
Emergence and Spread of B.1.1.7 Lineage in Primary Care and Clinical Impact in the Morbi-Mortality among Hospitalized Patients in Madrid, Spain

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Sequencing Analysis and Phylogenetic Analysis
2.3. Statistical analysis
3. Results
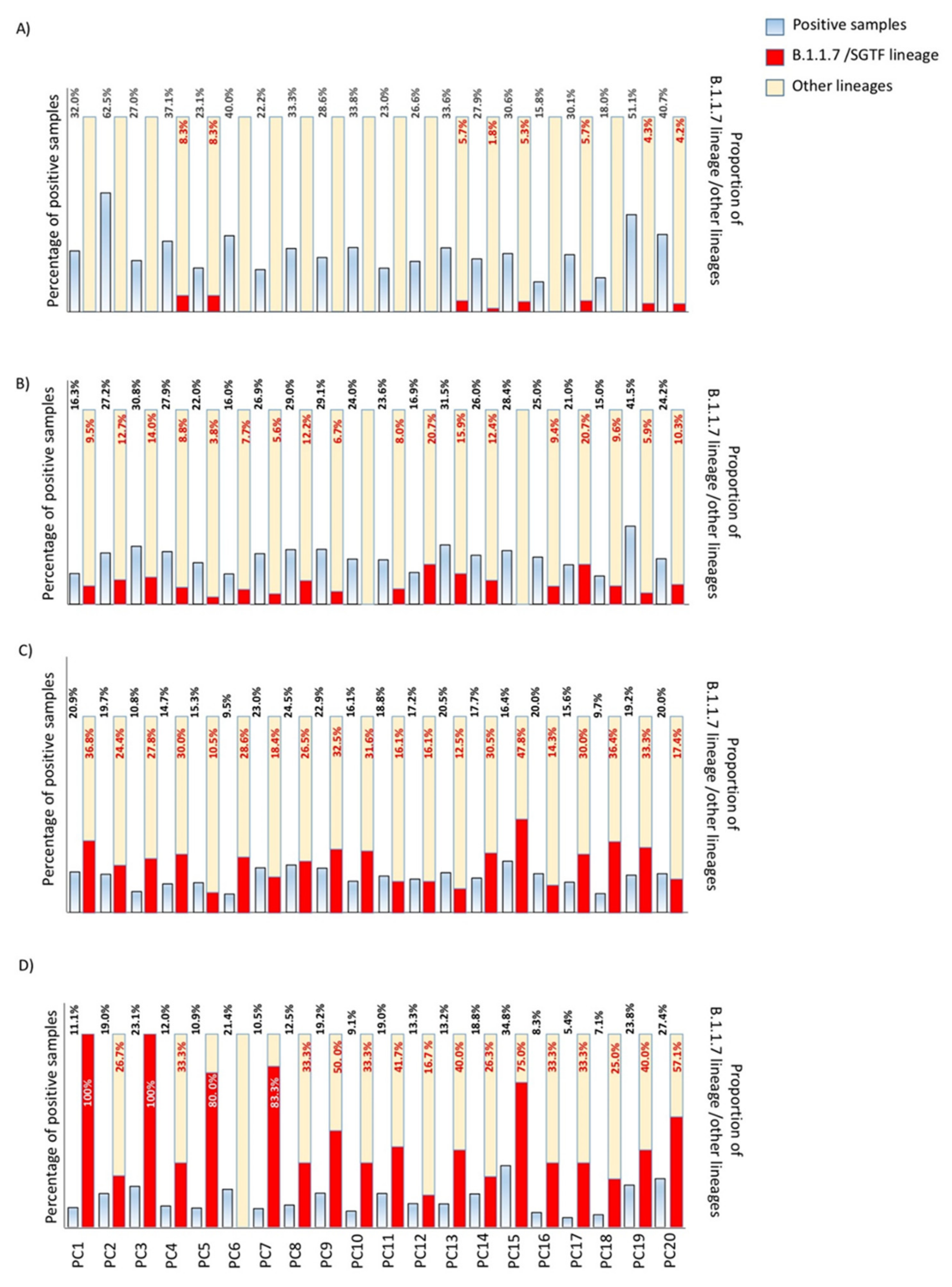
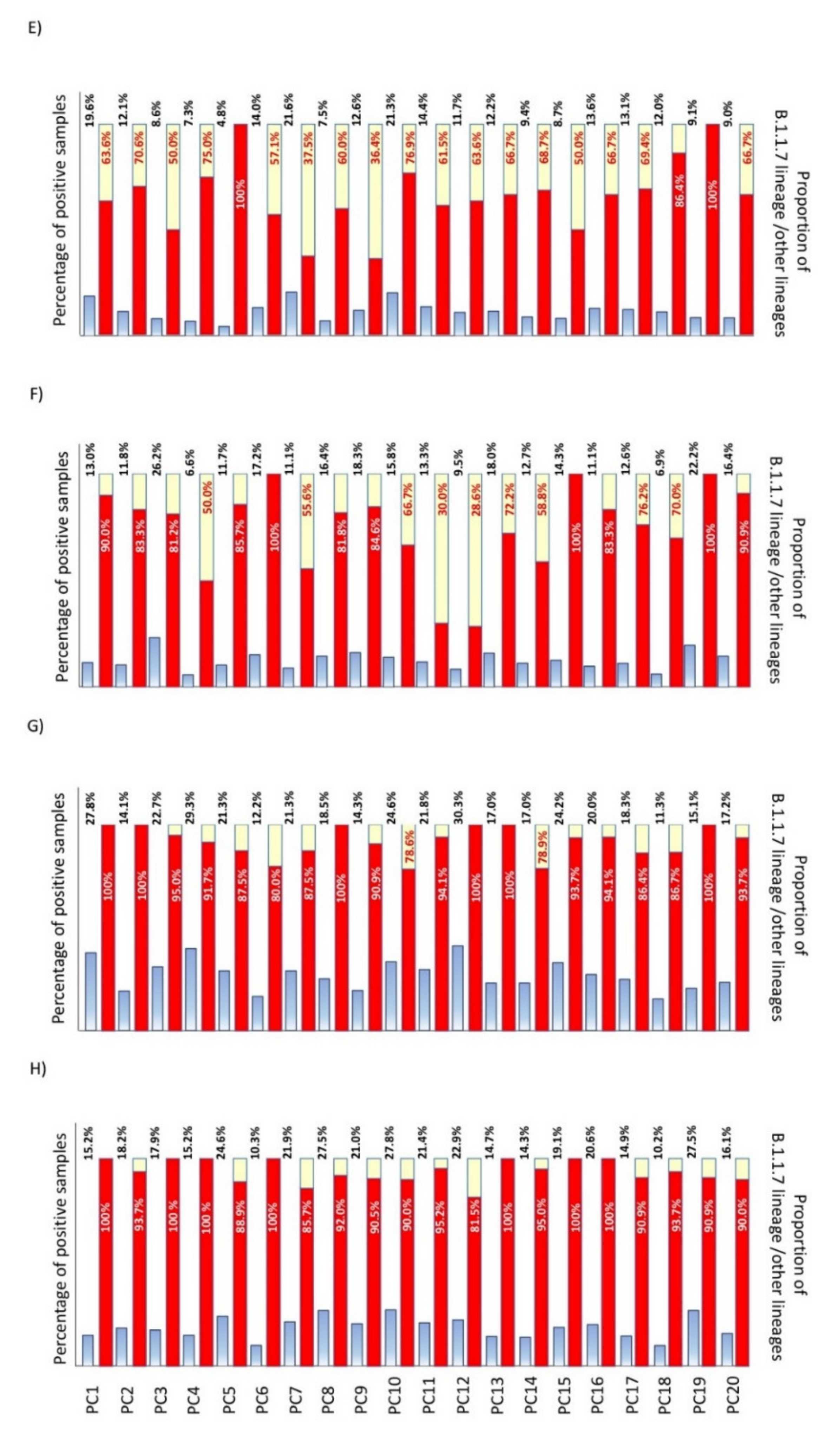
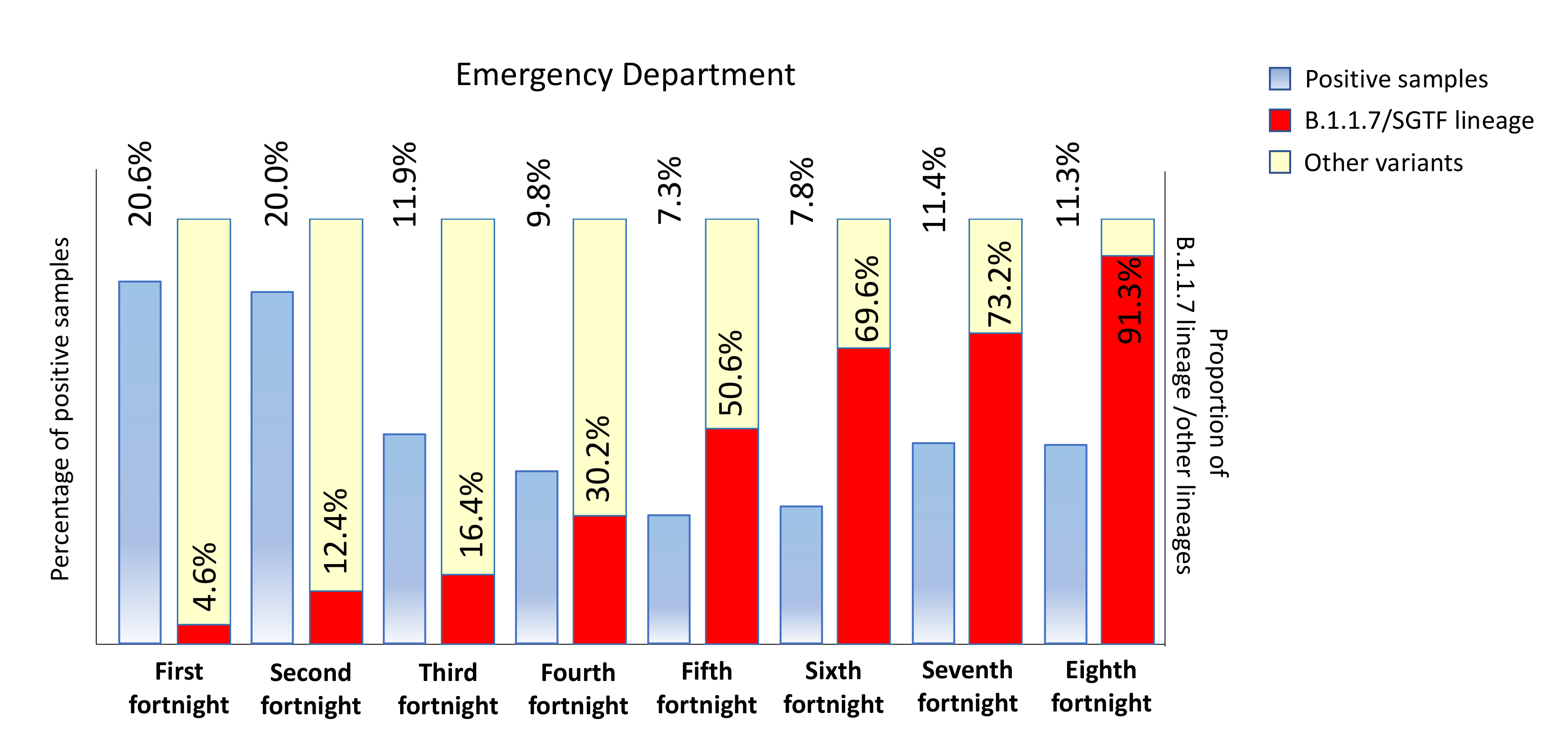
3.1. Replacement Rate of B.1.1.7/SGTF (Alpha Variant) in Outpatients in Our Healthcare Area
3.2. Burden of Disease in Hospitalized Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dow, A.W.; DiPiro, J.T.; Giddens, J.; Buckley, P.; Santen, S.A. Emerging From the COVID-19 Crisis With a Stronger Health Care Workforce. Acad. Med. 2020, 95, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Anon. Coronaviruses|NIH: National Institute of Allergy and Infectious Diseases. Available online: https://www.niaid.nih.gov/diseases-conditions/coronaviruses (accessed on 11 April 2021).
- Anon. Updated rapid risk assessment from ECDC on the risk related to the spread of new SARS-CoV-2 variants of concern in the EU/EEA—First update. Eurosurveillance 2021, 26. [Google Scholar] [CrossRef]
- Viedma, E.; Dahdouh, E.; González-Alba, J.M.; González-Bodi, S.; Martínez-García, L.; Lázaro-Perona, F.; Recio, R.; Rodríguez-Tejedor, M.; Folgueira, M.D.; Cantón, R.; et al. Genomic Epidemiology of SARS-CoV-2 in Madrid, Spain, during the First Wave of the Pandemic: Fast Spread and Early Dominance by D614G Variants. Microorganisms 2021, 9, 454. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, eabe8499-1468. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. medRxiv 2020. [Google Scholar] [CrossRef]
- Kirby, T. New variant of SARS-CoV-2 in UK causes surge of COVID-19. Lancet Respir. Med. 2021, 9, e20–e21. [Google Scholar] [CrossRef]
- Anon. Tracking the International Spread of SARS-CoV-2 Lineages B.1.1.7 and B.1.351/501Y-V2 SARS-CoV-2 Coronavirus / nCoV-2019 Genomic Epidemiology Virological. Available online: https://virological.org/t/tracking-the-international-spread-of-sars-cov-2-lineages-b-1-1-7-and-b-1-351-501y-v2/592 (accessed on 11 April 2021).
- Leung, K.; Shum, M.H.; Leung, G.M.; Lam, T.T.; Wu, J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eurosurveillance 2021, 26, 2002106. [Google Scholar] [CrossRef] [PubMed]
- Galloway, S.E.; Paul, P.; MacCannell, D.R.; Johansson, M.A.; Brooks, J.T.; MacNeil, A.; Slayton, R.B.; Tong, S.; Silk, B.J.; Armstrong, G.L.; et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage—United States, December 29, 2020–January 12, 2021. MMWR. Morb. Mortal. Wkly. Rep. 2021, 70, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef] [PubMed]
- Buenestado-Serrano, S.; Recio, R.; Campoy, P.J.S.; Catalán, P.; Folgueira, M.D.; Villa, J.; Gallego, I.M.; de la Cueva, V.M.; Meléndez, M.A.; Zayas, C.A.; et al. First confirmation of importation and transmission in Spain of the newly identified SARS-CoV-2 B.1.1.7 variant. Enferm. Infecc. Microbiol. Clín. 2021. [Google Scholar] [CrossRef] [PubMed]
- Anon. Artic Network. Available online: https://artic.network/ncov-2019 (accessed on 11 April 2021).
- Anon. PANGO Lineages. Available online: https://cov-lineages.org/pangolin_tutorial.html (accessed on 11 April 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- General de Sanidad; Consumo, D.E.S.Y. Centro de Coordinación de Alertas y Emergencias Sanitarias; Ministerio de Sanidad Gobierno de España: Madrid, Spain, 2020; pp. 1–2.
- Anon. COVID-19. Available online: https://cnecovid.isciii.es/covid19/ (accessed on 11 April 2021).
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nat. Cell Biol. 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J. Hum. Genet. 2020, 65, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Challen, R.; Brooks-Pollock, E.; Read, J.M.; Dyson, L.; Tsaneva-Atanasova, K.; Danon, L. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: Matched cohort study. BMJ 2021, 372, n579. [Google Scholar] [CrossRef] [PubMed]
- Frampton, D.; Rampling, T.; Cross, A. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B.1.1.7 lineage in London, UK: A whole-genome sequencing and hospital-based cohort study. Lancet Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Martina Patone, A.; Thomas, K.; Hatch, R. Analysis of severe outcomes associated with the SARS-CoV-2 Variant of Concern 202012/01 in England using ICNARC Case Mix Programme and QResearch databases. medRxiv 2021. [Google Scholar] [CrossRef]
- Funk, T.; Pharris, A.; Spiteri, G. Characteristics of SARS-CoV-2 variants of concern B.1.1.7, B.1.351 or P.1: Data from seven EU/EEA countries, weeks 38/2020 to 10/2021. Euro. Surveill. 2021, 26, 2100348. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J. Increased hazard of death in community-tested cases of SARS-CoV-2 Variant of Concern 202012/01. medRxiv 2021. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variable | Total of Patients | |||
|---|---|---|---|---|
| B.1.1.7/SGTF (n = 426) | No B.1.1.7 (n = 1129) | p | OR | |
| Age (median) | 68 | 71 | 0.004 | |
| IQR | (56–79) | (57–83) | ||
| Sex | ||||
| Women | 41.1% | 44.3% | 0.25 | |
| Men | 58.9% | 55.7% | ||
| ICU admission | 19.5% | 10.3% | 0.001 | 2.11 |
| (95%CI) | (15.83–23.57) | (8.56–12.19) | (1.55–2.87) | |
| IRCU admission | 8.7% | 4.4% | 0.001 | 2.05 |
| (95%CI) | (6.19–11.77) | (3.3–5.79) | (1.32–3.19) | |
| Death | 13.9% | 15.6% | 0.49 | 0.87 |
| (95%CI) | (10.52–17.95) | (13.47–17.87) | (0.62–1.23) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-García, L.; Espinel, M.A.; Abreu, M.; González-Alba, J.M.; Gijón, D.; McGee, A.; Cantón, R.; Galán, J.C.; Aranaz, J. Emergence and Spread of B.1.1.7 Lineage in Primary Care and Clinical Impact in the Morbi-Mortality among Hospitalized Patients in Madrid, Spain. Microorganisms 2021, 9, 1517. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071517
Martínez-García L, Espinel MA, Abreu M, González-Alba JM, Gijón D, McGee A, Cantón R, Galán JC, Aranaz J. Emergence and Spread of B.1.1.7 Lineage in Primary Care and Clinical Impact in the Morbi-Mortality among Hospitalized Patients in Madrid, Spain. Microorganisms. 2021; 9(7):1517. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071517
Chicago/Turabian StyleMartínez-García, Laura, Marco Antonio Espinel, Melanie Abreu, José María González-Alba, Desirèe Gijón, Amaranta McGee, Rafael Cantón, Juan Carlos Galán, and Jesús Aranaz. 2021. "Emergence and Spread of B.1.1.7 Lineage in Primary Care and Clinical Impact in the Morbi-Mortality among Hospitalized Patients in Madrid, Spain" Microorganisms 9, no. 7: 1517. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071517