
The Effects of Genetic Relatedness on the Preterm Infant Gut Microbiota


 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Enrollment and Sample Collection
2.2. Sample DNA Extraction and Sequencing
2.3. Bioinformatics and Statistics
3. Results
3.1. Clinical Characteristics of Samples
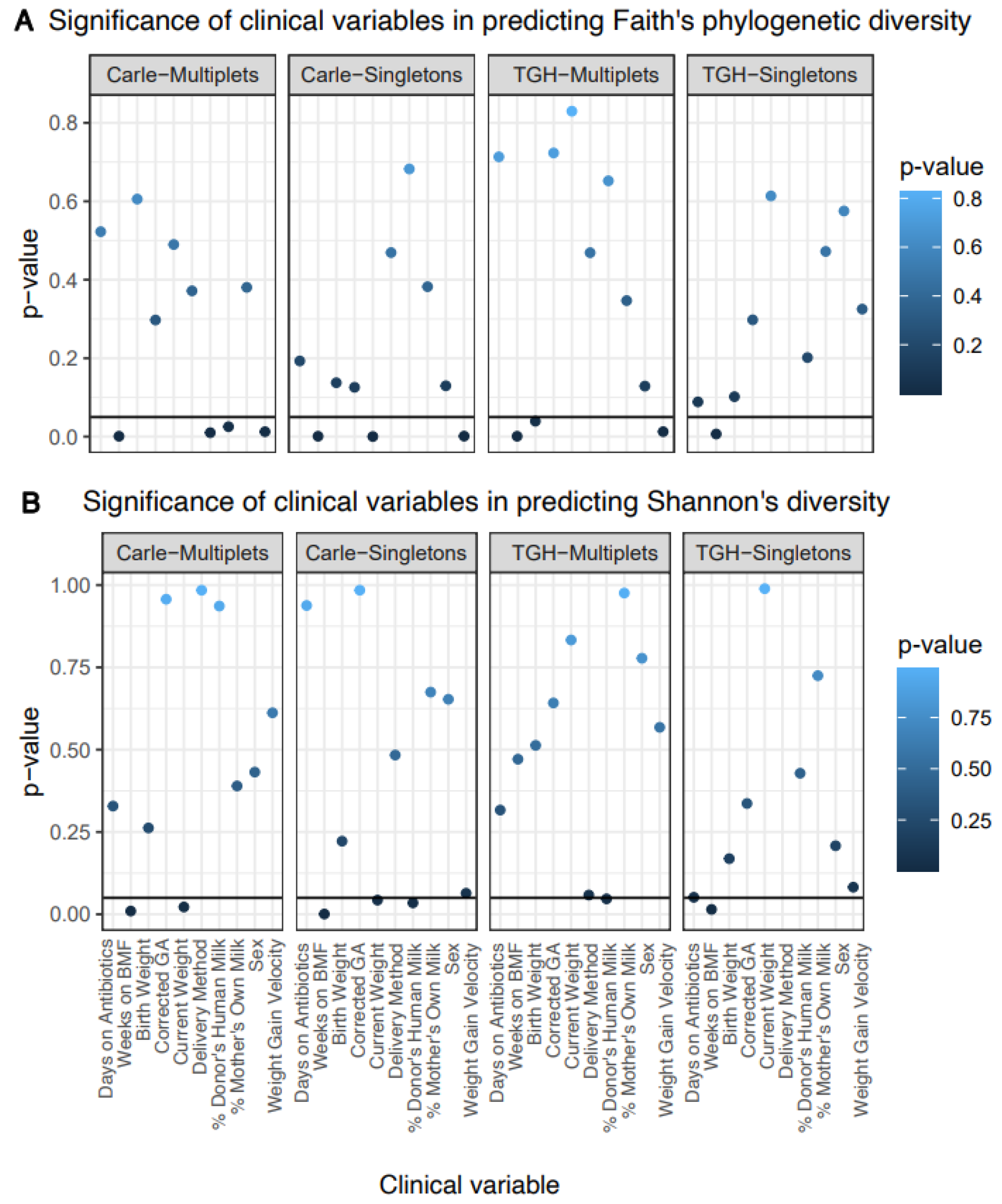
3.2. Gut Microbial Alpha Diversity
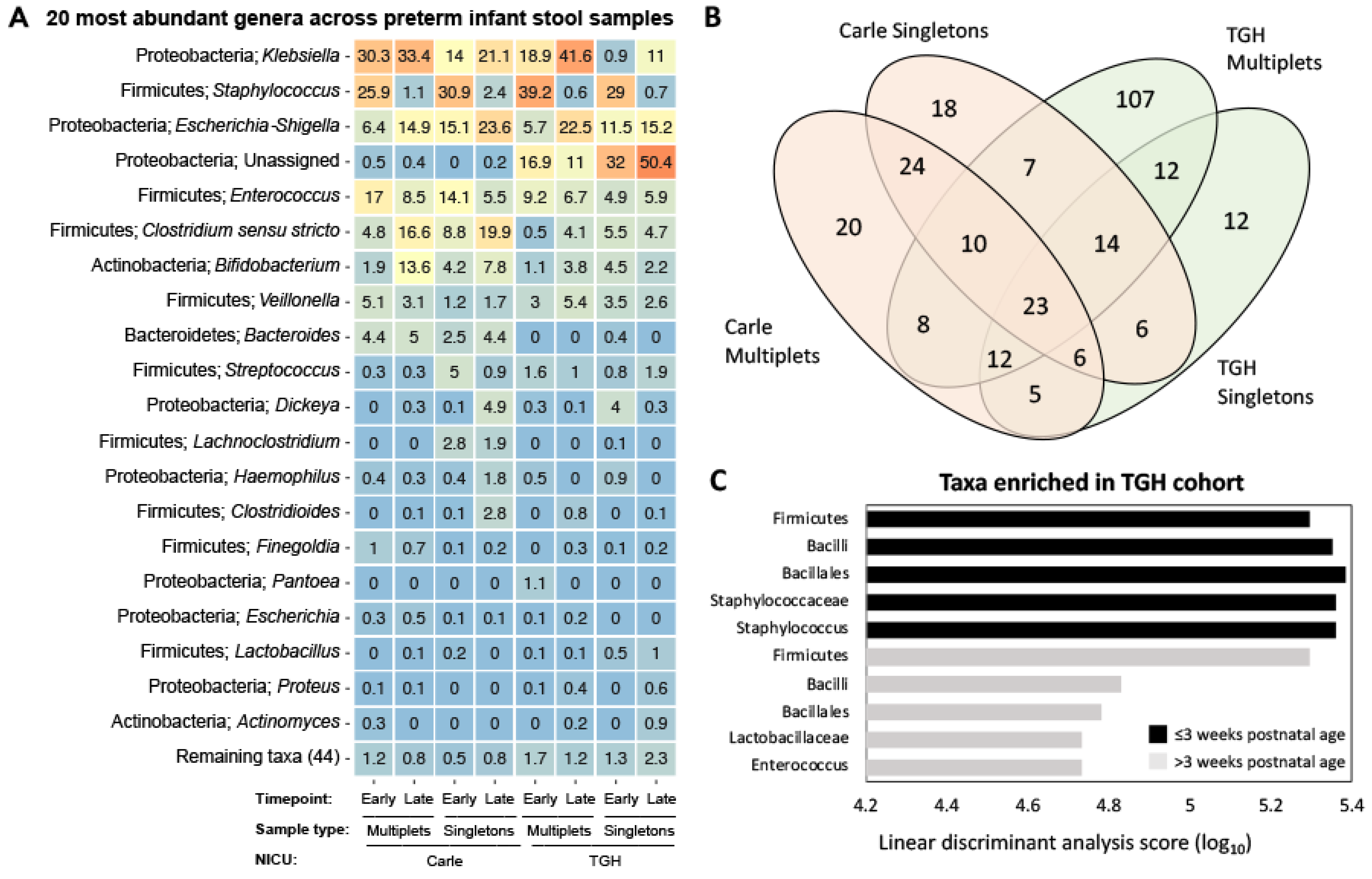
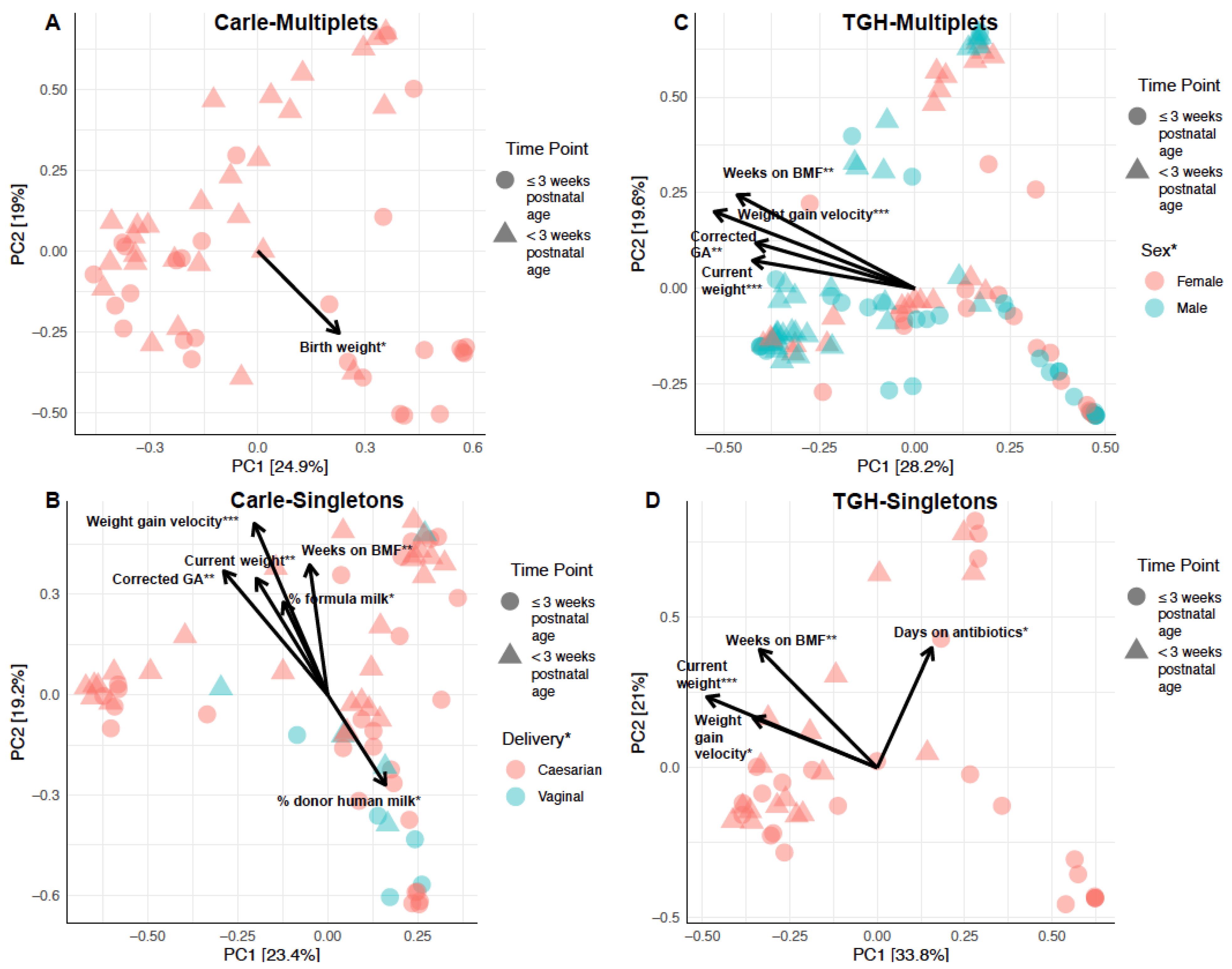
3.3. Gut Microbial Composition and Beta Diversity
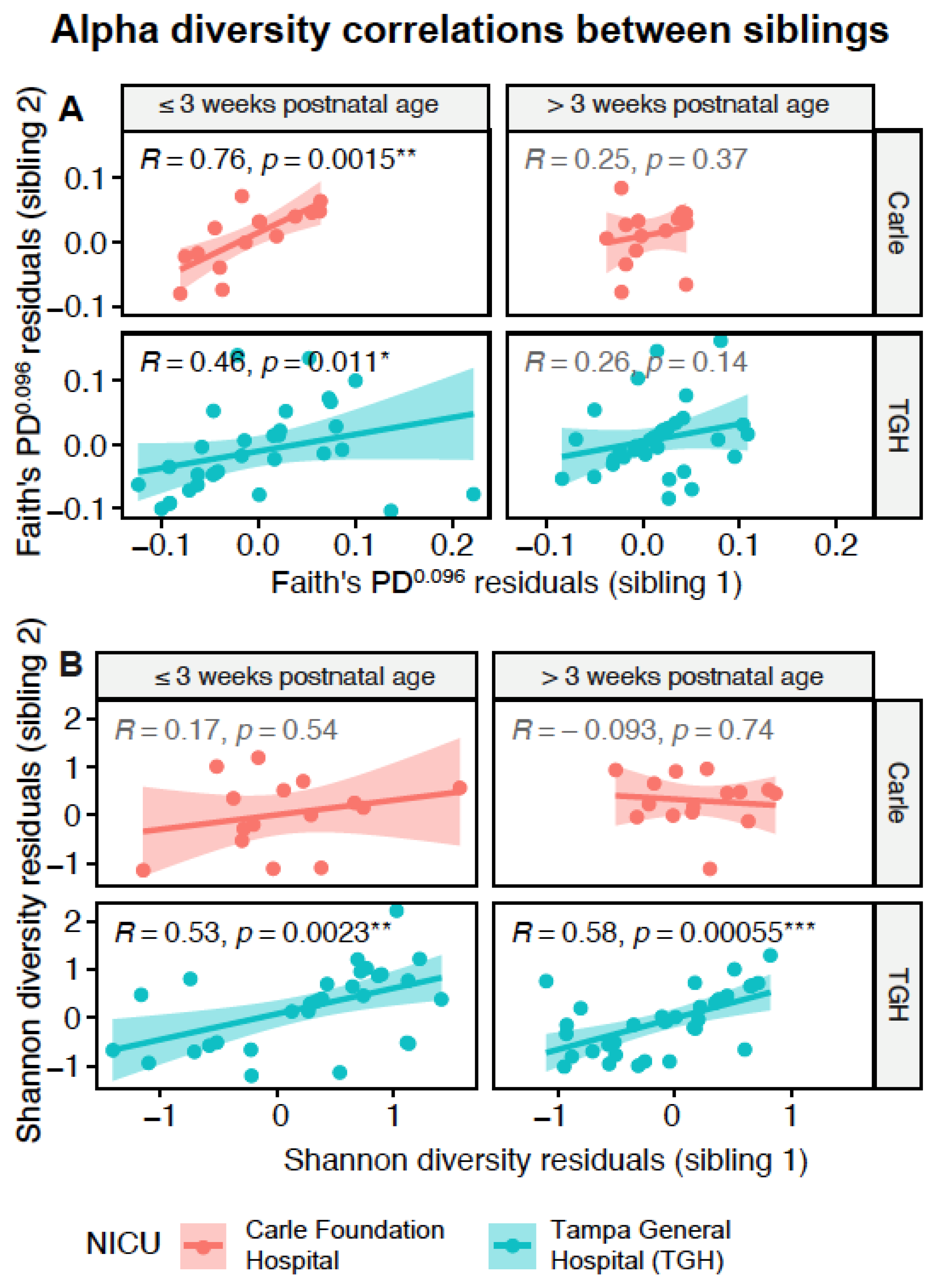
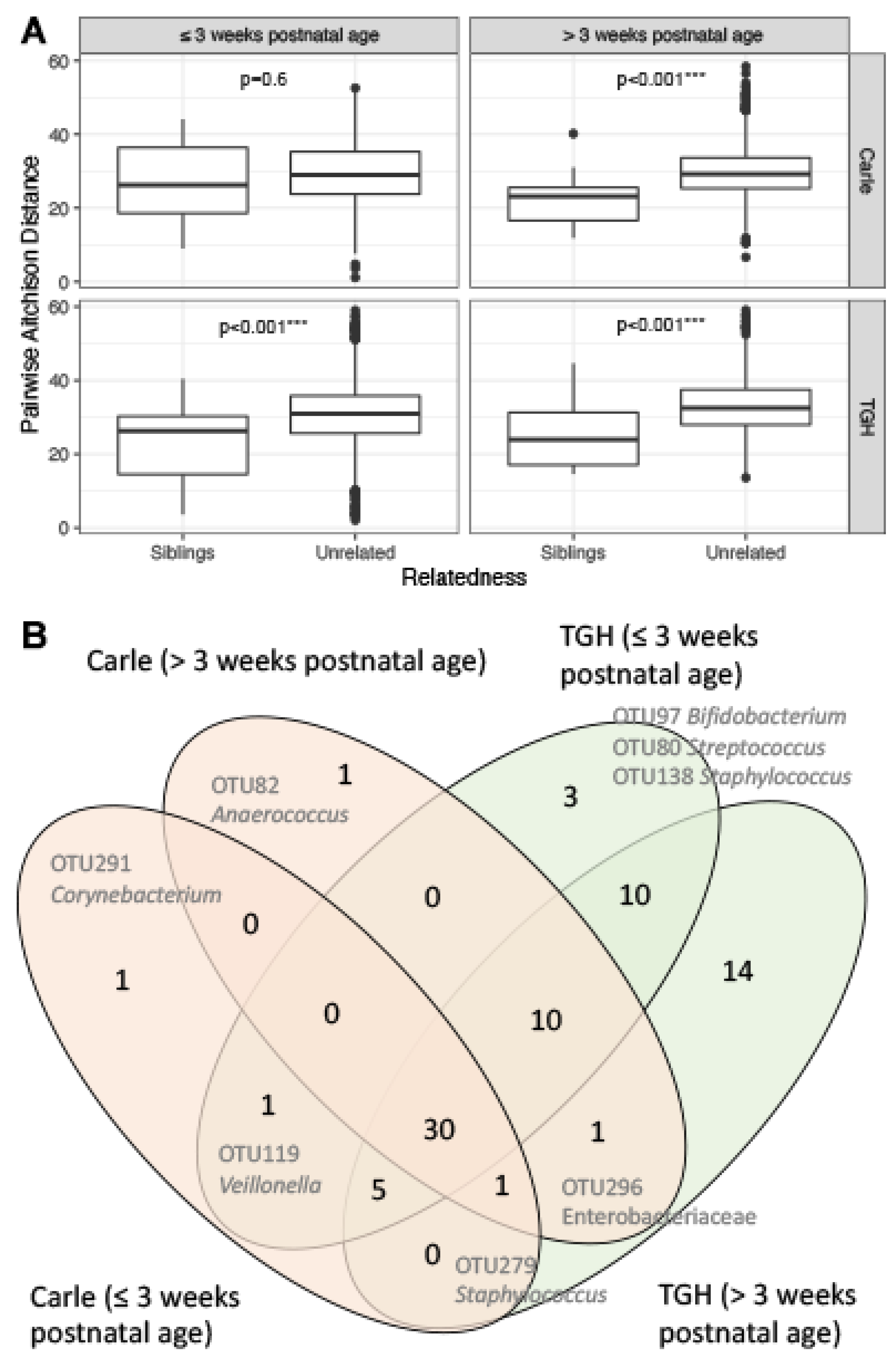
3.4. Gut Microbiota Similarities between Siblings
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hooper, L.V.; Wong, M.H.; Thelin, A.; Hansson, L.; Falk, P.G.; Gordon, J.I. Molecular analysis of commensal host-microbial relationships in the intestine. Science 2001, 291, 881–884. [Google Scholar] [CrossRef] [Green Version]
- Rautava, S.; Luoto, R.; Salminen, S.; Isolauri, E. Microbial contact during pregnancy, intestinal colonization and human disease. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 565–576. [Google Scholar] [CrossRef]
- Arboleya, S.; Martinez-Camblor, P.; Solis, G.; Suarez, M.; Fernandez, N.; de Los Reyes-Gavilan, C.G.; Gueimonde, M. Intestinal microbiota and weight-gain in preterm neonates. Front. Microbiol. 2017, 8, 183. [Google Scholar] [CrossRef]
- Younge, N.E.; Newgard, C.B.; Cotten, C.M.; Goldberg, R.N.; Muehlbauer, M.J.; Bain, J.R.; Stevens, R.D.; O’Connell, T.M.; Rawls, J.F.; Seed, P.C.; et al. Disrupted maturation of the microbiota and metabolome among extremely preterm infants with postnatal growth failure. Sci. Rep. 2019, 9, 8167. [Google Scholar] [CrossRef]
- Matamoros, S.; Gras-Leguen, C.; Le Vacon, F.; Potel, G.; de La Cochetiere, M.F. Development of intestinal microbiota in infants and its impact on health. Trends Microbiol. 2013, 21, 167–173. [Google Scholar] [CrossRef]
- Groer, M.W.; Luciano, A.A.; Dishaw, L.J.; Ashmeade, T.L.; Miller, E.; Gilbert, J.A. Development of the preterm infant gut microbiome: A research priority. Microbiome 2014, 2, 38. [Google Scholar] [CrossRef] [Green Version]
- Schwiertz, A.; Gruhl, B.; Lobnitz, M.; Michel, P.; Radke, M.; Blaut, M. Development of the intestinal bacterial composition in hospitalized preterm infants in comparison with breast-fed, full-term infants. Pediatr. Res. 2003, 54, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Morowitz, M.J.; Denef, V.J.; Costello, E.K.; Thomas, B.C.; Poroyko, V.; Relman, D.A.; Banfield, J.F. Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc. Natl. Acad. Sci. USA 2011, 108, 1128–1133. [Google Scholar] [CrossRef] [Green Version]
- Arboleya, S.; Ang, L.; Margolles, A.; Yiyuan, L.; Dongya, Z.; Liang, X.; Solis, G.; Fernandez, N.; de Los Reyes-Gavilan, C.G.; Gueimonde, M. Deep 16S rRNA metagenomics and quantitative PCR analyses of the premature infant fecal microbiota. Anaerobe 2012, 18, 378–380. [Google Scholar] [CrossRef] [Green Version]
- Arboleya, S.; Binetti, A.; Salazar, N.; Fernandez, N.; Solis, G.; Hernandez-Barranco, A.; Margolles, A.; de Los Reyes-Gavilan, C.G.; Gueimonde, M. Establishment and development of intestinal microbiota in preterm neonates. FEMS Microbiol. Ecol. 2012, 79, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.V.; Scholz, M.; Zolfo, M.; Taft, D.H.; Schibler, K.R.; Tett, A.; Segata, N.; Morrow, A.L. Metagenomic sequencing with strain-level resolution implicates uropathogenic E. coli in necrotizing enterocolitis and mortality in preterm infants. Cell Rep. 2016, 14, 2912–2924. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.J.; Marrs, E.C.; Nelson, A.; Lanyon, C.; Perry, J.D.; Embleton, N.D.; Cummings, S.P.; Berrington, J.E. Development of the preterm gut microbiome in twins at risk of necrotising enterocolitis and sepsis. PLoS ONE 2013, 8, e73465. [Google Scholar] [CrossRef]
- Bertelli, C.; Pillonel, T.; Torregrossa, A.; Prodhom, G.; Fischer, C.J.; Greub, G.; Giannoni, E. Bifidobacterium longum bacteremia in preterm infants receiving probiotics. Clin. Infect. Dis. 2015, 60, 924–927. [Google Scholar] [CrossRef] [Green Version]
- Mancano, M.A. Bacteremia in preterm infants receiving probiotics; throbbing headache associated with Enoxaparin use; DRESS reaction following isoniazid treatment; SIADH associated with Glimepiride; mania associated with Bortezomib administration. Hosp. Pharm. 2015, 50, 441–445. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.C.; Athalye-Jape, G.K.; Deshpande, G.C.; Simmer, K.N.; Patole, S.K. Probiotic supplementation and late-onset sepsis in preterm infants: A meta-analysis. Pediatrics 2016, 137, e20153684. [Google Scholar] [CrossRef] [Green Version]
- Staude, B.; Oehmke, F.; Lauer, T.; Behnke, J.; Gopel, W.; Schloter, M.; Schulz, H.; Krauss-Etschmann, S.; Ehrhardt, H. The microbiome and preterm birth: A change in paradigm with profound implications for pathophysiologic concepts and novel therapeutic strategies. Biomed. Res. Int. 2018, 2018, 7218187. [Google Scholar] [CrossRef]
- Gupta, R.W.; Tran, L.; Norori, J.; Ferris, M.J.; Eren, A.M.; Taylor, C.M.; Dowd, S.E.; Penn, D. Histamine-2 receptor blockers alter the fecal microbiota in premature infants. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 397–400. [Google Scholar] [CrossRef]
- Keski-Nisula, L.; Kyynarainen, H.R.; Karkkainen, U.; Karhukorpi, J.; Heinonen, S.; Pekkanen, J. Maternal intrapartum antibiotics and decreased vertical transmission of Lactobacillus to neonates during birth. Acta Paediatr. 2013, 102, 480–485. [Google Scholar] [CrossRef]
- Arboleya, S.; Sanchez, B.; Milani, C.; Duranti, S.; Solis, G.; Fernandez, N.; de los Reyes-Gavilan, C.G.; Ventura, M.; Margolles, A.; Gueimonde, M. Intestinal microbiota development in preterm neonates and effect of perinatal antibiotics. J. Pediatr. 2015, 166, 538–544. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, C.; Morrow, A.L.; Lagomarcino, A.J.; Altaye, M.; Taft, D.H.; Yu, Z.; Newburg, D.S.; Ward, D.V.; Schibler, K.R. Early empiric antibiotic use in preterm infants is associated with lower bacterial diversity and higher relative abundance of Enterobacter. J. Pediatr. 2014, 165, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Zhang, Z.; Morales, M.; Wang, Y.; Khafipour, E.; Friel, J. Feeding practice influences gut microbiome composition in very low birth weight preterm infants and the association with oxidative stress: A prospective cohort study. Free Radic. Biol. Med. 2019, 142, 146–154. [Google Scholar] [CrossRef]
- Yee, A.L.; Miller, E.; Dishaw, L.J.; Gordon, J.M.; Ji, M.; Dutra, S.; Ho, T.T.B.; Gilbert, J.A.; Groer, M. Longitudinal microbiome composition and stability correlate with increased weight and length of very-low-birth-weight infants. MSystems 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Zanella, A.; Silveira, R.C.; Roesch, L.F.W.; Corso, A.L.; Dobbler, P.T.; Mai, V.; Procianoy, R.S. Influence of own mother’s milk and different proportions of formula on intestinal microbiota of very preterm newborns. PLoS ONE 2019, 14, e0217296. [Google Scholar] [CrossRef]
- Brooks, B.; Firek, B.A.; Miller, C.S.; Sharon, I.; Thomas, B.C.; Baker, R.; Morowitz, M.J.; Banfield, J.F. Microbes in the neonatal intensive care unit resemble those found in the gut of premature infants. Microbiome 2014, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Dey, S.K.; Fisher, S.J. Preterm labor: One syndrome, many causes. Science 2014, 345, 760–765. [Google Scholar] [CrossRef] [Green Version]
- Nanthakumar, N.; Meng, D.; Goldstein, A.M.; Zhu, W.; Lu, L.; Uauy, R.; Llanos, A.; Claud, E.C.; Walker, W.A. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: An immature innate immune response. PLoS ONE 2011, 6, e17776. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Sung, J.; Lee, J.; Ko, G. Comparison of the gut microbiotas of healthy adult twins living in South Korea and the United States. Appl. Environ. Microbiol. 2011, 77, 7433–7437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tims, S.; Derom, C.; Jonkers, D.M.; Vlietinck, R.; Saris, W.H.; Kleerebezem, M.; de Vos, W.M.; Zoetendal, E.G. Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J. 2013, 7, 707–717. [Google Scholar] [CrossRef] [Green Version]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Costello, E.K.; Carlisle, E.M.; Bik, E.M.; Morowitz, M.J.; Relman, D.A. Microbiome assembly across multiple body sites in low-birthweight infants. mBio 2013, 4, e00782-13. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Bauer, L.L.; Chen, X.; Wang, M.; Kuhlenschmidt, T.B.; Kuhlenschmidt, M.S.; Fahey, G.C., Jr.; Donovan, S.M. Microbial composition and in vitro fermentation patterns of human milk oligosaccharides and prebiotics differ between formula-fed and sow-reared piglets. J. Nutr. 2012, 142, 681–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. CD-HIT: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Janssen, S.; McDonald, D.; Gonzalez, A.; Navas-Molina, J.A.; Jiang, L.; Xu, Z.Z.; Winker, K.; Kado, D.M.; Orwoll, E.; Manary, M.; et al. Phylogenetic placement of exact amplicon sequences improves associations with clinical information. MSystems 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Beck, J.; Brister, J.R.; Bolton, E.E.; Canese, K.; Comeau, D.C.; Funk, K.; Ketter, A.; Kim, S.; Kimchi, A.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2020, 48, D9–D16. [Google Scholar] [CrossRef] [Green Version]
- Drancourt, M.; Bollet, C.; Carlioz, A.; Martelin, R.; Gayral, J.P.; Raoult, D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J. Clin. Microbiol. 2000, 38, 3623–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Techn. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 3rd ed.; SAGE Publications, Inc.: Thousand Oaks, CA, USA, 2018; p. 608. [Google Scholar]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 48. [Google Scholar] [CrossRef]
- Akaike, H. Information theory and an extension of the maximum likelihood principle. In Selected Papers of Hirotugu Akaike; Parzen, E., Tanabe, K., Kitagawa, G., Eds.; Springer: New York, NY, USA, 1998; pp. 199–213. [Google Scholar] [CrossRef]
- Andersen, K.S.; Kirkegaard, R.H.; Karst, S.M.; Albertsen, M. Ampvis2: An R package to analyse and visualise 16S rRNA amplicon data. bioRxiv 2018, 299537. [Google Scholar] [CrossRef] [Green Version]
- Legendre, P.; Gallagher, E.D. Ecologically meaningful transformations for ordination of species data. Oecologia 2001, 129, 271–280. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Aitchison, J.; Barceló-Vidal, C.; Martín-Fernández, J.A.; Pawlowsky-Glahn, V. Logratio analysis and compositional distance. Math. Geol. 2000, 32, 271–275. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Good, I.J. The population frequencies of species and the estimation of population parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Granger, C.L.; Embleton, N.D.; Palmer, J.M.; Lamb, C.A.; Berrington, J.E.; Stewart, C.J. Maternal breastmilk, infant gut microbiome and the impact on preterm infant health. Acta Paediatr. 2020, 110, 2. [Google Scholar] [CrossRef] [PubMed]
- Hammond, P.J.; Flett, M.E.; De La Hunt, M. Fulminant necrotising enterocolitis immediately following change to low birth weight formula feeds. Eur. J. Pediatr. Surg. 2008, 18, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Mimouni, F.B.; Nathan, N.; Ziegler, E.E.; Lubetzky, R.; Mandel, D. The use of multinutrient human milk fortifiers in preterm infants: A systematic review of unanswered questions. Clin. Perinatol. 2017, 44, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Speer, C.P. Late-onset neonatal sepsis: Recent developments. Arch. Dis. Child. Fetal. Neonatal. Ed. 2015, 100, F257–F263. [Google Scholar] [CrossRef] [Green Version]
- Hufnagel, M.; Liese, C.; Loescher, C.; Kunze, M.; Proempeler, H.; Berner, R.; Krueger, M. Enterococcal colonization of infants in a neonatal intensive care unit: Associated predictors, risk factors and seasonal patterns. BMC Infect. Dis. 2007, 7, 107. [Google Scholar] [CrossRef] [Green Version]
- Shane, A.L.; Hansen, N.I.; Stoll, B.J.; Bell, E.F.; Sanchez, P.J.; Shankaran, S.; Laptook, A.R.; Das, A.; Walsh, M.C.; Hale, E.C.; et al. Methicillin-resistant and susceptible Staphylococcus aureus bacteremia and meningitis in preterm infants. Pediatrics 2012, 129, e914–e922. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.B.; Highlander, S.K.; Anderson, E.L.; Li, W.; Dayrit, M.; Klitgord, N.; Fabani, M.M.; Seguritan, V.; Green, J.; Pride, D.T.; et al. Library preparation methodology can influence genomic and functional predictions in human microbiome research. Proc. Natl. Acad. Sci. USA 2015, 112, 14024–14029. [Google Scholar] [CrossRef] [Green Version]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic determinants of the gut microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, K.M.; Mannino, F.L.; Gonzalez, A.; Chase, J.H.; Caporaso, J.G.; Knight, R.; Kelley, S.T. Bacterial diversity in two Neonatal Intensive Care Units (NICUs). PLoS ONE 2013, 8, e54703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, H.; Hakim, J.A.; Crossman, D.K.; Lefkowitz, E.J.; Morrow, C.D. Sharing of gut microbial strains between selected individual sets of twins cohabitating for decades. PLoS ONE 2019, 14, e0226111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youseff, N.; Reinhart, K.; Sakr, Y. The pros and cons of multicentre studies. Neth. J. Crit Care 2008, 12, 120–122. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carle Hospital n = 118 (44.7) | Tampa General Hospital n = 146 (55.3) | ||||||
|---|---|---|---|---|---|---|---|
| Cohort Type, n, (%) | Multiplet n = 54 (45.8) | Singleton n = 64 (54.2) | p1 | Multiplet n = 105 (71.9) | Singleton n = 41 (28.1) | p2 | p3 |
| Gestational age at delivery, weeks | 28.8 (2.3) | 28.9 (2.2) | 0.89 | 29.4 (2.2) | 28.5 (2.7) | 0.001 | 0.12 |
| Birth weight, grams | 1246.9 (443.1) | 1238.7 (399.2) | 0.93 | 1349.4 (390.5) | 1118.7 (199.3) | 0.0004 | 0.24 |
| Sex Female, n, (%) Male, n, (%) | 30 (55.6) 24 (44.4) | 42 (65.6) 22 (34.4) | 0.35 | 42 (40.0) 63 (60.0) | 15 (36.6) 26 (63.4) | 0.84 | 0.0006 |
| Type of delivery Vaginal delivery, n, (%) Cesarean delivery, n, (%) | 4 (7.4) 50 (92.6) | 10 (15.6) 54 (84.4) | 0.27 | 4 (3.8) 101 (96.2) | - 41 (100.0) | 0.48 | 0.007 |
| Number of placentas Monochorionic twins, n, (%) Dichorionic twins, n, (%) | 12 (22.2) 42 (77.8) | - - | - | 24 (22.9) 81 (77.1) | - - | - | 0.9 |
| Carle Hospital n = 118 (44.7%) | Tampa General Hospital n = 146 (55.3%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cohort Type, n, (%) | Multiplet n = 54 (45.8%) | Singleton n = 64 (54.2%) | Multiplet n = 105 (71.9%) | Singleton n = 41 (28.1%) | p * | ||||
| Early | Late | Early | Late | Early | Late | Early | Late | ||
| Corrected gestational age, weeks | 30.8 (2.0) | 33.2 (2.2) | 30.6 (2.1) | 33.1 (2.1) | 31.1 (2.1) | 33.9 (1.9) | 30.4 (2.4) | 33.0 (2.5) | 0.31 |
| Postnatal age, days | 13.7 (8.2) | 29.9 (9.0) | 11.8 (6.6) | 29.0 (8.0) | 10.1 (4.9) | 29.2 (7.3) | 13.0 (6.4) | 31.4 (5.0) | 0.62 |
| Current weight, grams | 1388.8 (441.4) | 1853.1 (597.9) | 1369.3 (438.2) | 1866.1 (574.2) | 1358.2 (376.7) | 1877.1 (463.5) | 1194.0 (283.5) | 1655.7 (260.7) | 0.5 |
| Weight gain velocity, g/d | 9.3 (9.7) | 20.9 (9.1) | 6.9 (14.0) | 21.2 (10.0) | −1.4 (12.3) | 17.8 (6.7) | 1.1 (15.5) | 17.1 (4.0) | 0.001 |
| Days on antibiotics, days | 0.5 (1.5) | 0.2 (1.0) | 0.7 (1.5) | 0.0 (0.2) | 2.7 (2.7) | 3.3 (3.7) | 4.2 (3.6) | 4.5 (3.7) | <0.0001 |
| Bovine milk fortifier, weeks | 1.4 (1.0) | 3.3 (1.8) | 1.1 (1.0) | 2.8 (2.1) | 0.9 (0.8) | 3.7 (1.1) | 1.5 (1.0) | 3.9 (0.8) | 0.16 |
| Proportion of mother’s own milk | 0.83 (0.28) | 0.77 (0.32) | 0.63 (0.37) | 0.56 (0.46) | 0.47 (0.42) | 0.53 (0.43) | 0.86 (0.29) | 0.81 (0.36) | 0.15 |
| Proportion of donor’s human milk | 0.16 (0.24) | 0.13 (0.26) | 0.36 (0.38) | 0.20 (0.34) | 0.34 (0.42) | 0.15 (0.30) | 0.11 (0.28) | 0.00 (0.00) | 0.31 |
| Proportion of formula | 0.01 (0.07) | 0.10 (0.16) | 0.01 (0.07) | 0.24 (0.36) | 0.19 (0.37) | 0.32 (0.40) | 0.04 (0.09) | 0.19 (0.36) | 0.005 |
| OTU | Class | Family/Genus |
|---|---|---|
| OTU190 | Actinobacteria | Bifidobacterium |
| OTU221 | Actinobacteria | Bifidobacterium |
| OTU210 | Actinobacteria | Bifidobacterium |
| OTU147 | Actinobacteria | Bifidobacterium |
| OTU52 | Actinobacteria | Bifidobacterium |
| OTU233 | Bacilli | Staphylococcus |
| OTU117 | Bacilli | Staphylococcus |
| OTU176 | Bacilli | Streptococcus |
| OTU6 | Bacteroidia | Bacteroides |
| OTU200 | Clostridia | Anaerococcus |
| OTU178 | Clostridia | Clostridium sensu stricto |
| OTU28 | Clostridia | Clostridium sensu stricto |
| OTU187 | Clostridia | Clostridium sensu stricto |
| OTU45 | Erysipelotrichia | (Clostridium) innocuum group |
| OTU283 | Gammaproteobacteria | Pasteurellaceae |
| OTU284 | Gammaproteobacteria | Enterobacteriaceae |
| OTU195 | Gammaproteobacteria | Enterobacteriaceae |
| OTU141 | Gammaproteobacteria | Enterobacteriaceae |
| OTU162 | Gammaproteobacteria | Enterobacteriaceae |
| OTU222 | Gammaproteobacteria | Enterobacteriaceae |
| OTU173 | Gammaproteobacteria | Enterobacteriaceae |
| OTU58 | Gammaproteobacteria | Citrobacter |
| OTU246 | Gammaproteobacteria | Citrobacter |
| OTU272 | Gammaproteobacteria | Escherichia |
| OTU234 | Gammaproteobacteria | Escherichia |
| OTU105 | Gammaproteobacteria | Escherichia-Shigella |
| OTU148 | Gammaproteobacteria | Escherichia-Shigella |
| OTU24 | Gammaproteobacteria | Escherichia-Shigella |
| OTU10 | Gammaproteobacteria | Klebsiella |
| OTU133 | Negativicutes | Veillonella |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, S.J.; Aguilar-Lopez, M.; Wetzel, C.; Dutra, S.V.O.; Bray, V.; Groer, M.W.; Donovan, S.M.; Ho, T. The Effects of Genetic Relatedness on the Preterm Infant Gut Microbiota. Microorganisms 2021, 9, 278. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020278
Lim SJ, Aguilar-Lopez M, Wetzel C, Dutra SVO, Bray V, Groer MW, Donovan SM, Ho T. The Effects of Genetic Relatedness on the Preterm Infant Gut Microbiota. Microorganisms. 2021; 9(2):278. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020278
Chicago/Turabian StyleLim, Shen Jean, Miriam Aguilar-Lopez, Christine Wetzel, Samia V. O. Dutra, Vanessa Bray, Maureen W. Groer, Sharon M. Donovan, and Thao Ho. 2021. "The Effects of Genetic Relatedness on the Preterm Infant Gut Microbiota" Microorganisms 9, no. 2: 278. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020278