamhawy, S.; Sisso, E.M.; Foley, J.J.; Arnone, J.T. Functionally Related Genes Cluster into Genomic Regions That Coordinate Transcription at a Distance in Saccharomyces cerevisiae. Msphere 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Li, D.; Zhang, Y.; Killeen, K.; Groutas, W.; Calderone, R. Repurposing an inhibitor of ribosomal biogenesis with broad anti-fungal activity. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Dorsaz, S.; Snäkä, T.; Favre-Godal, Q.; Maudens, P.; Boulens, N.; Furrer, P.; Ebrahimi, S.N.; Hamburger, M.; Allémann, E.; Gindro, K. Identification and mode of action of a plant natural product targeting human fungal pathogens. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar]
- Harcus, D.; Dignard, D.; Lépine, G.; Askew, C.; Raymond, M.; Whiteway, M.; Wu, C. Comparative xylose metabolism among the ascomycetes C. albicans, S. stipitis and S. cerevisiae. PLoS ONE 2013, 8, e80733. [Google Scholar] [CrossRef] [Green Version]
- Arnone, J.T.; McAlear, M.A. Adjacent gene pairing plays a role in the coordinated expression of ribosome biogenesis genes MPP10 and YJR003C in Saccharomyces cerevisiae. Eukar. Cell 2011, 10, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Arnone, J.T.; Arace, J.R.; Soorneedi, A.R.; Citino, T.T.; Kamitaki, T.L.; McAlear, M.A. Dissecting the cis and trans elements that regulate adjacent-gene coregulation in Saccharomyces cerevisiae. Eukar. Cell 2014, 13, 738–748. [Google Scholar] [CrossRef] [Green Version]
- Wade, C.H.; Umbarger, M.A.; McAlear, M.A. The budding yeast rRNA and ribosome biosynthesis (RRB) regulon contains over 200 genes. Yeast 2006, 23, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Rendsvig, J.K.; Workman, C.T.; Hoof, J.B. Bidirectional histone-gene promoters in Aspergillus: Characterization and application for multi-gene expression. Fungal Biol. Biotechnol. 2019, 6, 1–14. [Google Scholar] [CrossRef]
- Xu, Z.; Wei, W.; Gagneur, J.; Perocchi, F.; Clauder-Münster, S.; Camblong, J.; Guffanti, E.; Stutz, F.; Huber, W.; Steinmetz, L.M. Bidirectional promoters generate pervasive transcription in yeast. Nature 2009, 457, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.A.; Mitra, R.D.; Hughes, J.D.; Church, G.M. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat. Genet. 2000, 26, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Cadena, P.; Sternberg, P.W. Enhancer sharing promotes neighborhoods of transcriptional regulation across eukaryotes. G3 Genes Genom. Genet. 2016, 6, 4167–4174. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Process | Gene Family Size | Singletons | Clusters | p-Value |
|---|---|---|---|---|
| Biofilm formation | 149 | 132 | 17 | 2.53 × 10−4 |
| Carbohydrate metabolic process | 191 | 168 | 23 | 8.19 × 10−4 |
| Cell adhesion | 68 | 61 | 7 | 2.94 × 10−5 |
| Cell budding | 46 | 46 | 0 | n.s |
| Cell cycle | 477 | 375 | 102 | 1.02 × 10−4 |
| Cell development | 113 | 110 | 3 | 6.03 × 10−1 |
| Cell wall organization | 160 | 160 | 0 | n.s. |
| Cellular homeostasis | 205 | 183 | 22 | 9.99 × 10−3 |
| Cellular protein modification process | 584 | 452 | 132 | 3.92 × 10−3 |
| Cellular respiration | 89 | 85 | 4 | 1.19 × 10−1 |
| Conjugation | 63 | 63 | 0 | n.s. |
| Cytokinesis | 91 | 87 | 4 | 1.36 × 10−1 |
| Cytoskeleton organization | 184 | 157 | 27 | 5.38 × 10−6 |
| DNA metabolic process | 344 | 290 | 54 | 3.24 × 10−3 |
| Filamentous growth | 607 | 470 | 138 | 9.55 × 10−3 |
| Generation of precursor metabolites and energy | 140 | 119 | 21 | 4.34 × 10−7 |
| Growth of unicellular organism as a thread of attached cells | 88 | 84 | 4 | 1.11 × 10−1 |
| Hyphal growth | 97 | 91 | 6 | 3.50 × 10−2 |
| Interspecies interaction between organisms | 343 | 300 | 43 | 1.58 × 10−1 |
| Lipid metabolic process | 286 | 249 | 37 | 1.44 × 10−2 |
| Nucleus organization | 56 | 52 | 4 | 3.68 × 10−3 |
| Organelle organization | 1053 | 682 | 371 | 6.68 × 10−3 |
| Pathogenesis | 275 | 239 | 36 | 7.33 × 10−3 |
| Protein catabolic process | 220 | 196 | 24 | 1.44 × 10−2 |
| Protein folding | 82 | 82 | 0 | n.s. |
| Pseudohyphal growth | 41 | 39 | 2 | 1.78 × 10−2 |
| Regulation of biological process | 1500 | 859 | 641 | 6.73 × 10−1 |
| Response to chemical | 804 | 591 | 213 | 1.19 × 10−1 |
| Response to drug | 406 | 343 | 63 | 5.53 × 10−2 |
| Response to stress | 860 | 614 | 246 | 6.24 × 10−2 |
| Ribosome biogenesis | 305 | 277 | 28 | 5.29 × 10−2 |
| RNA metabolic process | 774 | 551 | 221 | 1.23 × 10−3 |
| Signal transduction | 219 | 203 | 16 | 3.85 × 10−1 |
| Translation | 245 | 207 | 38 | 2.45 × 10−5 |
| Transport | 1060 | 698 | 362 | 5.45 × 10−2 |
| Transposition | 4 | 4 | 0 | n.s. |
| Vesicle-mediated transport | 320 | 290 | 30 | 6.62 × 10−1 |
| Vitamin metabolic process | 40 | 34 | 6 | 8.64 × 10−7 |
| Molecular Process | Family | Singletons | Clustered Set |
|---|---|---|---|
| Biofilm formation | 0.282 | 0.266 | 0.376 |
| Carbohydrate metabolic process | 0.212 | 0.224 | 0.101 |
| Cell adhesion | 0.274 | 0.283 | 0.370 |
| Cell cycle | 0.258 | 0.256 | 0.260 |
| Cell development | 0.216 | 0.215 | 0.433 |
| Cellular homeostasis | 0.283 | 0.266 | 0.429 |
| Cellular protein modification process | 0.289 | 0.276 | 0.331 |
| Cellular respiration | 0.246 | 0.250 | 0.293 |
| Cytokinesis | 0.299 | 0.300 | 0.100 |
| Cytoskeleton organization | 0.241 | 0.262 | 0.116 |
| DNA metabolic process | 0.242 | 0.260 | 0.148 |
| Filamentous growth | 0.281 | 0.295 | 0.237 |
| Generation of precursor metabolites and energy | 0.237 | 0.231 | 0.287 |
| Growth of unicellular organism as a thread of attached cells | 0.280 | 0.301 | 0.017 |
| Hyphal growth | 0.308 | 0.306 | 0.287 |
| Interspecies interaction between organisms | 0.348 | 0.368 | 0.182 |
| Lipid metabolic process | 0.345 | 0.327 | 0.481 |
| Nucleus organization | 0.540 | 0.525 | 0.772 |
| Organelle organization | 0.285 | 0.310 | 0.240 |
| Pathogenesis | 0.363 | 0.381 | 0.243 |
| Protein catabolic process | 0.328 | 0.329 | 0.352 |
| Pseudohyphal growth | 0.260 | 0.262 | −0.438 |
| Regulation of biological process | 0.295 | 0.304 | 0.282 |
| Response to chemical | 0.269 | 0.296 | 0.196 |
| Response to drug | 0.290 | 0.298 | 0.253 |
| Response to stress | 0.316 | 0.302 | 0.229 |
| Ribosome biogenesis | 0.680 | 0.673 | 0.736 |
| RNA metabolic process | 0.416 | 0.415 | 0.470 |
| Signal transduction | 0.288 | 0.279 | 0.372 |
| Translation | 0.525 | 0.531 | 0.582 |
| Transport | 0.264 | 0.271 | 0.250 |
| Vesicle-mediated transport | 0.319 | 0.313 | 0.368 |
| Vitamin metabolic process | 0.258 | 0.227 | 0.641 |
| Molecular Process | Divergent | Tandem | Convergent |
|---|---|---|---|
| Biofilm formation | 3 | 6 | 0 |
| Carbohydrate metabolic process | 2 | 3 | 1 |
| Cell adhesion | 0 | 2 | 0 |
| Cell cycle | 17 | 29 | 8 |
| Cell development | 1 | 1 | 0 |
| Cellular homeostasis | 3 | 5 | 2 |
| Cellular protein modification process | 15 | 38 | 16 |
| Cellular respiration | 1 | 1 | 0 |
| Cytokinesis | 0 | 2 | 0 |
| Cytoskeleton organization | 5 | 4 | 4 |
| DNA metabolic process | 9 | 11 | 5 |
| Filamentous growth | 20 | 40 | 15 |
| Generation of precursor metabolites and energy | 1 | 2 | 1 |
| Growth of unicellular organism as a thread of attached cells | 0 | 2 | 0 |
| Hyphal growth | 0 | 2 | 0 |
| Interspecies interaction between organisms | 6 | 12 | 5 |
| Lipid metabolic process | 4 | 14 | 1 |
| Nucleus organization | 0 | 1 | 0 |
| Organelle organization | 62 | 89 | 52 |
| Pathogenesis | 7 | 9 | 5 |
| Protein catabolic process | 1 | 4 | 3 |
| Pseudohyphal growth | 0 | 1 | 0 |
| Regulation of biological process | 87 | 180 | 94 |
| Response to chemical | 22 | 63 | 26 |
| Response to drug | 4 | 20 | 4 |
| Response to stress | 30 | 66 | 33 |
| Ribosome biogenesis | 6 | 6 | 2 |
| RNA metabolic process | 50 | 43 | 27 |
| Signal transduction | 5 | 1 | 2 |
| Translation | 5 | 8 | 6 |
| Transport | 16 | 46 | 27 |
| Vesicle-mediated transport | 5 | 8 | 3 |
| Vitamin metabolic process | 2 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asfare, S.; Eldabagh, R.; Siddiqui, K.; Patel, B.; Kaba, D.; Mullane, J.; Siddiqui, U.; Arnone, J.T. Systematic Analysis of Functionally Related Gene Clusters in the Opportunistic Pathogen, Candida albicans. Microorganisms 2021, 9, 276. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020276
Asfare S, Eldabagh R, Siddiqui K, Patel B, Kaba D, Mullane J, Siddiqui U, Arnone JT. Systematic Analysis of Functionally Related Gene Clusters in the Opportunistic Pathogen, Candida albicans. Microorganisms. 2021; 9(2):276. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020276
Chicago/Turabian StyleAsfare, Sarah, Reem Eldabagh, Khizar Siddiqui, Bharvi Patel, Diellza Kaba, Julie Mullane, Umar Siddiqui, and James T. Arnone. 2021. "Systematic Analysis of Functionally Related Gene Clusters in the Opportunistic Pathogen, Candida albicans" Microorganisms 9, no. 2: 276. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020276