
Bacterial Microbiota of Field-Collected Helicoverpa zea (Lepidoptera: Noctuidae) from Transgenic Bt and Non-Bt Cotton

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Season 1
2.1.1. Insect Collection, Identification and Sample Preparation
2.1.2. Quantification of Cultivable Bacteria
2.1.3. Denaturing Gradient Gel Electrophoresis
2.2. Season 2
2.2.1. Insect Collection
2.2.2. Quantification of Cultivable Bacteria
2.2.3. DNA Extraction
2.2.4. NGS Library Preparation and Sequencing
2.2.5. Bioinformatics
2.2.6. Statistical Analyses
3. Results
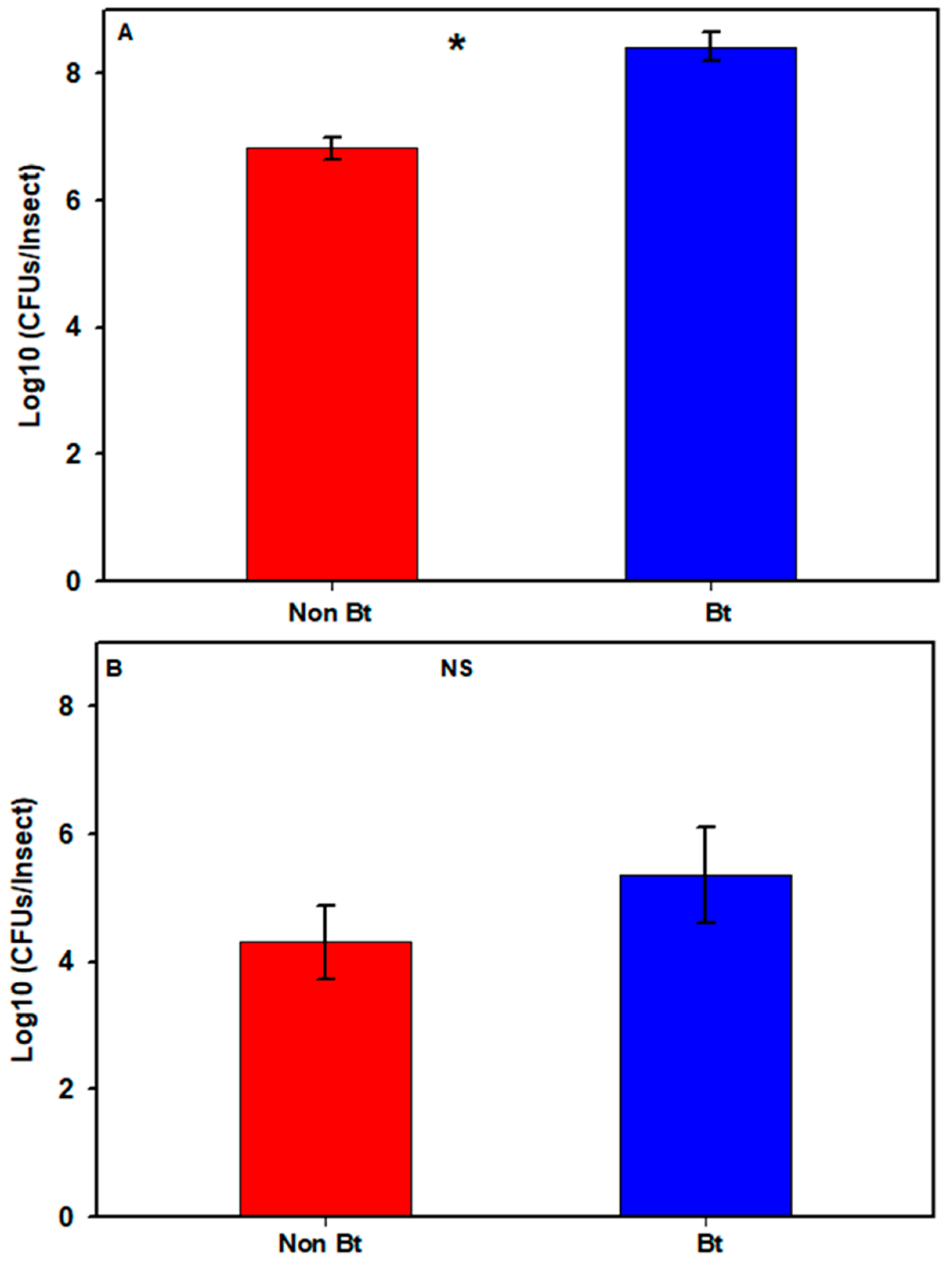
3.1. Cultivable Bacteria: Non-Bt versus Bt Cotton
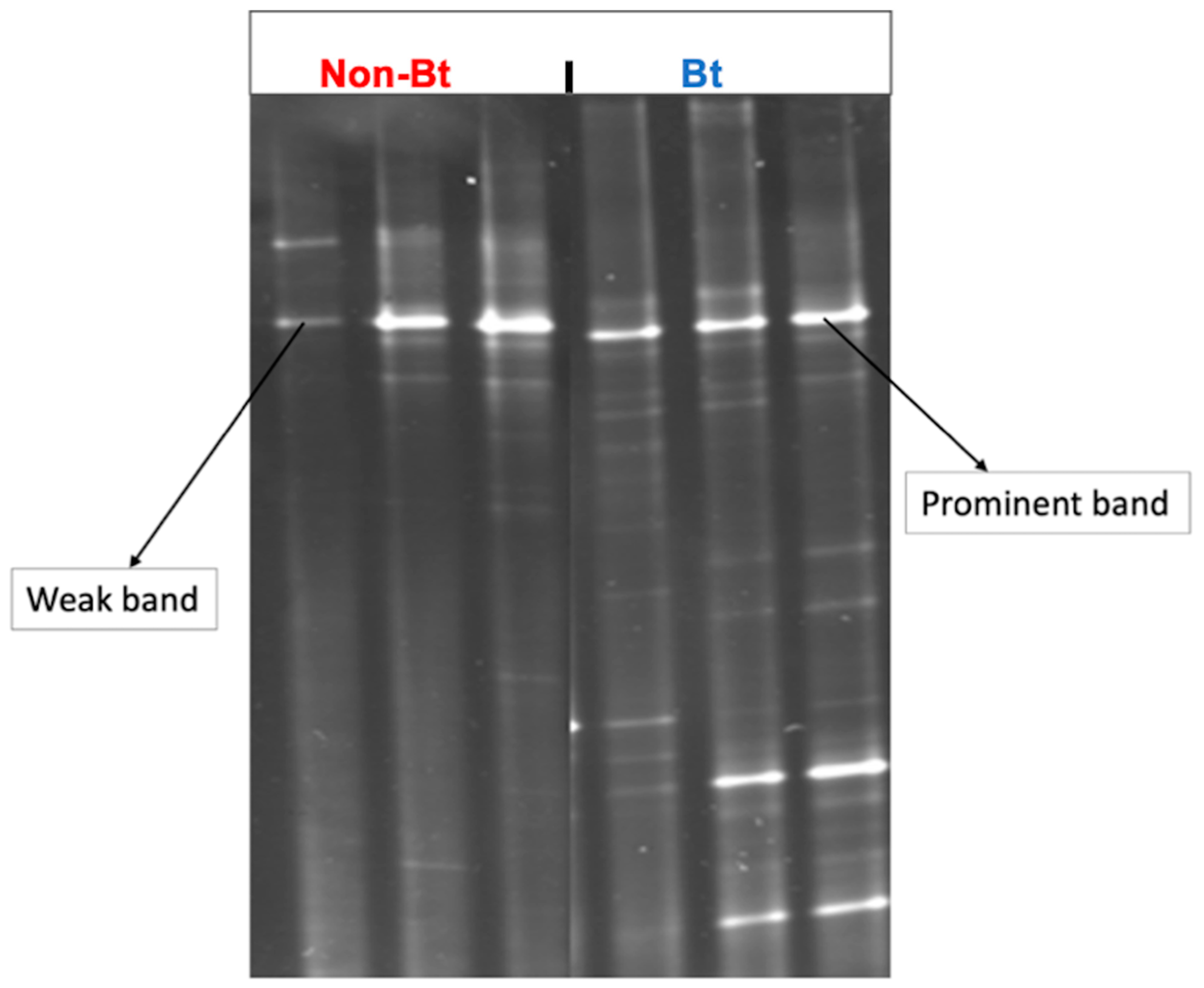
3.2. DGGE Analysis: Non-Bt versus Bt Cotton
3.3. Overall Structure of Bacterial Communities Associated with Bollworms in Season 2
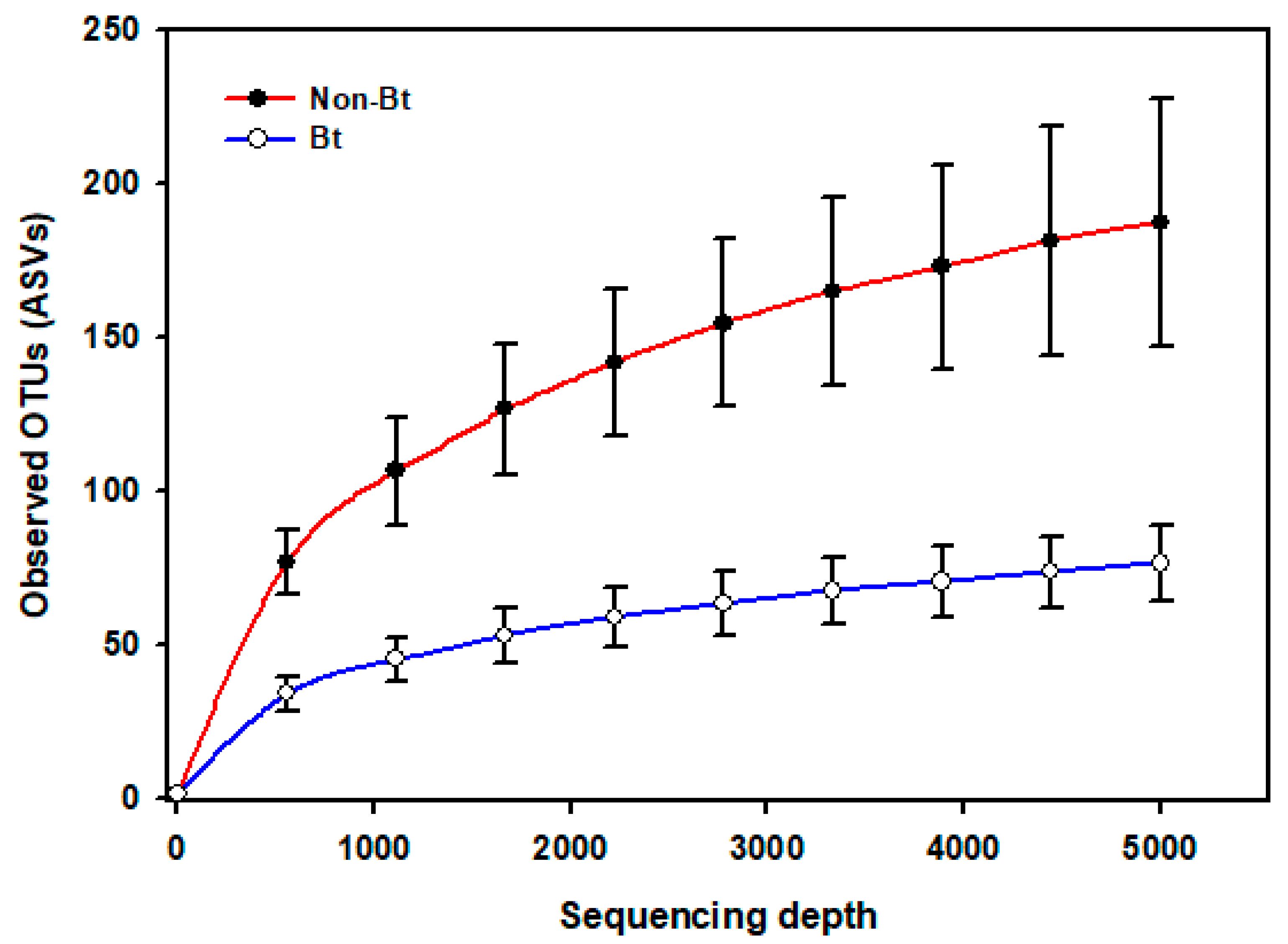
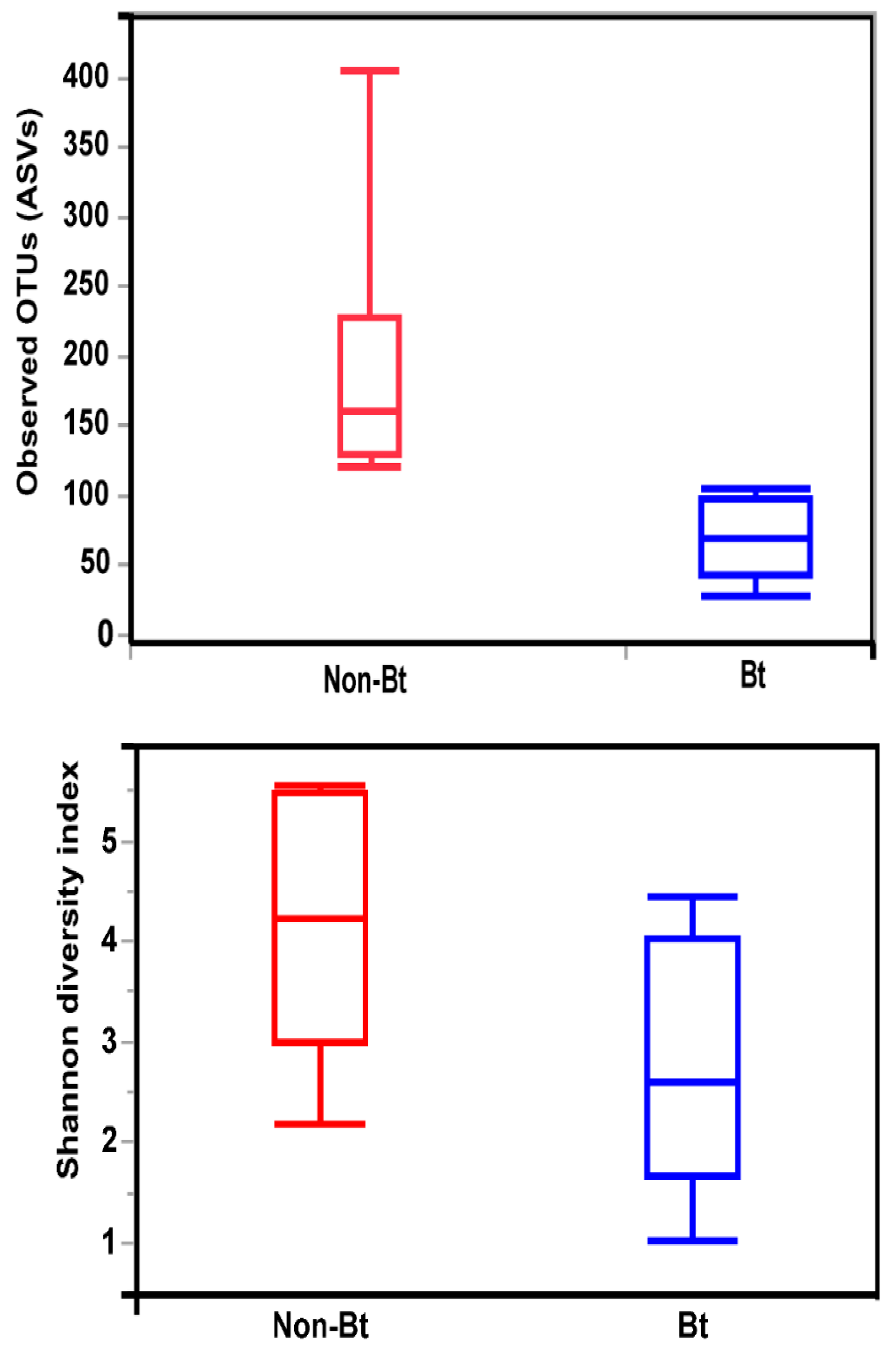
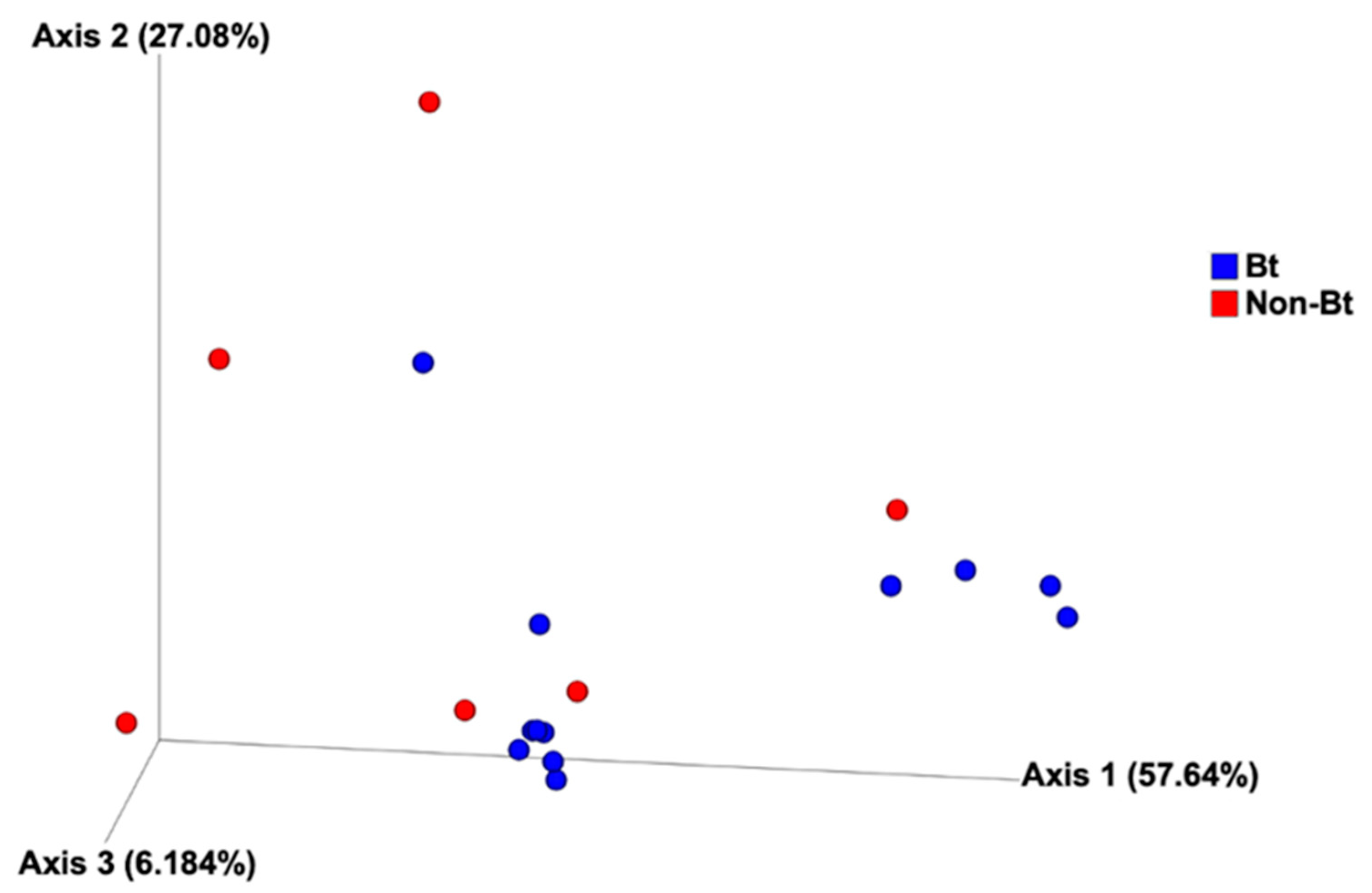
3.4. Alpha and Beta Diversity
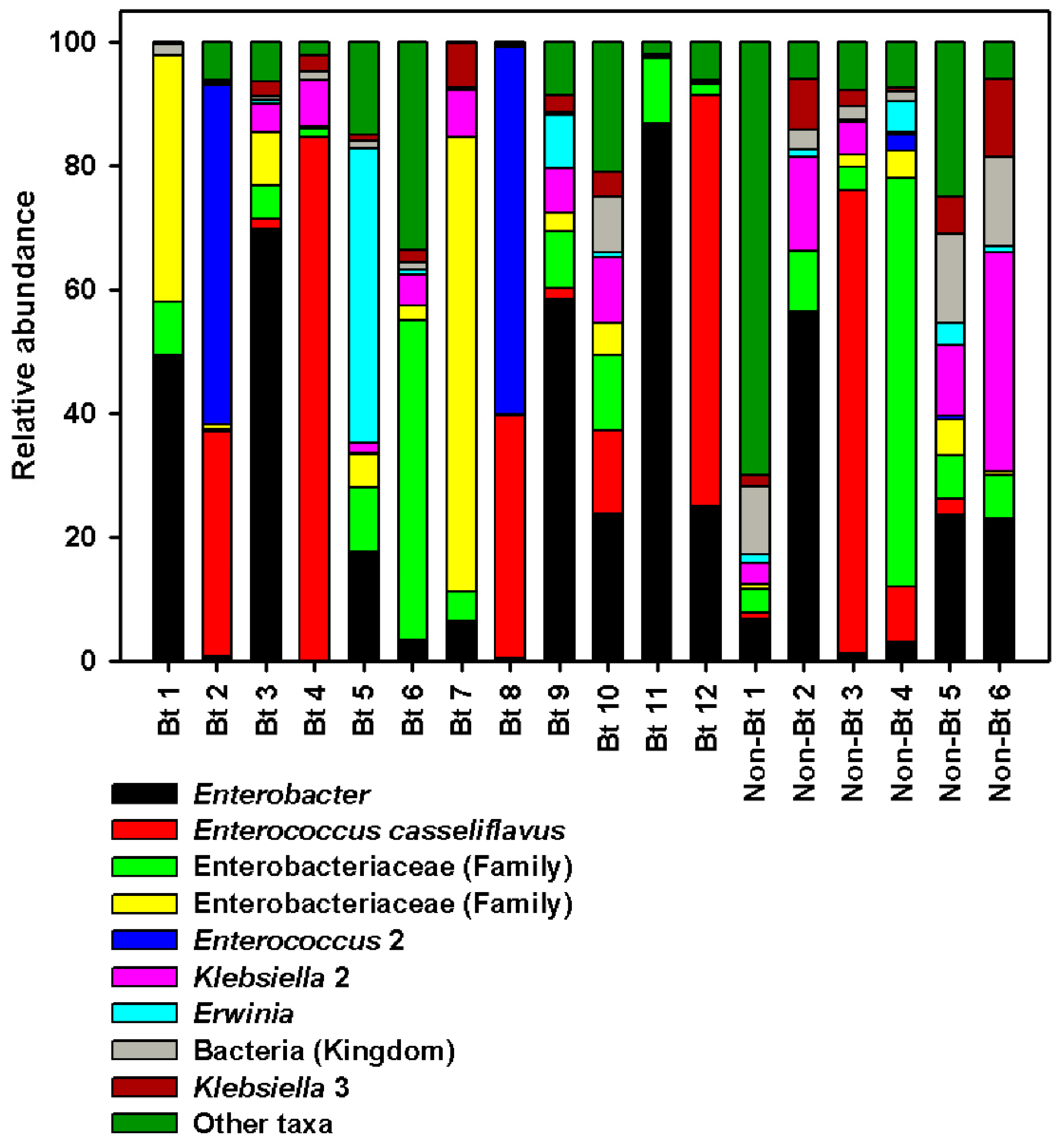
3.5. Microbial Community Composition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Emani, C. (Ed.) Transgenic Cotton for Agronomical Useful Traits; Springer International Publishing: New York, NY, USA, 2016; pp. 201–216. [Google Scholar]
- Reisig, D.D.; Huseth, A.S.; Bacheler, J.S.; Aghaee, M.-A.; Braswell, L.; Burrack, H.J.; Flanders, K.; Greene, J.K.; Herbert, D.A.; Jacobson, A.; et al. Long-term empirical and observational evidence of practical Helicoverpa zea resistance to cotton with pyramided Bt toxins. J. Econ. Entomol. 2018, 111, 1824–1833. [Google Scholar] [CrossRef] [PubMed]
- USDA. U.S. Department of Agriculture—National Agricultural Statistical Service Agricultural Survey (USDA NASS). 2017. Available online: http://quickstats.nass.usda.gov/ (accessed on 11 March 2019).
- Schnepf, E.; Crickmore, N.; Van Rie, J.; Lereclus, D.; Baum, J.; Feitelson, J.; Zeigler, D.R.; Dean, D.H. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol. Mol. Biol. Rev. 1998, 62, 775–806. [Google Scholar] [CrossRef] [Green Version]
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef]
- Trapero, C.; Wilson, I.W.; Stiller, W.N.; Wilson, L.J. Enhancing integrated pest management in GM cotton systems using host plant resistance. Front. Plant Sci. 2016, 7, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaneo, M.G.; Yafuso, C.; Schmidt, C.; Huang, C.-Y.; Rahman, M.; Olson, C.; Ellers-Kirk, C.; Orr, B.J.; Marsh, S.E.; Antilla, L.; et al. Farm-scale evaluation of the impacts of transgenic cotton on biodiversity, pesticide use, and yield. Proc. Natl. Acad. Sci. USA 2006, 103, 7571–7576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.I.; Luttrell, R.G.; Young, S.Y. Susceptibilities of Helicoverpa zea and Heliothis virescens (Lepidoptera: Noctuidae) populations to Cry1Ac insecticidal protein. J. Econ. Entomol. 2006, 99, 164–175. [Google Scholar] [CrossRef]
- Storer, N.P.; Babcock, J.M.; Schlenz, M.; Meade, T.; Thompson, G.D.; Bing, J.W.; Huckaba, R.M. Discovery and characterization of field resistance to Bt maize: Spodoptera frugiperda in Puerto Rico. J. Econom. Entomol. 2010, 103, 1031–1038. [Google Scholar] [CrossRef]
- Tabashnik, B.E.; Carrière, Y. Field-evolved resistance to Bt cotton: Bollworm in the U.S. and pink bollworm in India. Southwest Entomol. 2010, 35, 417–424. [Google Scholar] [CrossRef]
- Jin, L.; Wang, J.; Guan, F.; Zhang, J.; Yu, S.; Liu, S.; Xue, Y.; Li, L.; Wu, S.; Wang, X.; et al. Dominant point mutation in a tetraspanin gene associated with field-evolved resistance of cotton bollworm to transgenic Bt cotton. Proc. Natl. Acad. Sci. USA 2018, 115, 11760–11765. [Google Scholar] [CrossRef] [Green Version]
- Tabashnik, B.E.; Brévault, T.; Carrière, Y. Insect resistance to Bt crops: Lessons from the first billion acres. Nat. Biotechnol. 2013, 31, 510–521. [Google Scholar] [CrossRef]
- Tabashnik, B.E.; Mota-Sanchez, D.; Whalon, M.E.; Hollingworth, R.M.; Carrière, Y. Defining terms for proactive management of resistance to Bt crops and pesticides. J. Econ. Entomol. 2014, 107, 496–507. [Google Scholar] [CrossRef]
- Caccia, S.; Moar, W.J.; Chandrashekhar, J.; Oppert, C.; Anilkumar, K.J.; Jurat-Fuentes, J.L.; Ferré, J. Association of Cry1Ac toxin resistance in Helicoverpa zea (Boddie) with increased alkaline phosphatase levels in the midgut lumen. Appl. Environ. Microbiol. 2012, 78, 5690–5698. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wei, J.; Ni, X.; Zhang, J.; Jurat-Fuentes, J.L.; Fabrick, J.A.; Carrière, Y.; Tabashnik, B.E.; Li, X. Decreased Cry1Ac activation by midgut proteases associated with Cry1Ac resistance in Helicoverpa zea. Pest. Manag. Sci. 2018, 75, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Lawrie, R.D.; Mitchell Iii, R.D.; Deguenon, J.M.; Ponnusamy, L.; Reisig, D.; Pozo-Valdivia, A.D.; Kurtz, R.W.; Roe, R.M. Multiple known mechanisms and a possible role of an enhanced immune system in Bt-resistance in a field population of the bollworm, Helicoverpa zea: Differences in gene expression with RNAseq. Int. J. Mol. Sci. 2020, 21, 6528. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.M.; Moran, N.A.; Hunter, M.S. Variation in resistance to parasitism in aphids is due to symbionts not host genotype. Proc. Natl. Acad. Sci. USA 2005, 102, 12795–12800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopal, R. Beneficial interactions between insects and gut bacteria. Indian J. Microbiol. 2009, 49, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Vilanova, C.; Baixeras, J.; Latorre, A.; Porcar, M. The generalist inside the specialist: Gut bacterial communities of two insect species feeding on toxic plants are dominated by Enterococcus sp. Front. Microbiol. 2016, 7, 1005. [Google Scholar] [CrossRef] [PubMed]
- Deguenon, J.M.; Travanty, N.; Zhu, J.; Carr, A.; Denning, S.; Reiskind, M.H.; Watson, D.W.; Michael Roe, R.; Ponnusamy, L. Exogenous and endogenous microbiomes of wild-caught Phormia regina (Diptera: Calliphoridae) flies from a suburban farm by 16S rRNA gene sequencing. Sci. Rep. 2019, 9, 20365. [Google Scholar] [CrossRef] [PubMed]
- Kodama, R.; Nakasuji, Y. Further studies on the pathogenic mechanism of bacterial diseases in gnotobiotic silkworm larvae. Osaka Inst. Ferment Annu. Rep. 1971, 5, 1–9. [Google Scholar]
- Santo Domingo, J.W.; Kaufman, M.G.; Klug, M.J.; Holben, W.E.; Harris, D.; Tiedje, J.M. Influence of diet on the structure and function of the bacterial hindgut community of crickets. Mol. Ecol. 1998, 7, 761–767. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Hayatsu, M.; Hosokawa, T.; Nagayama, A.; Tago, K.; Fukatsu, T. Symbiont-mediated insecticide resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 8618–8622. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Guo, Z.; Riegler, M.; Xi, Z.; Liang, G.; Xu, Y. Gut symbiont enhances insecticide resistance in a significant pest, the oriental fruit fly Bactrocera dorsalis (Hendel). Microbiome 2017, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Zheng, D.; Zhong, H.; Qin, B.; Gurr, G.M.; Vasseur, L.; Lin, H.; Bai, J.; He, W.; You, M. DNA sequencing reveals the midgut microbiota of diamondback moth, Plutella xylostella (L.) and a possible relationship with insecticide resistance. PLoS ONE 2013, 8, e68852. [Google Scholar] [CrossRef]
- Gadad, H.; Vastrad, A.; Krishnaraj, P. Gut bacteria mediated insecticide resistance in Spodoptera litura (Fab.). J. Exp. Zool. India 2016, 1099–1102. [Google Scholar] [CrossRef]
- Xia, X.; Sun, B.; Gurr, G.M.; Vasseur, L.; Xue, M.; You, M. Gut microbiota mediate insecticide resistance in the diamondback moth, Plutella xylostella (L.). Front. Microbiol. 2018, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Broderick, N.A.; Raffa, K.F.; Handelsman, J. Midgut bacteria required for Bacillus thuringiensis insecticidal activity. Proc. Natl. Acad. Sci. USA 2006, 103, 15196–15199. [Google Scholar] [CrossRef] [Green Version]
- Caccia, S.; Di Lelio, I.; La Storia, A.; Marinelli, A.; Varricchio, P.; Franzetti, E.; Banyuls, N.; Tettamanti, G.; Casartelli, M.; Giordana, B.; et al. Midgut microbiota and host immunocompetence underlie Bacillus thuringiensis killing mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, 9486–9491. [Google Scholar] [CrossRef] [Green Version]
- Raymond, B.; Johnston, P.R.; Wright, D.J.; Ellis, R.J.; Crickmore, N.; Bonsall, M.B. A mid-gut microbiota is not required for the pathogenicity of Bacillus thuringiensis to diamond back moth larvae. Environ. Microbiol. 2009, 11, 2556–2563. [Google Scholar] [CrossRef]
- Frankenhuyzen, K.V.; Liu, Y.; Tonon, A. Interactions between Bacillus thuringiensis subsp. kurstaki HD-1 and midgut bacteria in larvae of gypsy moth and spruce budworm. J. Invertebr. Pathol. 2010, 103, 124–131. [Google Scholar] [CrossRef]
- Wang, J.; Peiffer, M.; Hoover, K.; Rosa, C.; Zeng, R.; Felton, G.W. Helicoverpa zea gut-associated bacteria indirectly induce defenses in tomato by triggering a salivary elicitor (s). N. Phytol. 2017, 214, 1294–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neunzig, H.H. The eggs and early-instar larvae of Heliothis zea and Heliothis virescens (Lepidoptera: Noctuidae). Ann. Entomol. Soc. Am. 1964, 57, 98–102. [Google Scholar] [CrossRef]
- Toennisson, A.; Burrack, H. Commonly Confused Caterpillars: Distinguishing Tobacco Budworms from Corn Earworm. NC State Extension. Available online: https://tobacco.ces.ncsu.edu/2017/04/commonly-confused-caterpillars-distinguishing-tobacco-budworms-from-corn-earworm/ (accessed on 8 August 2018).
- Capinera, J.L. Corn earworm, Helicoverpa zea (Boddie) (Lepidoptera: Noctuidae). IFAS Extension, University of Florida. EENY-145 (IN302). Available online: https://edis.ifas.ufl.edu/pdffiles/IN/IN30200.pdf (accessed on 8 August 2018).
- Naghili, H.; Tajik, H.; Mardani, K.; Rouhani, S.M.R.; Ehsani, A.; Zare, P. Validation of drop plate technique for bacterial enumeration by parametric and nonparametric tests. Vet. Res. Forum 2013, 4, 179–183. [Google Scholar]
- Hoben, H.; Somasegaran, P. Comparison of the pour, spread, and drop plate methods for enumeration of Rhizobium spp. in inoculants made from presterilized peat. Appl. Environ. Microbiol. 1982, 44, 1246–1247. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Ponnusamy, L.; Gonzalez, A.; Van Treuren, W.; Weiss, S.; Parobek, C.M.; Juliano, J.J.; Knight, R.; Roe, R.M.; Apperson, C.S.; Meshnick, S.R. Diversity of Rickettsiales in the microbiome of the lone star tick, Amblyomma americanum. Appl. Environ. Microbiol. 2014, 80, 354–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnusamy, L.; Xu, N.; Stav, G.; Wesson, D.M.; Schal, C.; Apperson, C.S. Diversity of bacterial communities in container habitats of mosquitoes. Microb. Ecol. 2008, 56, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]
- McManus, R.; Ravenscraft, A.; Moore, W. Bacterial associates of a Gregarious riparian beetle with explosive defensive chemistry. Front. Microbiol. 2018, 9, 2361. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Eenviron. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. GigaScience 2013, 2, 2–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Muyzer, G.; Smalla, K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek 1998, 73, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Dhammi, A.; Ponnusamy, L.; Kakuman, M.; Roe, R.M. Global Analysis of the Tobacco Budworm-Cotton Microbiome. In Proceedings of the Beltwide Cotton Conferences, New Orleans, LA, USA, 6–8 January 2014. [Google Scholar]
- Gracy, R.G.; Malathi, V.M.; Jalali, S.K.; Jose, V.L.; Thulasi, A. Variatio’n in larval gut bacteria between insecticide-resistant and -susceptible populations of Helicoverpa armigera (Hübner) (Lepidoptera: Noctuidae). Phytoparasitica 2016, 44, 477–490. [Google Scholar] [CrossRef]
- Paniagua Voirol, L.R.; Frago, E.; Kaltenpoth, M.; Hilker, M.; Fatouros, N.E. Bacterial symbionts in Lepidoptera: Their diversity, transmission, and impact on the host. Front. Microbiol. 2018, 9, 556. [Google Scholar] [CrossRef]
- Wang, A.; Yao, Z.; Zheng, W.; Zhang, H. Bacterial communities in the gut and reproductive organs of Bactrocera minax (Diptera: Tephritidae) based on 454 pyrosequencing. PLoS ONE 2014, 9, e106988. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Gurr, G.M.; Vasseur, L.; Zheng, D.; Zhong, H.; Qin, B.; Lin, J.; Wang, Y.; Song, F.; Li, Y.; et al. Metagenomic sequencing of diamondback moth gut microbiome unveils key holobiont adaptations for herbivory. Front. Microbiol. 2017, 8, 663. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, N.; Martens, R.; Tebbe, C.C. Origin and diversity of metabolically active gut bacteria from laboratory-bred larvae of Manduca sexta (Sphingidae, Lepidoptera, Insecta). Appl. Environ. Microbiol. 2008, 74, 7189–7196. [Google Scholar] [CrossRef] [Green Version]
- Thakur, A.; Dhammi, P.; Saini, H.S.; Kaur, S. Pathogenicity of bacteria isolated from gut of Spodoptera litura (Lepidoptera: Noctuidae) and fitness costs of insect associated with consumption of bacteria. J. Invertebr. Pathol. 2015, 127, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Broderick, N.A.; Raffa, K.F.; Goodman, R.M.; Handelsman, J. Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl. Environ. Microbiol. 2004, 70, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takatsuka, J.; Kunimi, Y. Intestinal bacteria affect growth of Bacillus thuringiensis in larvae of the oriental tea tortrix, Homona magnanima diakonoff (Lepidoptera: Tortricidae). J. Invertebr. Pathol. 2000, 76, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Shu, C.; Crickmore, N.; Liu, C.; Xiang, W.; Song, F.; Zhang, J. Cultivable gut bacteria of scarabs (Coleoptera: Scarabaeidae) inhibit Bacillus thuringiensis multiplication. Environ. Entomol. 2014, 43, 612–616. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deguenon, J.M.; Dhammi, A.; Ponnusamy, L.; Travanty, N.V.; Cave, G.; Lawrie, R.; Mott, D.; Reisig, D.; Kurtz, R.; Roe, R.M. Bacterial Microbiota of Field-Collected Helicoverpa zea (Lepidoptera: Noctuidae) from Transgenic Bt and Non-Bt Cotton. Microorganisms 2021, 9, 878. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9040878
Deguenon JM, Dhammi A, Ponnusamy L, Travanty NV, Cave G, Lawrie R, Mott D, Reisig D, Kurtz R, Roe RM. Bacterial Microbiota of Field-Collected Helicoverpa zea (Lepidoptera: Noctuidae) from Transgenic Bt and Non-Bt Cotton. Microorganisms. 2021; 9(4):878. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9040878
Chicago/Turabian StyleDeguenon, Jean M., Anirudh Dhammi, Loganathan Ponnusamy, Nicholas V. Travanty, Grayson Cave, Roger Lawrie, Dan Mott, Dominic Reisig, Ryan Kurtz, and R. Michael Roe. 2021. "Bacterial Microbiota of Field-Collected Helicoverpa zea (Lepidoptera: Noctuidae) from Transgenic Bt and Non-Bt Cotton" Microorganisms 9, no. 4: 878. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9040878