
Molecular Analysis of SARS-CoV-2 Circulating in Bangladesh during 2020 Revealed Lineage Diversity and Potential Mutations

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Sequence Metadata Retrieval and Data Curation
2.2. Phylogenetic Analysis
2.3. Evaluation of Deduced Amino Acid Substitutions
3. Results
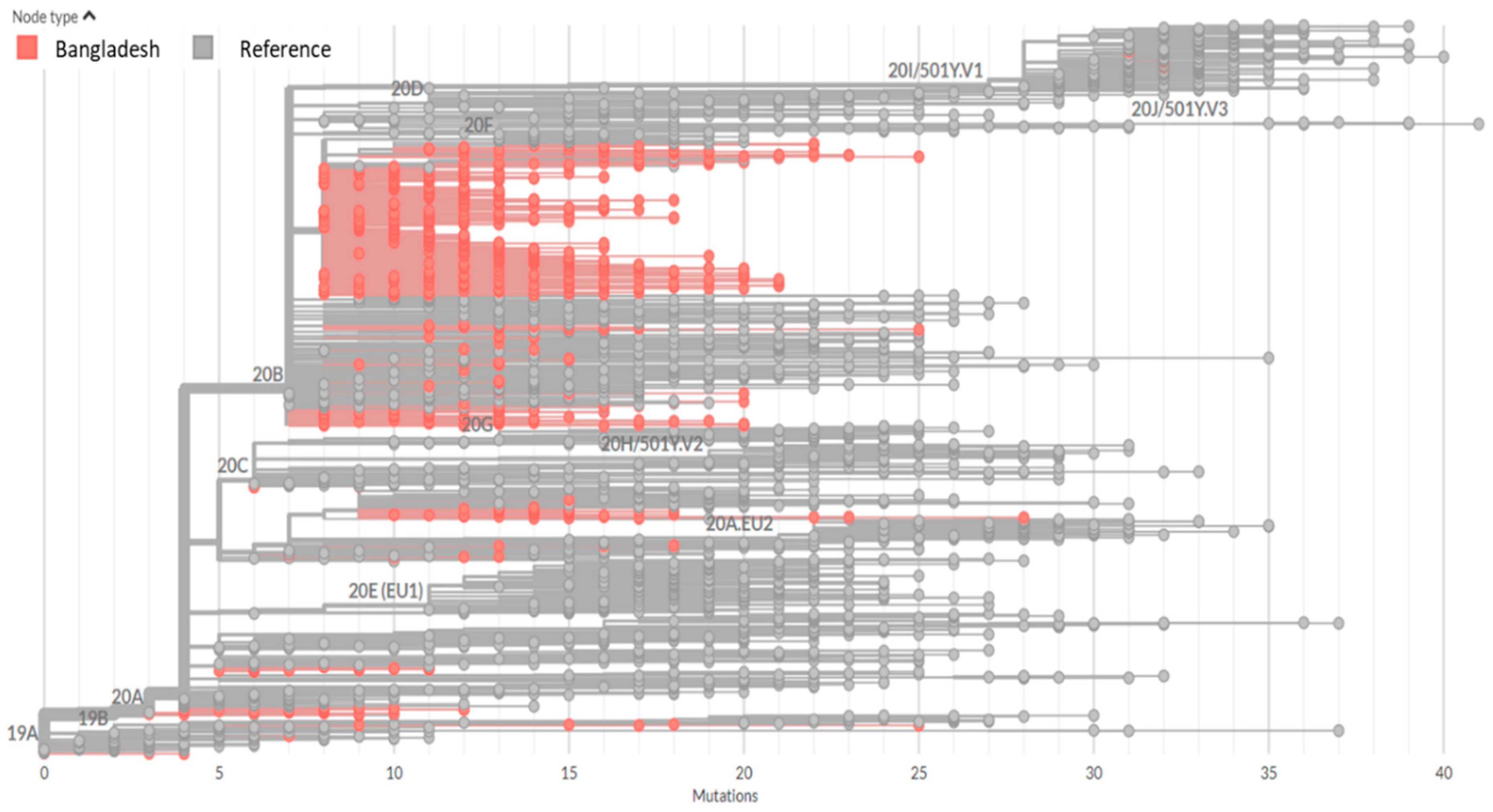
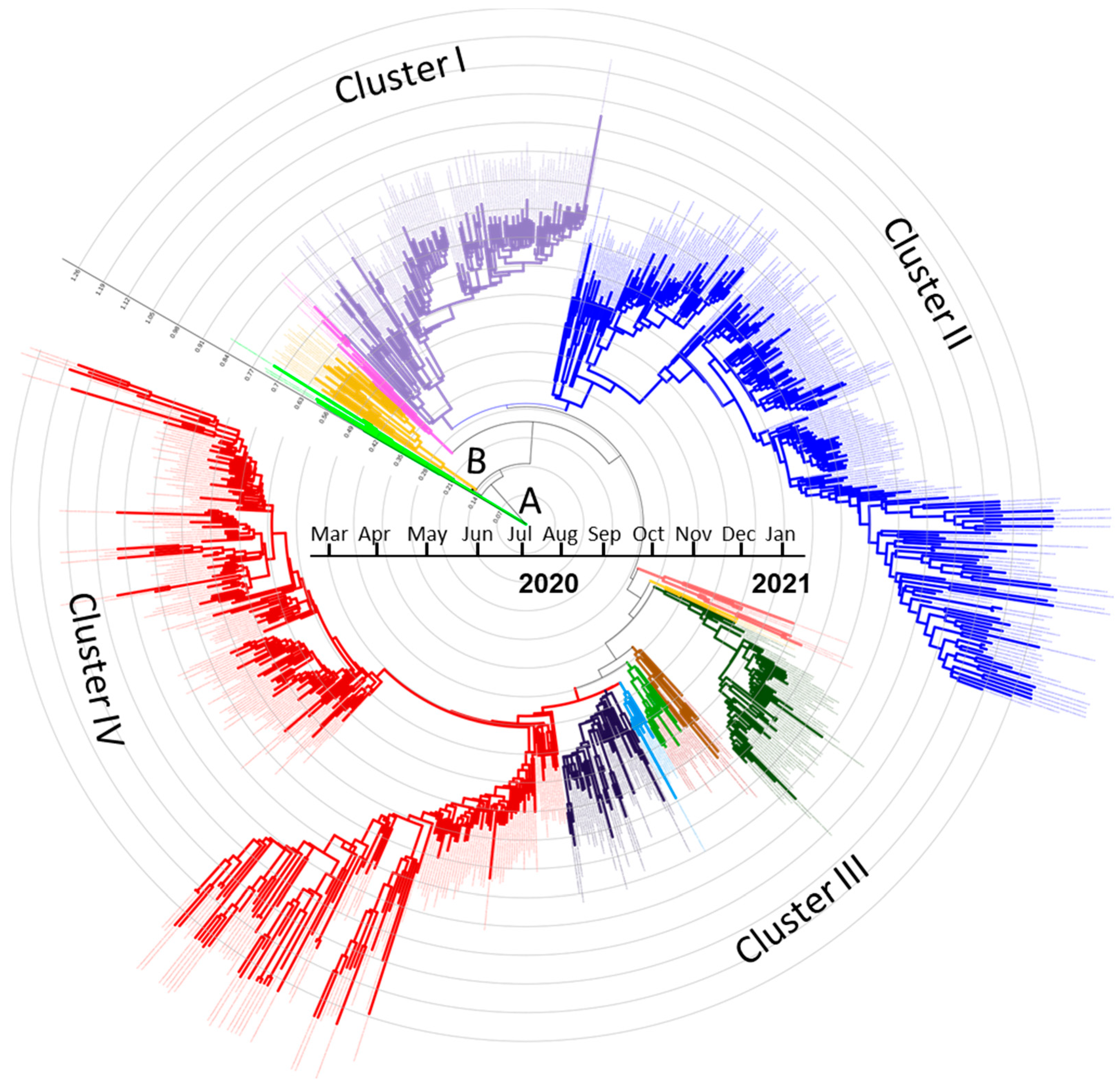
3.1. Phylogenetic Clusters and Evolutionary Relationship Analysis
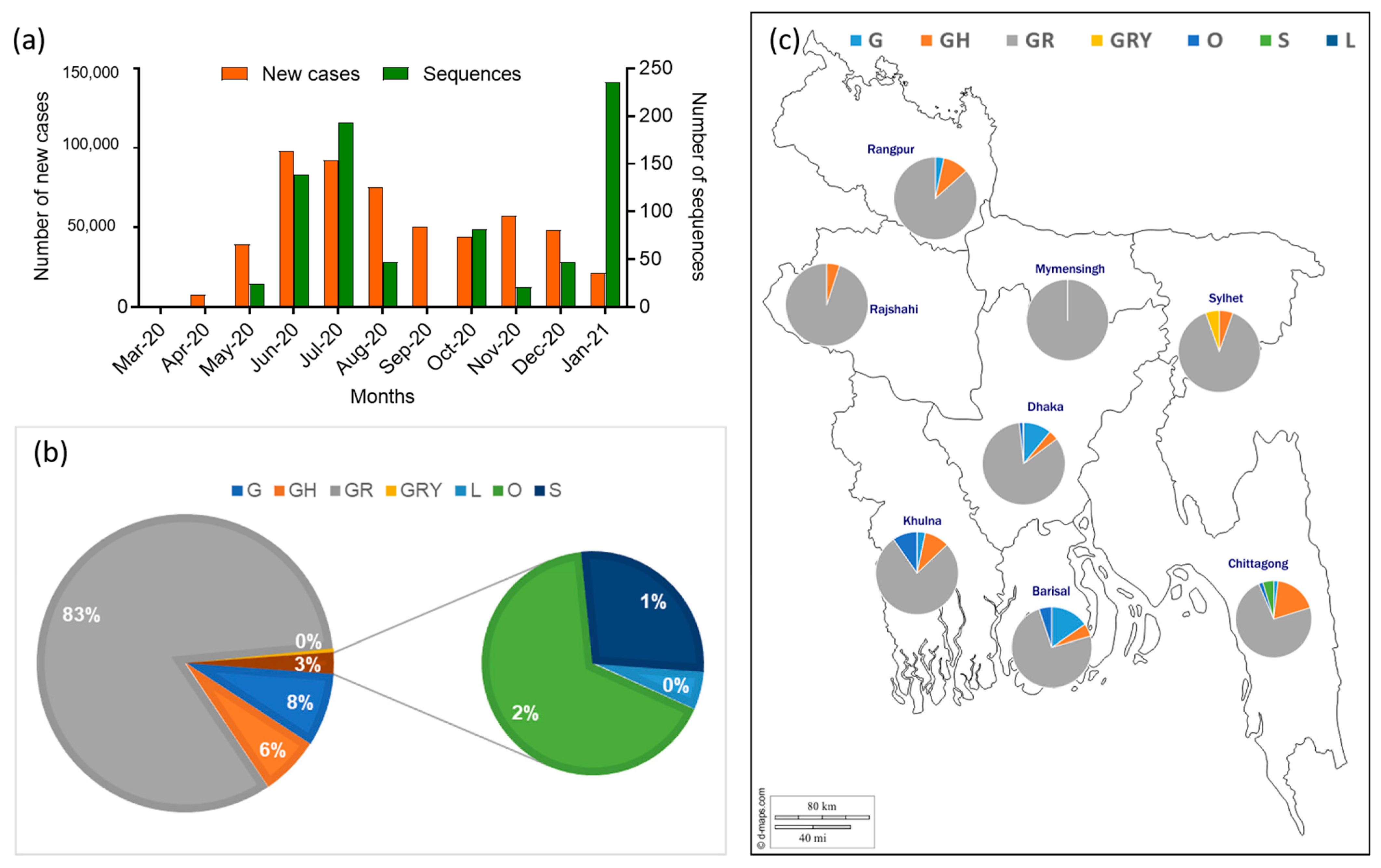
3.2. Clades and Lineage Distribution of Bangladeshi SARS-CoV-2 Strains
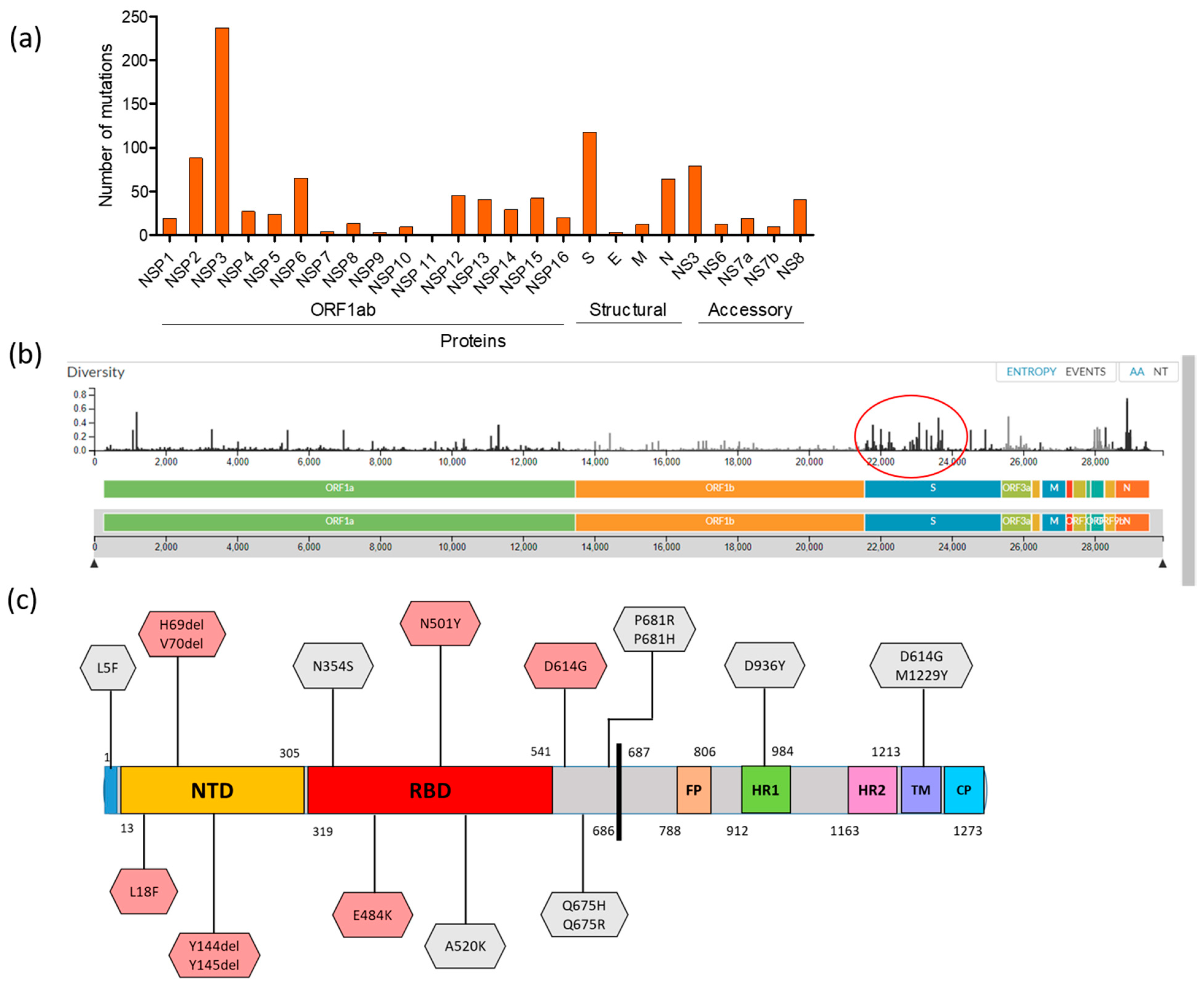
3.3. Mutational Dimension and Variant Determination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.; To, K.K.; Tse, H.; Jin, D.Y.; Yuen, K.Y. Interspecies transmission and emergence of novel viruses: Lessons from bats and birds. Trends Microbiol. 2013, 21, 544–555. [Google Scholar] [CrossRef]
- Chan, J.F.; Lau, S.K.; To, K.K.; Cheng, V.C.; Woo, P.C.; Yuen, K.Y. Middle East respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef] [Green Version]
- Cheng, V.C.; Lau, S.K.; Woo, P.C.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 2007, 20, 660–694. [Google Scholar] [CrossRef] [Green Version]
- Helmy, Y.A.; Fawzy, M.; Elaswad, A.; Sobieh, A.; Kenney, S.P. The COVID-19 Pandemic: A comprehensive review of taxonomy, genetics, epidemiology, diagnosis, treatment, and control. J. Clin. Med. 2020, 9, 1225. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hu, J.; Wang, J.; Han, Y.; Hu, Y.; Wen, J.; Li, Y.; Ji, J.; Ye, J.; Zhang, Z.; et al. Genome organization of the SARS-CoV. Genom. Proteom. Bioinform. 2003, 1, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294.e1289. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Callaway, E. ‘A bloody mess’: Confusion reigns over naming of new COVID variants. Nature 2021, 589, 339. [Google Scholar] [CrossRef] [PubMed]
- Burki, T. Understanding variants of SARS-CoV-2. Lancet 2021, 397, 462. [Google Scholar] [CrossRef]
- Lu, J.; du Plessis, L.; Liu, Z.; Hill, V.; Kang, M.; Lin, H.; Sun, J.; François, S.; Kraemer, M.U.G.; Faria, N.R.; et al. Genomic epidemiology of SARS-CoV-2 in Guangdong province, China. Cell 2020, 181, 997–1003.e1009. [Google Scholar] [CrossRef] [PubMed]
- Banu, S.; Jolly, B.; Mukherjee, P.; Singh, P.; Khan, S.; Zaveri, L.; Shambhavi, S.; Gaur, N.; Reddy, S.; Kaveri, K.; et al. A distinct phylogenetic cluster of Indian severe acute respiratory syndrome coronavirus 2 isolates. Open Forum Infect. Dis. 2020, 7, ofaa434. [Google Scholar] [CrossRef] [PubMed]
- Candido, D.S.; Claro, I.M. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 2020, 369, 1255–1260. [Google Scholar] [CrossRef]
- Lemoine, F.; Correia, D.; Lefort, V.; Doppelt-Azeroual, O.; Mareuil, F.; Cohen-Boulakia, S.; Gascuel, O. NGPhylogeny.fr: New generation phylogenetic services for non-specialists. Nucleic Acids Res. 2019, 47, W260–W265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemoine, F.; Domelevo Entfellner, J.B.; Wilkinson, E.; Correia, D.; Dávila Felipe, M.; De Oliveira, T.; Gascuel, O. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 2018, 556, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Junier, T.; Zdobnov, E.M. The Newick utilities: High-throughput phylogenetic tree processing in the UNIX shell. Bioinformatics 2010, 26, 1669–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, N.; Dey, S.K. Phylogenetic and whole genome analysis of first seven SARS-CoV-2 isolates in Bangladesh. Future Virol. 2020, 15. [Google Scholar] [CrossRef]
- Parvez, M.S.A.; Rahman, M.M.; Morshed, M.N.; Rahman, D.; Anwar, S.; Hosen, M.J. Genetic analysis of SARS-CoV-2 isolates collected from Bangladesh: Insights into the origin, mutational spectrum and possible pathomechanism. Comput. Biol. Chem. 2021, 90, 107413. [Google Scholar] [CrossRef] [PubMed]
- Shishir, T.A.; Naser, I.B. In silico comparative genomics of SARS-CoV-2 to determine the source and diversity of the pathogen in Bangladesh. PLoS ONE 2021, 16, e0245584. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e819. [Google Scholar] [CrossRef]
- Conceicao, C.; Thakur, N. The SARS-CoV-2 Spike protein has a broad tropism for mammalian ACE2 proteins. PLoS Biol. 2020, 18, e3001016. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020, 182, 1295–1310.e1220. [Google Scholar] [CrossRef]
- Wise, J. Covid-19: The E484K mutation and the risks it poses. BMJ 2021, 372, n359. [Google Scholar] [CrossRef] [PubMed]
- Jangra, S.; Ye, C.; Rathnasinghe, R.; Stadlbauer, D.; Krammer, F.; Simon, V.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Schotsaert, M. The E484K mutation in the SARS-CoV-2 spike protein reduces but does not abolish neutralizing activity of human convalescent and post-vaccination sera. medRxiv 2021. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Grant, O.C.; Montgomery, D.; Ito, K.; Woods, R.J. Analysis of the SARS-CoV-2 spike protein glycan shield reveals implications for immune recognition. Sci. Rep. 2020, 10, 14991. [Google Scholar] [CrossRef]
- Badaoui, B.; Sadki, K.; Talbi, C.; Salah, D.; Tazi, L. Genetic diversity and genomic epidemiology of SARS-CoV-2 in Morocco. Biosaf. Health 2021. [Google Scholar] [CrossRef]
- Maitra, A.; Sarkar, M.C.; Raheja, H.; Biswas, N.K.; Chakraborti, S.; Singh, A.K.; Ghosh, S.; Sarkar, S.; Patra, S.; Mondal, R.K.; et al. Mutations in SARS-CoV-2 viral RNA identified in Eastern India: Possible implications for the ongoing outbreak in India and impact on viral structure and host susceptibility. J. Biosci. 2020, 45, 76. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clades | Lineages | Number of Strains | Percentage | Time of Introduction | Present Status |
|---|---|---|---|---|---|
| G | A | 63 | 7.9% | 07-03-2020 | 14-07-2020 |
| B | |||||
| B.1 | |||||
| GH | B.1 | 51 | 6.4% | 21-03-2020 | 23-11-2020 |
| B.1.159 | |||||
| B.1.260 | |||||
| B.1.36 | |||||
| B.1.36.16 | |||||
| GR | B.1.1.10 | 656 | 82.9% | 18-04-2020 | 31-01-2021 Circulating |
| B.1.1.103 | |||||
| B.1.1.107 | |||||
| B.1.1.12 | |||||
| B.1.1.127 | |||||
| B.1.1.128 | |||||
| B.1.1.145 | |||||
| B.1.1.175 | |||||
| B.1.1.220 | |||||
| B.1.1.25 | |||||
| B.1.1.250 | |||||
| B.1.1.256 | |||||
| B.1.1.269 | |||||
| B.1.1.279 | |||||
| B.1.1.296 | |||||
| B.1.1.304 | |||||
| B.1.1.316 | |||||
| B.1.1.59 | |||||
| B.1.1.74 | |||||
| B.1.1.80 | |||||
| GRY | B.1.1.7 | 3 | 0.3% | 31-12-2020 | 31-01-2021 Circulating |
| L | B | 1 | 0.1% | 11-05-2020 | Not seen to date |
| O | B | 12 | 1.5% | 20-03-2020 | 15-07-2020 |
| B.1 | |||||
| B.1.1.25 | |||||
| B.1.1.316 | |||||
| B.1.1.74 | |||||
| B.1.36.16 | |||||
| B.40 | |||||
| S | A | 5 | 0.6% | 03-05-2020 | 13-05-2020 |
| 7 clades | 28 | 791 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parvin, R.; Afrin, S.Z.; Begum, J.A.; Ahmed, S.; Nooruzzaman, M.; Chowdhury, E.H.; Pohlmann, A.; Paul, S.K. Molecular Analysis of SARS-CoV-2 Circulating in Bangladesh during 2020 Revealed Lineage Diversity and Potential Mutations. Microorganisms 2021, 9, 1035. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9051035
Parvin R, Afrin SZ, Begum JA, Ahmed S, Nooruzzaman M, Chowdhury EH, Pohlmann A, Paul SK. Molecular Analysis of SARS-CoV-2 Circulating in Bangladesh during 2020 Revealed Lineage Diversity and Potential Mutations. Microorganisms. 2021; 9(5):1035. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9051035
Chicago/Turabian StyleParvin, Rokshana, Sultana Zahura Afrin, Jahan Ara Begum, Salma Ahmed, Mohammed Nooruzzaman, Emdadul Haque Chowdhury, Anne Pohlmann, and Shyamal Kumar Paul. 2021. "Molecular Analysis of SARS-CoV-2 Circulating in Bangladesh during 2020 Revealed Lineage Diversity and Potential Mutations" Microorganisms 9, no. 5: 1035. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9051035