
Annexin Induces Cellular Uptake of Extracellular Vesicles and Delays Disease in Escherichia coli O157:H7 Infection


Abstract
:1. Introduction
2. Methods
2.1. Cell Cultures
2.2. Generation of Fluorescent Blood Cell-Derived Extracellular Vesicles
2.3. Generation of Fluorescent HeLa Cell-Derived EVs
2.4. Extracellular Vesicle Uptake
2.5. Quantification of Extracellular Vesicle Uptake by Cells
2.6. Phosphatidylserine Exposure on RAW264.7 Cells
2.7. Quantification of Phosphatidylserine Expression on Cells
2.8. Mice
2.9. Bacterial Strain
2.10. Escherichia coli O157:H7 Infection Protocol in Mice
2.11. Detection of Phosphatidylserine-Positive Murine Extracellular Vesicles
2.12. Treatment with anxA5
2.13. Blood Collection
2.14. Blood Urea Nitrogen Measurement
2.15. Quantification of anxA5 Concentration in Mouse Plasma
2.16. Quantification of Platelet-Derived Murine EVs
2.17. Immunofluorescence Staining of Murine Kidney
2.18. Stx2 Detection in Mouse Kidneys
2.19. Interaction of anxA5 with Stx2
2.20. Interaction of anxA5 with E. coli O157 Lipopolysaccharide
2.21. Cytotoxicity/Viability Assay
2.22. Statistical Analysis
3. Results
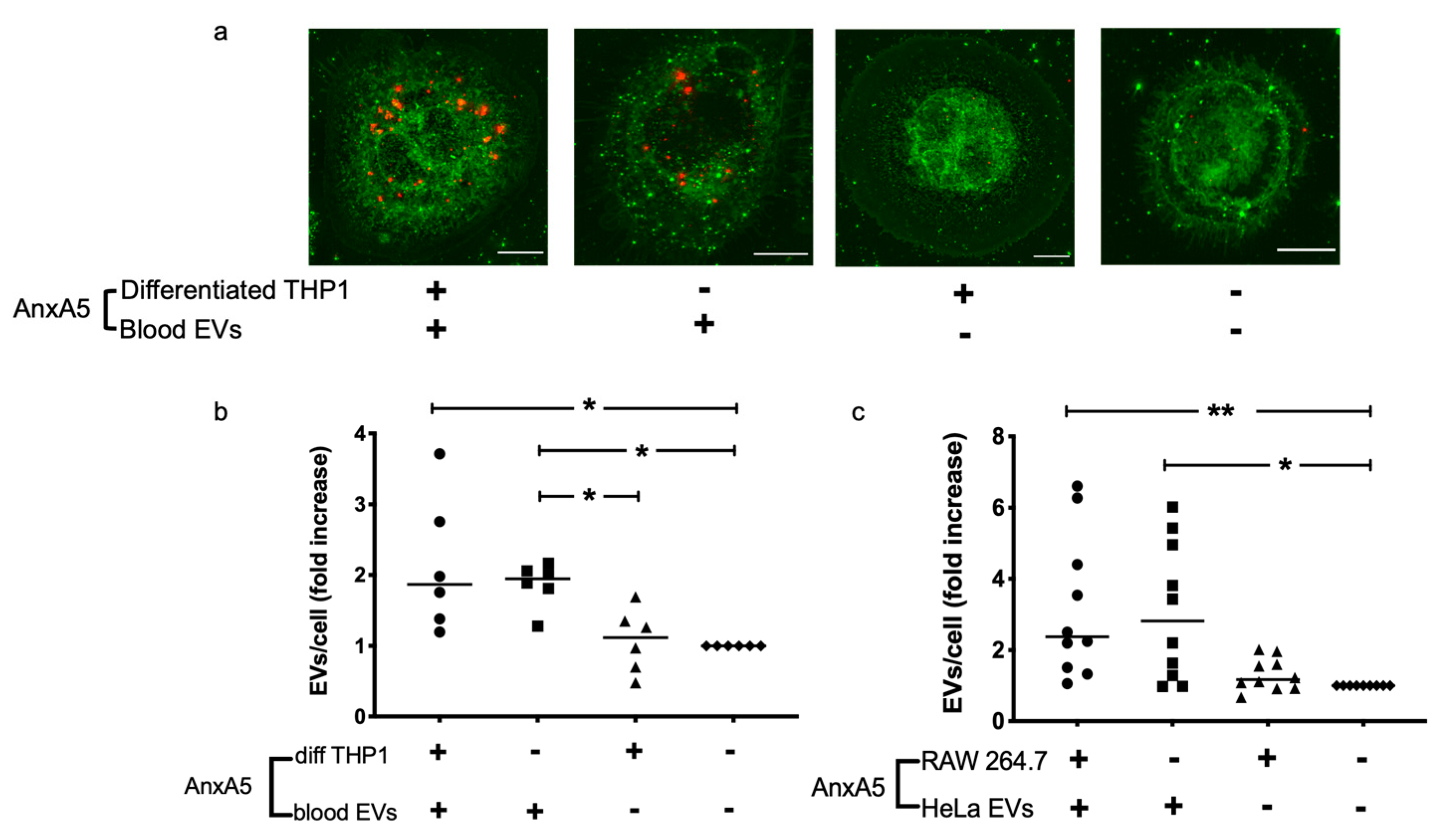
3.1. AnxA5 Increases Cellular Uptake of Extracellular Vesicles
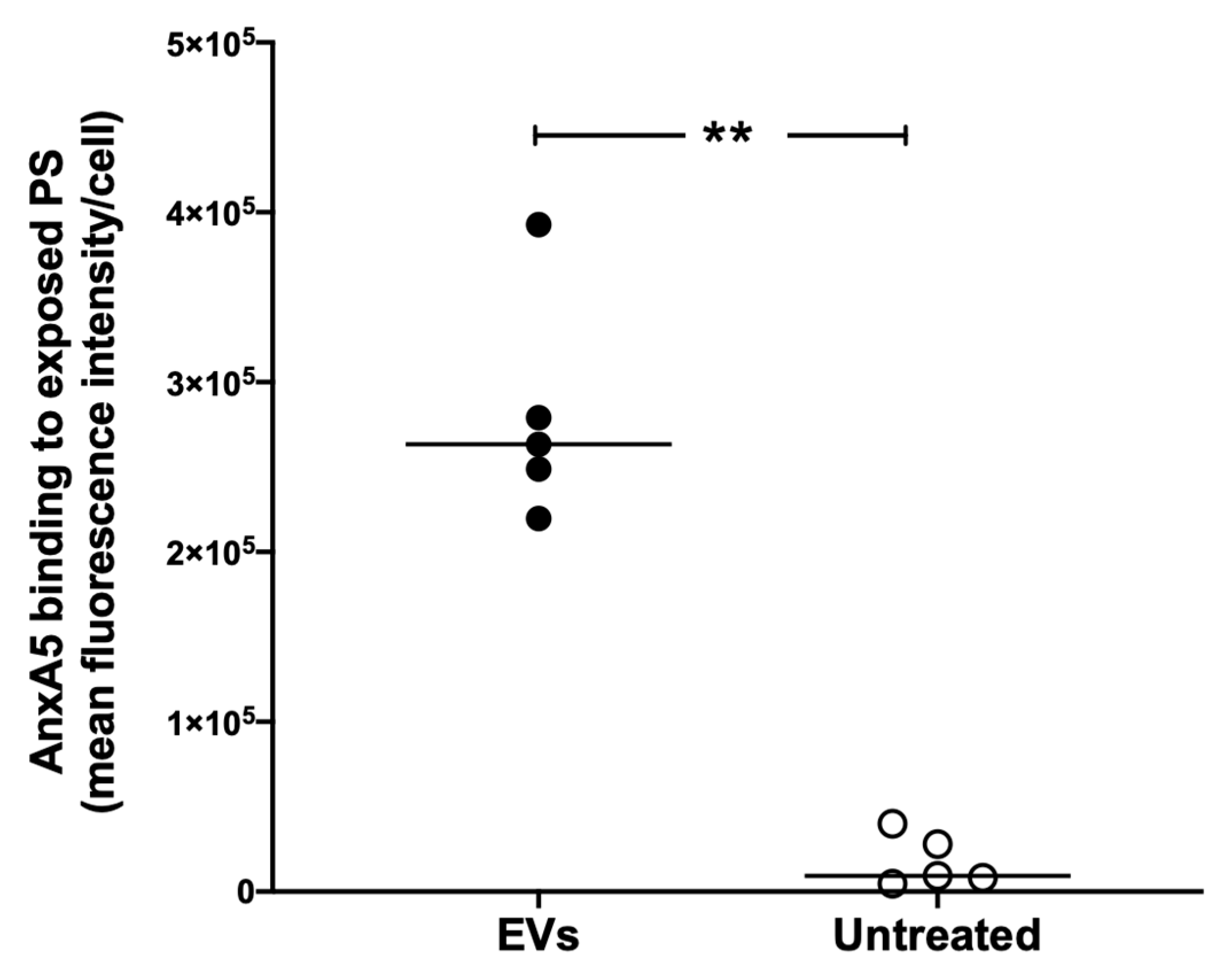
3.2. Extracellular Vesicles Induce Excess anxA5 Binding to RAW264.7 Cells
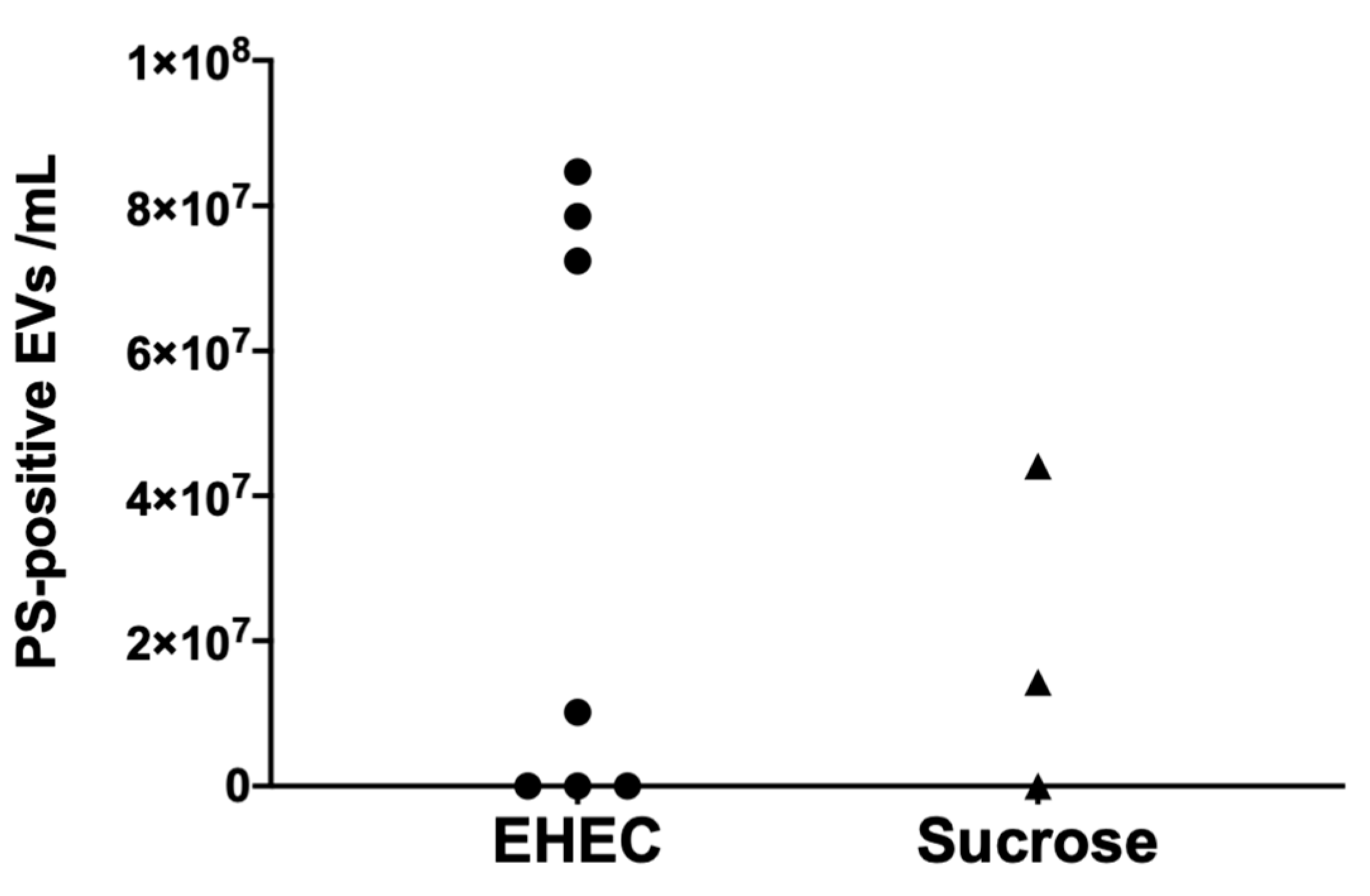
3.3. Murine Extracellular Vesicles Express Phosphatidylserine
3.4. AnxA5 Treatment Delayed Disease Symptoms in Murine E. coli O157:H7 Infection
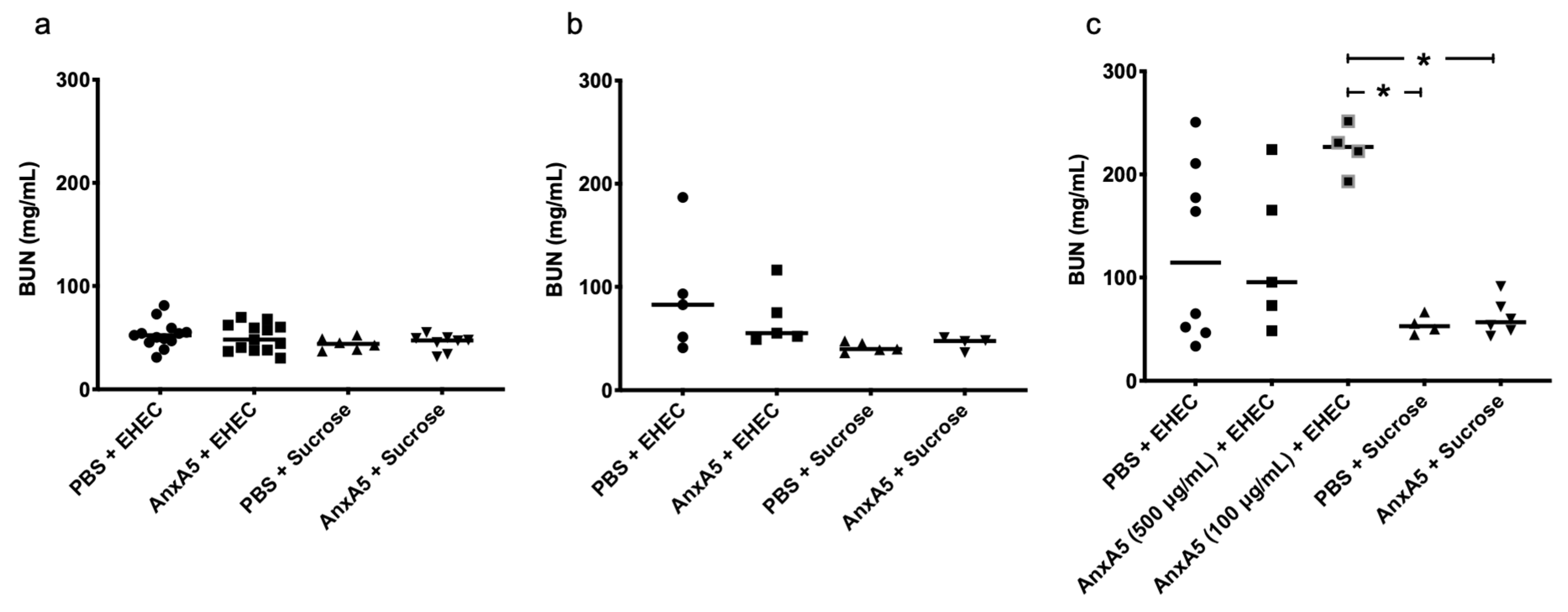
3.5. Blood Urea Nitrogen Levels in E. coli O157:H7-Infected Mice
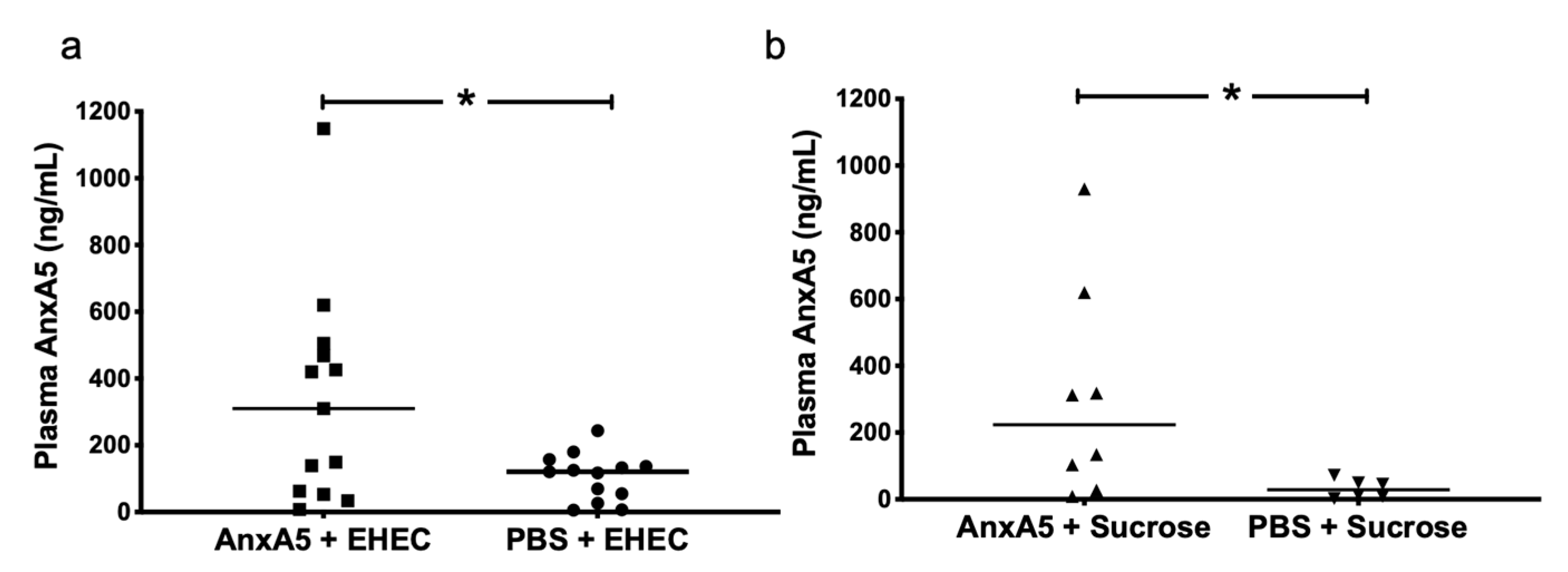
3.6. AnxA5 Detection in Mouse Plasma
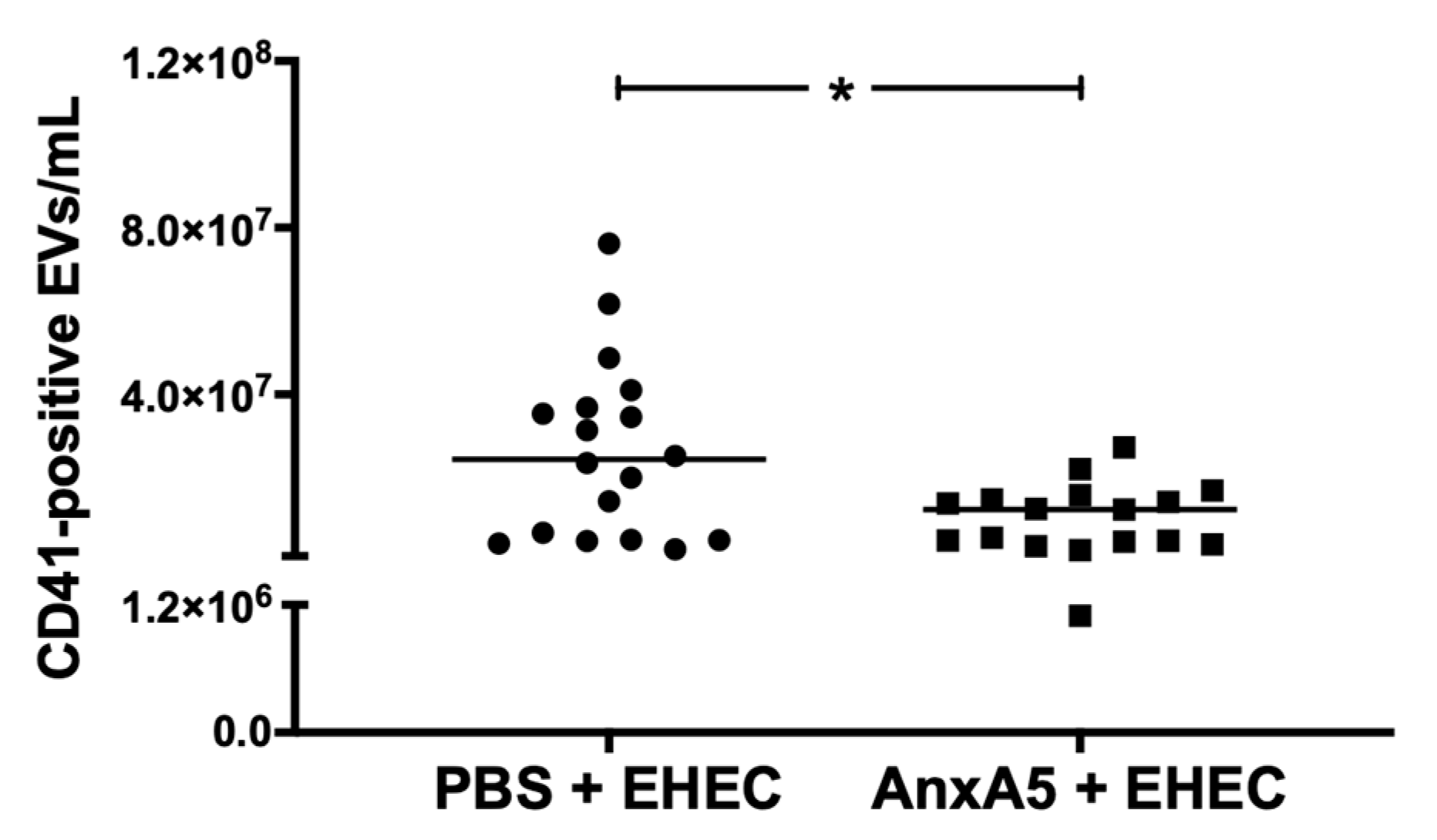
3.7. AnxA5 Lowered Circulating Platelet-Derived Extracellular Vesicles
3.8. Fibrinogen Deposition in Murine Kidneys
3.9. Stx2 Was Not Detectable in Murine Kidneys
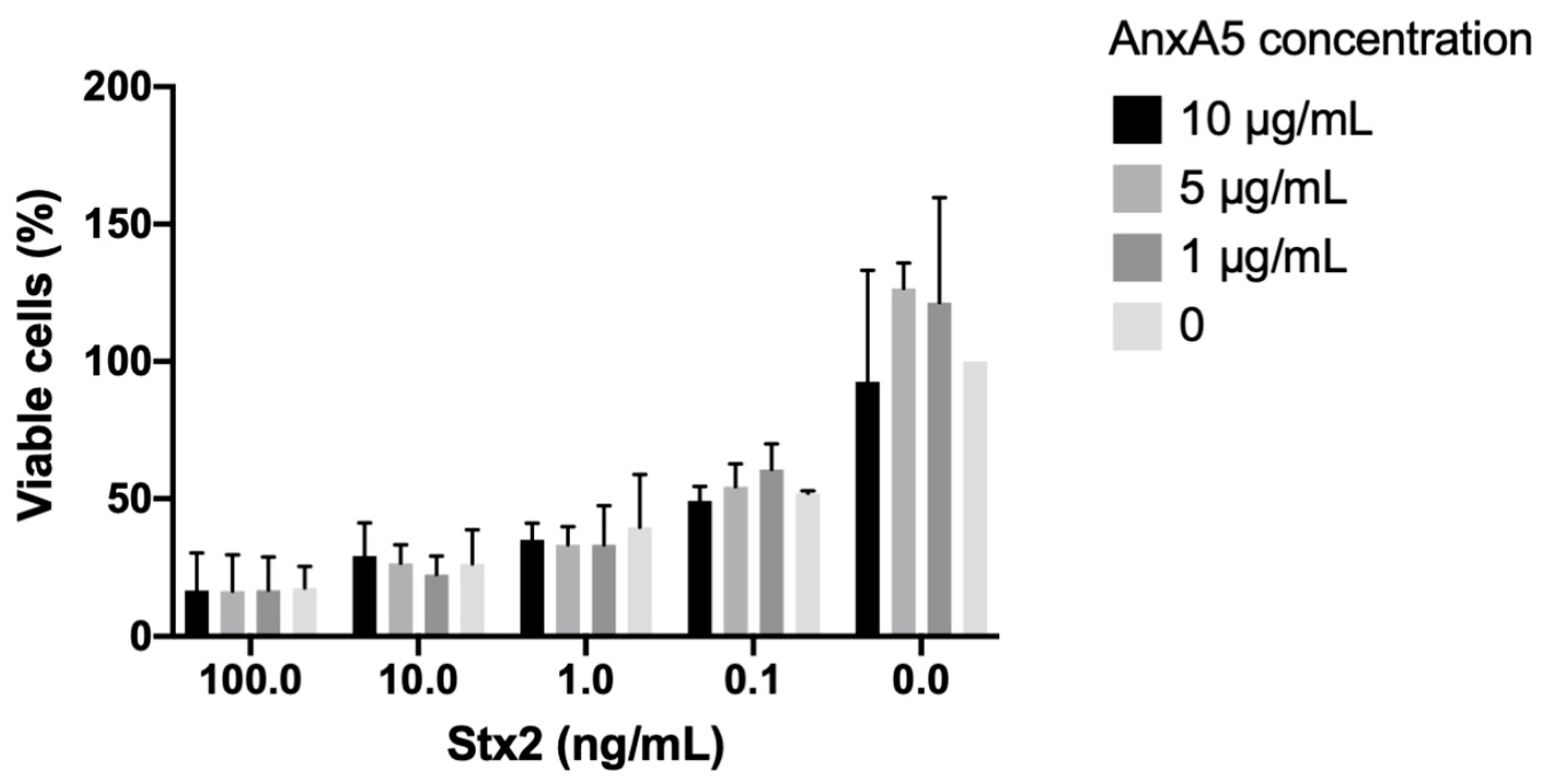
3.10. AnxA5 Did Not Bind to Stx2 or O157LPS and Did Not Protect HeLa Cells from Stx2-Induced Cytotoxicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lizarbe, M.A.; Barrasa, J.I.; Olmo, N.; Gavilanes, F.; Turnay, J. Annexin-phospholipid interactions. Functional implications. Int. J. Mol. Sci. 2013, 14, 2652–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meers, P.; Daleke, D.; Hong, K.; Papahadjopoulos, D. Interactions of annexins with membrane phospholipids. Biochemistry 1991, 30, 2903–2908. [Google Scholar] [CrossRef]
- Stewart, S.E.; Ashkenazi, A.; Williamson, A.; Rubinsztein, D.C.; Moreau, K. Transbilayer phospholipid movement facilitates the translocation of annexin across membranes. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.C.; Chipot, C.; Scheuring, S. Annexin-V stabilizes membrane defects by inducing lipid phase transition. Nat. Commun. 2020, 11, 230. [Google Scholar] [CrossRef]
- Bouter, A.; Gounou, C.; Bérat, R.; Tan, S.; Gallois, B.; Granier, T.; d’Estaintot, B.L.; Pöschl, E.; Brachvogel, B.; Brisson, A.R. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat. Commun. 2011, 2, 270. [Google Scholar] [CrossRef] [Green Version]
- Gerke, V.; Moss, S.E. Annexins: From structure to function. Physiol. Rev. 2002, 82, 331–371. [Google Scholar] [CrossRef]
- Hayes, M.J.; Rescher, U.; Gerke, V.; Moss, S.E. Annexin-actin interactions. Traffic 2004, 5, 571–576. [Google Scholar] [CrossRef]
- Rogers, M.A.; Buffolo, F.; Schlotter, F.; Atkins, S.K.; Lee, L.H.; Halu, A.; Blaser, M.C.; Tsolaki, E.; Higashi, H.; Luther, K.; et al. Annexin A1-dependent tethering promotes extracellular vesicle aggregation revealed with single-extracellular vesicle analysis. Sci. Adv. 2020, 6, eabb1244. [Google Scholar] [CrossRef] [PubMed]
- Futter, C.E.; White, I.J. Annexins and endocytosis. Traffic 2007, 8, 951–958. [Google Scholar] [CrossRef]
- Creutz, C. The annexins and exocytosis. Science 1992, 258, 924–931. [Google Scholar] [CrossRef]
- Patel, D.M.; Ahmad, S.F.; Weiss, D.G.; Gerke, V.; Kuznetsov, S.A. Annexin A1 is a new functional linker between actin filaments and phagosomes during phagocytosis. J. Cell Sci. 2011, 124, 578–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, R.; Romisch, J.; Paques, E.P. The crystal and molecular structure of human annexin V, an anticoagulant protein that binds to calcium and membranes. EMBO J. 1990, 9, 3867–3874. [Google Scholar] [CrossRef] [PubMed]
- Leikina, E.; Melikov, K.; Sanyal, S.; Verma, S.K.; Eun, B.; Gebert, C.; Pfeifer, K.; Lizunov, V.A.; Kozlov, M.M.; Chernomordik, L.V. Extracellular annexins and dynamin are important for sequential steps in myoblast fusion. J. Cell Biol. 2013, 200, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Boersma, H.H.; Kietselaer, B.L.; Stolk, L.M.; Bennaghmouch, A.; Hofstra, L.; Narula, J.; Heidendal, G.A.; Reutelingsperger, C.P. Past, present, and future of annexin A5: From protein discovery to clinical applications. J. Nucl. Med. 2005, 46, 2035–2050. [Google Scholar]
- Dachary-Prigent, J.; Freyssinet, J.M.; Pasquet, J.M.; Carron, J.C.; Nurden, A.T. Annexin V as a probe of aminophospholipid exposure and platelet membrane vesiculation: A flow cytometry study showing a role for free sulfhydryl groups. Blood 1993, 81, 2554–2565. [Google Scholar] [CrossRef] [Green Version]
- Ståhl, A.L.; Vaziri-Sani, F.; Heinen, S.; Kristoffersson, A.C.; Gydell, K.H.; Raafat, R.; Gutierrez, A.; Beringer, O.; Zipfel, P.F.; Karpman, D. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 2008, 111, 5307–5315. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020, 21, 585–606. [Google Scholar] [CrossRef] [PubMed]
- Mir, B.; Goettsch, C. Extracellular vesicles as delivery vehicles of specific cellular cargo. Cells 2020, 9, 1601. [Google Scholar] [CrossRef]
- Ståhl, A.L.; Sartz, L.; Nelsson, A.; Békássy, Z.D.; Karpman, D. Shiga toxin and lipopolysaccharide induce platelet-leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS ONE 2009, 4, e6990. [Google Scholar] [CrossRef] [Green Version]
- McKee, M.L.; O’Brien, A.D. Investigation of enterohemorrhagic Escherichia coli O157:H7 adherence characteristics and invasion potential reveals a new attachment pattern shared by intestinal E. coli. Infect. Immun. 1995, 63, 2070–2074. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, A.D.; Newland, J.W.; Miller, S.F.; Holmes, R.K.; Smith, H.W.; Formal, S.B. Shiga-like toxin-converting phages from Escherichia coli strains that cause hemorrhagic colitis or infantile diarrhea. Science 1984, 226, 694–696. [Google Scholar] [CrossRef]
- Karpman, D.; Loos, S.; Tati, R.; Arvidsson, I. Haemolytic uraemic syndrome. J. Intern. Med. 2017, 281, 123–148. [Google Scholar] [CrossRef]
- Ståhl, A.L.; Arvidsson, I.; Johansson, K.E.; Chromek, M.; Rebetz, J.; Loos, S.; Kristoffersson, A.C.; Békássy, Z.D.; Mörgelin, M.; Karpman, D. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. 2015, 11, e1004619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon Toledo, C.; Rogers, T.J.; Svensson, M.; Tati, R.; Fischer, H.; Svanborg, C.; Karpman, D. Shiga toxin-mediated disease in MyD88-deficient mice infected with Escherichia coli O157:H7. Am. J. Pathol. 2008, 173, 1428–1439. [Google Scholar] [CrossRef] [Green Version]
- Arvidsson, I.; Rebetz, J.; Loos, S.; Herthelius, M.; Kristoffersson, A.C.; Englund, E.; Chromek, M.; Karpman, D. Early terminal complement blockade and C6 deficiency are protective in enterohemorrhagic Escherichia coli-infected mice. J. Immunol. 2016, 197, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.D.; Melton, A.R.; Schmitt, C.K.; McKee, M.L.; Batts, M.L.; Griffin, D.E. Profile of Escherichia coli O157:H7 pathogen responsible for hamburger-borne outbreak of hemorrhagic colitis and hemolytic uremic syndrome in Washington. J. Clin. Microbiol. 1993, 31, 2799–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Békássy, Z.D.; Calderon Toledo, C.; Leoj, G.; Kristoffersson, A.; Leopold, S.R.; Perez, M.T.; Karpman, D. Intestinal damage in enterohemorrhagic Escherichia coli infection. Pediatric Nephrol. 2011, 26, 2059–2071. [Google Scholar] [CrossRef] [Green Version]
- Calderon Toledo, C.; Arvidsson, I.; Karpman, D. Cross-reactive protection against enterohemorrhagic Escherichia coli infection by enteropathogenic E. coli in a mouse model. Infect. Immun. 2011, 79, 2224–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.; Ansari, D.; Pawłowski, K.; Zhou, Q.; Sasor, A.; Welinder, C.; Kristl, T.; Bauden, M.; Rezeli, M.; Jiang, Y.; et al. Proteomic analyses identify prognostic biomarkers for pancreatic ductal adenocarcinoma. Oncotarget 2018, 9, 9789–9807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutelingsperger, C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Arvidsson, I.; Ståhl, A.L.; Hedström, M.M.; Kristoffersson, A.C.; Rylander, C.; Westman, J.S.; Storry, J.R.; Olsson, M.L.; Karpman, D. Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J. Immunol. 2015, 194, 2309–2318. [Google Scholar] [CrossRef] [Green Version]
- Willysson, A.; Ståhl, A.L.; Gillet, D.; Barbier, J.; Cintrat, J.C.; Chambon, V.; Billet, A.; Johannes, L.; Karpman, D. Shiga toxin uptake and sequestration in extracellular vesicles is mediated by its B-subunit. Toxins 2020, 12, 449. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Hering, U.; Zschörnig, O.; Arnold, K. Annexin V interaction with phosphatidylserine-containing vesicles at low and neutral pH. Biochemistry 1997, 36, 8189–8194. [Google Scholar] [CrossRef] [PubMed]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef]
- Umbrecht-Jenck, E.; Demais, V.; Calco, V.; Bailly, Y.; Bader, M.F.; Chasserot-Golaz, S. S100A10-mediated translocation of annexin-A2 to SNARE proteins in adrenergic chromaffin cells undergoing exocytosis. Traffic 2010, 11, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Rentero, C.; Blanco-Muñoz, P.; Meneses-Salas, E.; Grewal, T.; Enrich, C. Annexins-coordinators of cholesterol homeostasis in endocytic pathways. Int. J. Mol. Sci. 2018, 19, 1444. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, C.; Shah, N.; Ishii, M.; Annamalai, B.; Brandon, C.; Rodgers, J.; Nowling, T.; Rohrer, B. Mechanisms of extracellular vesicle uptake in stressed retinal pigment epithelial cell monolayers. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165608. [Google Scholar] [CrossRef]
- Yu, C.; Muñoz, L.E.; Mallavarapu, M.; Herrmann, M.; Finnemann, S.C. Annexin A5 regulates surface αvβ5 integrin for retinal clearance phagocytosis. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; Lopez, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liu, C.; Wang, H.; Wang, L.; Xiao, F.; Guo, Z.; Zhang, H. Surface phosphatidylserine is responsible for the internalization on microvesicles derived from hypoxia-induced human bone marrow mesenchymal stem cells into human endothelial cells. PLoS ONE 2016, 11, e0147360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, D.J.; Speth, J.M.; Penke, L.R.; Wettlaufer, S.H.; Swanson, J.A.; Peters-Golden, M. Mechanisms and modulation of microvesicle uptake in a model of alveolar cell communication. J. Biol. Chem. 2017, 292, 20897–20910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Genderen, H.O.; Kenis, H.; Hofstra, L.; Narula, J.; Reutelingsperger, C.P. Extracellular annexin A5: Functions of phosphatidylserine-binding and two-dimensional crystallization. Biochim. Biophys. Acta 2008, 1783, 953–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenis, H.; van Genderen, H.; Bennaghmouch, A.; Rinia, H.A.; Frederik, P.; Narula, J.; Hofstra, L.; Reutelingsperger, C.P. Cell surface-expressed phosphatidylserine and annexin A5 open a novel portal of cell entry. J. Biol. Chem. 2004, 279, 52623–52629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, A.P.; Mackman, N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Ravassa, S.; Bennaghmouch, A.; Kenis, H.; Lindhout, T.; Hackeng, T.; Narula, J.; Hofstra, L.; Reutelingsperger, C. Annexin A5 down-regulates surface expression of tissue factor: A novel mechanism of regulating the membrane receptor repertoir. J. Biol. Chem. 2005, 280, 6028–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesh, V.L.; Burris, J.A.; Owens, J.W.; Gordon, V.M.; Wadolkowski, E.A.; O’Brien, A.D.; Samuel, J.E. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 1993, 61, 3392–3402. [Google Scholar] [CrossRef] [Green Version]
- Ewing, M.M.; de Vries, M.R.; Nordzell, M.; Pettersson, K.; de Boer, H.C.; van Zonneveld, A.J.; Frostegård, J.; Jukema, J.W.; Quax, P.H. Annexin A5 therapy attenuates vascular inflammation and remodeling and improves endothelial function in mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Jang, J.H.; Choi, E.J.; Kim, Y.S.; Lee, E.J.; Jung, I.D.; Han, H.D.; Wu, T.C.; Hung, C.F.; Kang, T.H.; et al. Annexin A5 increases survival in murine sepsis model by inhibiting HMGB1-mediated pro-inflammation and coagulation. Mol. Med. 2016, 22, 424–436. [Google Scholar] [CrossRef] [Green Version]
- Arnold, P.; Lu, X.; Amirahmadi, F.; Brandl, K.; Arnold, J.M.; Feng, Q. Recombinant human annexin A5 inhibits proinflammatory response and improves cardiac function and survival in mice with endotoxemia. Crit. Care Med. 2014, 42, e32–e41. [Google Scholar] [CrossRef]
- Ake, J.A.; Jelacic, S.; Ciol, M.A.; Watkins, S.L.; Murray, K.F.; Christie, D.L.; Klein, E.J.; Tarr, P.I. Relative nephroprotection during Escherichia coli O157:H7 infections: Association with intravenous volume expansion. Pediatrics 2005, 115, e673–e680. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EVs/Cells | THP1 | RAW 264.7 |
|---|---|---|
| Whole blood | + a | - |
| HeLa b | - | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tontanahal, A.; Arvidsson, I.; Karpman, D. Annexin Induces Cellular Uptake of Extracellular Vesicles and Delays Disease in Escherichia coli O157:H7 Infection. Microorganisms 2021, 9, 1143. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9061143
Tontanahal A, Arvidsson I, Karpman D. Annexin Induces Cellular Uptake of Extracellular Vesicles and Delays Disease in Escherichia coli O157:H7 Infection. Microorganisms. 2021; 9(6):1143. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9061143
Chicago/Turabian StyleTontanahal, Ashmita, Ida Arvidsson, and Diana Karpman. 2021. "Annexin Induces Cellular Uptake of Extracellular Vesicles and Delays Disease in Escherichia coli O157:H7 Infection" Microorganisms 9, no. 6: 1143. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9061143